 Nengqing Liu
Nengqing Liu Xiaojun Wen
Xiaojun Wen Zhanhui Ou1
Zhanhui Ou1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 10 August 2023
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1177019
X-Linked Alport Syndrome (XLAS) is an X-linked, dominant, hereditary nephropathy mainly caused by mutations in the COL4A5 gene, found on chromosome Xq22. In this study, we reported a pedigree with XLAS caused by a COL4A5 mutation. This family gave birth to a boy with XLAS who developed hematuria and proteinuria at the age of 1 year. We used next-generation sequencing (NGS) to identify mutations in the proband and his parents and confirmed the results using Sanger sequencing. This testing showed there was a single nucleotide missense variation, c.3659G>A (p.Gly1220Asp) (NM_033380.3), in the COL4A5 gene. To prevent the inheritance of the syndrome, we used eight embryos for trophoblast biopsy after assisted reproductive technology treatment, and whole genome amplification (WGA) was performed using multiple annealing and looping-based amplification cycles (MALBAC). Embryos were subjected to Preimplantation Genetic Testing (PGT) procedures, including Sanger sequencing, NGS-based single nucleotide polymorphism (SNP) haplotype linkage analysis, and chromosomal copy number variation (CNV) analysis. The results showed that three embryos (E1, E2, and E4) were free of CNV and genetic variation in the COL4A5 gene. Embryo E1 (4AA) was transferred after consideration of the embryo growth rate, morphology, and PGT results. Prenatal diagnosis in the second trimester showed that the fetus had a normal karyotype and did not carry the COL4A5 mutation (c.3659G>A). Ultimately, a healthy boy was born and did not carry the pathogenic COL4A5 mutation, which indicated that PGT prevented the intergenerational transmission of the causative mutation of XLAS.
Alport syndrome (AS) is the second rare hereditary renal failure syndrome, after autosomal dominant polycystic kidney disease, with an incidence of approximately 0.043% (1). The main clinical features are hematuria, proteinuria, progressive renal failure, end-stage renal disease, hearing loss, and eye abnormalities. The syndrome occurs via three inheritance modes: X-linked dominant inheritance, autosomal dominant inheritance, and autosomal recessive inheritance (2). The X-linked dominant version of AS (XLAS) involves mutation of the COL4A5 gene and is the most common type, accounting for more than 80% of cases (3). The COL4A5 gene is located on chromosome Xq22.3 and has a total length of 277.7 kb, with 53 exons (NM_033380.2). It encodes the type IV collagen α5 [α5(IV)] chain, which is the main component of the glomerular basement membrane (GBM), cochlea, and critical structural components of the eye. The ClinVar database details more than 1,800 variants of COL4A5, including deletions, frameshift mutations, nonsense mutations, splicing, and missense mutations. However, 70% of the variants are single nucleotide missense mutations. Mutations in the COL4A5 gene may disrupt the triple-helix folding and reduce the secretion of type IV collagen, ultimately resulting in defects in the basement membrane (4). There are significant sex differences in the clinical manifestations of XLAS patients, and male patients have more severe course and symptoms. Male patients with XLAS exhibit proteinuria and hematuria in early childhood, and more than 90% develop end-stage renal failure before the age of 40, with a median age of 25 years. Some patients may occur varying degrees of hearing loss and ocular changes (5). Male patients also have a solid genotype-phenotype association (6, 7), with truncating variants causing the most severe phenotype and the youngest age for the development of ESRD. Splicing variants have a moderate phenotype, and missense variants have a mild phenotype. Female XLAS patients have more extensive and milder clinical manifestations, mainly hematuria. There is a lack of apparent genotype-phenotype association, and only 15% of women will develop ESRD by age 60 (8–10).
People with XLAS face a significant lifetime risk of kidney failure, sensorineural deafness, and ocular abnormalities (11). Effective intervention for XLAS depends upon early diagnosis, and blocking the spread of the COL4A5 mutation is a fundamental solution. Preimplantation genetic testing (PGT) is defined as a type of assisted reproductive technology that is used to analyze the DNA of oocytes (polar bodies) or embryos (cleavage stage or blastocyst) to confirm genetic abnormalities (12). Preimplantation genetic testing is designed to block intergenerational transmission of disease-causing variants to avoid the need for therapeutic termination of pregnancy. The testing includes PGT for aneuploidy (PGT-A), PGT for chromosomal structural rearrangement (PGT-SR), and PGT for single genes/single gene defects (PGT-M) (13). Here, we reported an XLAS family in which a c.3659G>A (p.Gly1220Asp) mutation in the COL4A5 gene was identified in the wife and child. The couple underwent genetic counselling and PGT-M to have a healthy child. Based on multiple annealing and looping-based amplification cycles (MALBAC), next-generation sequencing (NGS) technology, and single nucleotide polymorphism (SNP) haplotype analysis, we were able to help the couple to have a healthy child. That proved the PGT-M was a viable way to block the inheritance of XLAS by the next generation.
In 2017, a couple had a boy admitted to the Department of Pediatrics, Zhongshan Bo'ai Hospital when he was one year old due to hematuria and proteinuria. Alport syndrome (AS) was suspected from the clinical manifestations and renal pathology biopsy results. In 2020, the boy underwent genetic testing, and the results showed that he had an X-linked hemizygous COL4A5 mutation, c.3659G>A (p.Gly1220Asp) (NM_033380.3). The couple visited the Zhongshan Bo'ai Hospital Reproductive Centre for genetic and fertility counselling to have another child. Clinical evaluation, sample collection, pedigree analysis, PGT, and assisted reproductive technology (ART) were performed at the Reproductive Center of Zhongshan Bo'ai Hospital. This study was approved by the internal ethics committee of Zhongshan Bo'ai Hospital, and informed consent was obtained from the couple.
Genomic DNA (gDNA) was isolated from the couple's and their children's peripheral blood using the TIANamp Blood DNA kit (Tiangen Biotechnology, Cat# DP348-02). Polymerase Chain Reaction (PCR) amplification and Sanger sequencing were conducted to validate the mutation sites in COL4A5. The forward (GGGTAGATTTGGGATTTGGT) and reverse (CTACCACTCACGGACATACC) primers (synthesized by Guangzhou IGE Biotechnology) were designed using Primer 5.0 software to cover the coding exons and flanking introns of COL4A5, for PCR amplification of the mutation. A VeritiPro™ 96-well thermal cycler (Thermo Fisher Scientific, Cat# 7394) was used for Quantitative-PCR (Q-PCR) analysis with SYBR® Premix Ex Taq™ II (Takara Biotechnology, Cat# RR820A). Sanger sequencing data were analyzed by ChromasPro software (Yikon Genomics). Database and literature surveys were conducted on the mutation sites. The type IV collagen α5 chain and type IV collagen 3D protein structures were assembled using PyMOL software.
Paired-end sequencing library preparation was performed on the genomic DNA of the proband using a DNA sample preparation kit (NEBNext). The amplified DNA was captured using a GenCap capture kit. The enrichment libraries were sequenced on an Illumina HiSeq X ten sequencer for paired reads of 150 bp. After sequencing, the raw data were saved in FASTQ format for bioinformatics analysis as follows: Firstly, Illumina sequencing adapters and low-quality reads (<80 bp) were filtered by Cutadapt. After quality control, the clean reads were mapped to the UCSC hg19 human reference genome using BWA. Duplicated reads were removed using Picard tools, and mapping reads were used for variation detection. Secondly, the SNP and InDel variants were detected using GATK HaplotypeCaller, and GATK VariantFiltration was used to filter the variants. Five steps were used to select the potential pathogenic mutations in the downstream analysis: (I) there should be more than five mutation reads, and the mutation ratio should be no less than 30%; (II) mutations were removed if their frequency was more than 5% in 1,000 g, ESP6500, and our in-house database; (III) mutations were dropped if they existed in the InNormal database (MyGenostics); (IV) synonymous mutations were removed; (V) after I, II and III, if the mutations were synonymous and they were reported in HGMD, they were not removed. After these five steps, the remaining mutations should be pathogenic.
Ovarian stimulation was performed using the standard long protocol of gonadotropin-releasing hormone (GnRH) agonist administration (long GnRH agonist protocol). At day 20 + 1 of the menstrual cycle, a GnRH agonist was used for initial down-regulation. Gonadotropins (mostly recFSH or HMG) were administered on day two of the subsequent cycle to promote adequate follicle development. Ovulation was triggered by HCG injection when follicles reached 18 mm in diameter. Oocytes were then extracted after 36 h using an ultrasound-guided follicular puncture. Sperm samples were prepared using the density gradient technique, and swim-up sperm were selected for intracytoplasmic sperm injection (ICSI). In accordance with program standards, embryos were produced using ICSI and cultivated up until the blastocyst stage. We observed the formation and morphology of blastocysts on day five and day six and scored them using Gardner's scoring system. The blastocysts with a Gardner score of 3BB or higher were considered suitable for embryo biopsy. The blastocyst trophoblasts were cut with a laser to obtain four to six trophectoderm (TE) cells for PGT-M.
We transferred TE cells to a 0.2 µl PCR tube that contained 4.5 µl of lysis buffer (Yikon Genomics, Suzhou, China) for whole genome amplification (WGA). The WGA of each embryo biopsy sample was performed using a MALBAC WGA kit (Yikon Genomics, XK-028-24, Suzhou, China), following the manufacturer's instructions. The collected cells were initially lysed in a lysis buffer, followed by MALBAC pre-amplification and exponential amplification, to obtain 2–5 μg of DNA.
The WGA product was used as the template for library preparation. The NGS library building kit (Yikon Genomics, KT100804248, Suzhou, China) performed enzyme digestion, terminal repair, joint connection, DNA purification, PCR enrichment, and PCR product purification. Libraries were sequenced on the MiSeq Dx platform (Illumina, California, United States). Raw data was automatically filtered to generate a FASTQ file in which the Q30 value was greater than 90%. The CNV analysis of the FASTQ files was performed to report any deletions or duplications of more than 4 Mb for each embryo. Valid reads of more than 1 Mb and CV (1,000 K_bin_size) were considered to be within the acceptable range. High-frequency SNP sites in the 2 Mb regions upstream and downstream of the pathogenic gene and within the gene were screened as genetic markers. These sites were submitted to the https://www.ampliseq.com/ website to design a primer pool. Using WGA products and gDNA as templates, SNP library preparation was carried out under the instructions for the NGS library preparation general kit (Yikon Genomics, KT100804424, Suzhou, China). That included multiple specific amplification of upstream and downstream SNP panels of mutant genes, purification of amplification products, enzymatic hydrolysis of non-target fragments, purification of enzymatic hydrolysis products, PCR enrichment library construction, PCR product purification and the establishment of linkage relationships by family pedigrees, to diagnose embryo pathogenicity. After creating the SNP library, a Nextseq550 sequencer (Illumina, California, United States) was used to complete the sequencing. CNV and SNP data were analyzed using ChromGo bioinformatics analysis software (Yikon Genomics, Suzhou, China).
When the child was one year old, he developed gross hematuria for no apparent reason. His body temperature, respiration, and blood pressure were average. There was no oedema of the face and lower limbs, no percussion pain in the kidney area, and no other abnormalities found during the physical examination. Urine testing showed 3+ occult blood and 1+ urine protein. The 24 h urine protein quantification was 213.46 mg/kg, the urine protein/creatinine ratio was 38.21 mg/g, and other liver and kidney functions, myocardial enzymes, electrolytes, and complements were normal. Colour Doppler ultrasonography showed no obvious abnormalities in the kidneys, bladder, and renal blood vessels. Urine microbial cultures were negative. Hearing and fundus examinations showed no abnormalities. A pathological kidney biopsy showed negative tissue immunofluorescence staining of α3 and α5 (IV) in the basement membranes, which suggested α3 and α5 chain deletion, and a basement membrane diameter of 180 nm. AS was clinically suspected. The boy's father was asymptomatic, and his mother only showed intermittent isolated hematuria. Peripheral blood samples from the proband and his parents were collected for pedigree analysis. The NGS results showed that the proband and his mother had the c.3659G>A (p.Gly1220Asp) (NM_033380.3) mutation in the COL4A5 gene, but this was not identified in his father (Figures 1A,B). We used the American College of Medical Genetics and Genomics (ACMG/AMP) Sequence Variation Interpretation Standards and Guidelines to classify the mutation as follows.
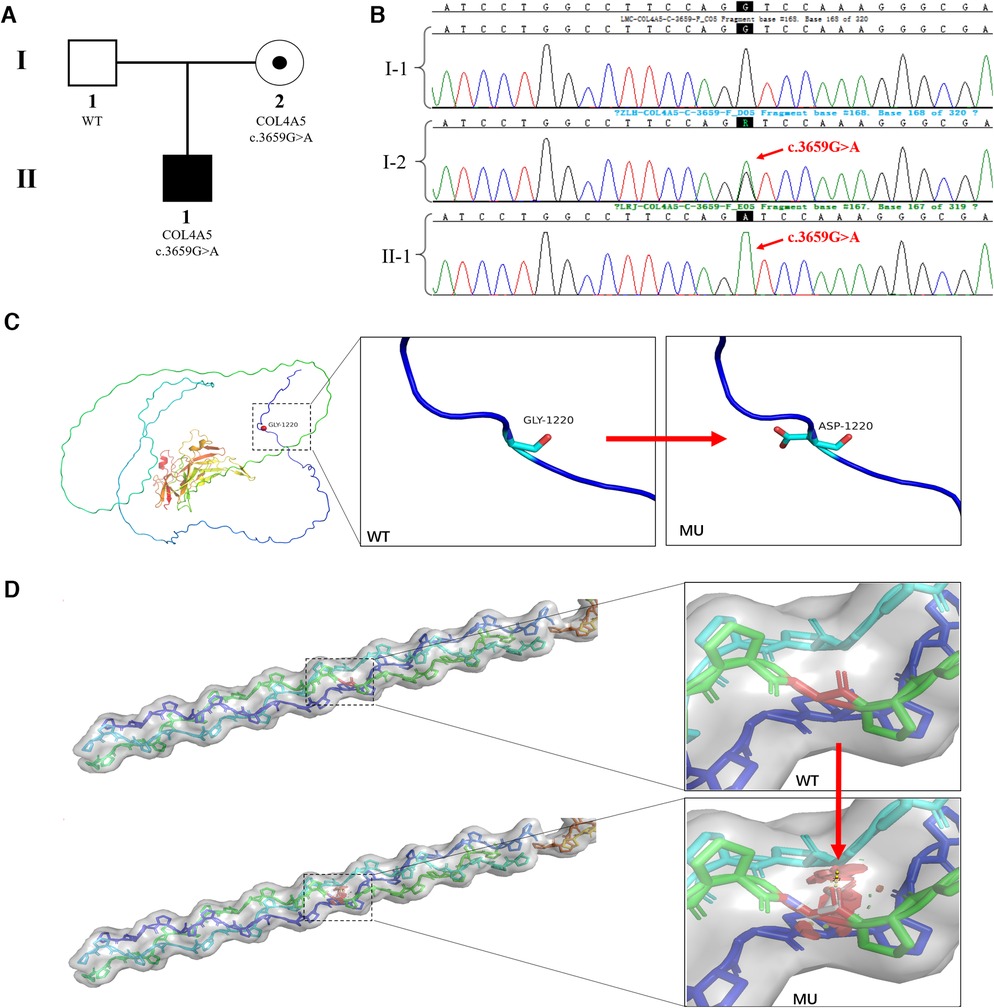
Figure 1. COL4A5 mutations and identification of affected families. (A) Pedigree of an XLAS-affected family: The proband (II-1), a 1-year-old boy, was identified as having XLAS caused by a pathogenic COL4A5 mutation; the mother (I-2) was also identified as having the same mutation but no clinical manifestations; the father was normal. (B) Sanger sequencing confirmed the hemizygous mutation of COL4A5 (c.3659G>A (p.Gly1220Asp) (NM_033380.3) in the proband (II-1) and his mother (I-2); the father did not have any mutations in the COL4A5 gene. (C) The crystal structure of the α5 chain of type IV collagen. The glycine at position 1,220 in the protein's primary structure is replaced by aspartic acid when the c.3659G>A mutation occurs. (D) Steric hindrance is introduced into the tertiary structure of type IV collagen due to the c.3659G>A (p.Gly1220Asp) mutation.
The c.3659G>A mutation in the COL4A5 gene was absent from population databases (PM2).
The region where the mutation was located was an essential part of this protein, and the amino acid sequence of different species was highly conserved. The mutation interrupted the Gly-X-Y domain of the collagen gene (PM1) (Figures 1C,D).
Computational evidence was used to predict the damaging effect of the mutation on the protein (PP3). The p.Gly1220Asp mutated protein has been found in AS patients (14) (PP4).
The mutation complied with the interpretation rules of PM2, PM1, PP3, and PP4 and was classified as “Category 2- Likely Pathogenic”.
This mutation inherited in an X-linked dominant manner was the cause of XLAS. The karyotype analysis of the couple showed normal karyotypes.
We obtained 20 oocytes, under ultrasound guidance, after ovulation induction. These metaphase II (MII) oocytes were subsequently inseminated by ICSI, and all were fertilized normally, as indicated by the presence of two pronuclei. Eight blastocysts were obtained on the fifth day after insemination and scored by the Gardner Grade (Table 1). All blastocysts underwent trophectoderm biopsy on day 6 to collect four to six cells. After cell lysis, WGA was performed using the MALBAC two-step method, and the products were purified. The purified products were detected by amplification of target fragments and Sanger sequencing. The results showed that the c.3659G>A mutation was present in the COL4A5 gene in embryos E3, E5, and E8, while no mutation was found in E1, E2, E4, E6, or E7 (Figure 2A).
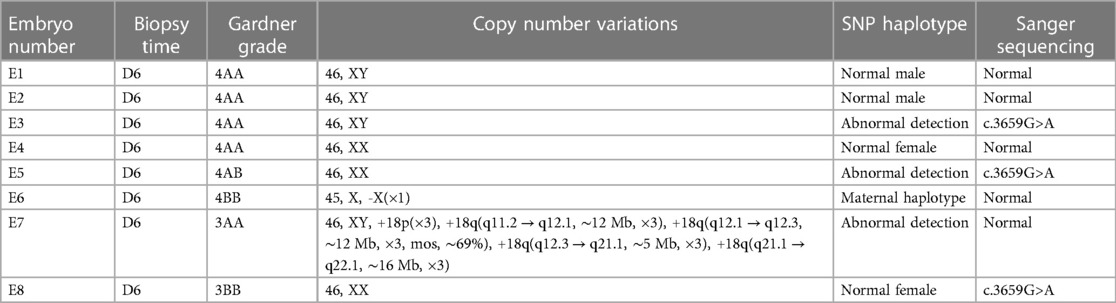
Table 1. Summary of detection results.
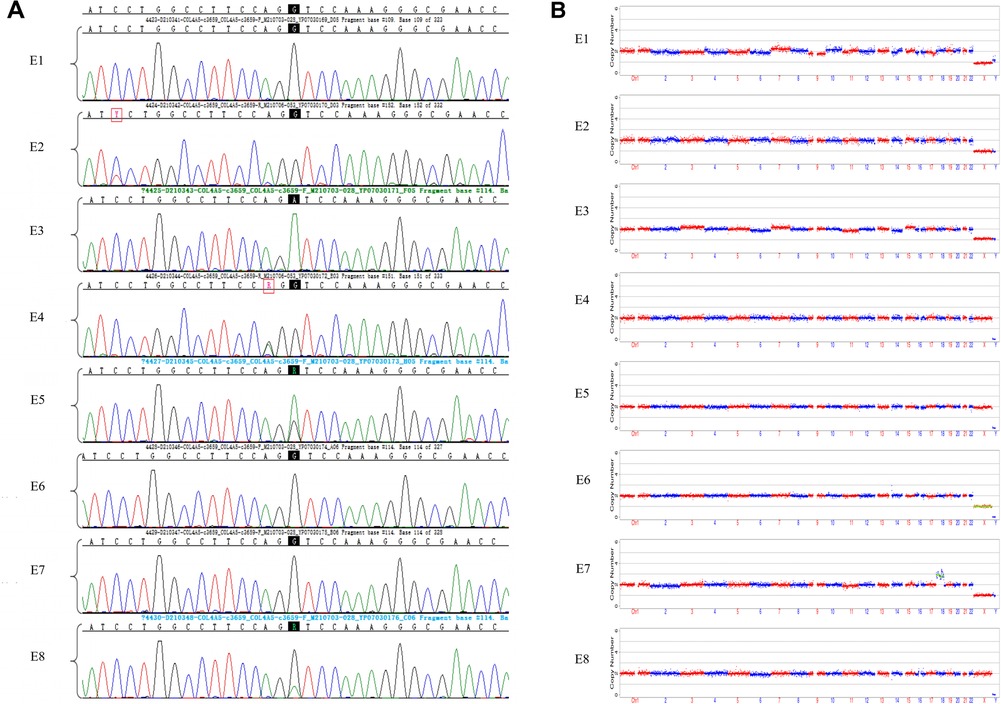
Figure 2. Results of sanger sequencing and CNV analysis of WGA products from embryonic trophoblast biopsy cells. (A) Sanger sequencing chromatograms of eight embryos. Mutation sites are shown with a black background. The E1, E2, E4, E6, and E7 embryos did not inherit the COL4A5 pathogenic mutation (c.3659G>A), while E3, E5, and E8 were found to have the mutation; (B) CNVs in eight embryos, at low NGS sequencing depth. NGS analysis of embryos E1, E2, E3, E4, E5, and E8 did not reveal any large CNVs or copy number abnormalities. Both E6 and E7 had copy number abnormalities.
The NGS-based CNV sequencing of WGA purified products on the Illumina MiseqDx platform was used to detect large CNVs of more than 4 Mb and 30%–70% mosaicism (larger than 10 Mb). No CNVs larger than 4 Mb or aneuploidy were found in embryos E1, E2, E3, E4, E5, or E8. However, E6 had a monosomy of the X chromosome, and E7 was aneuploid (Figure 2B).
Allelic dropout (ADO) may occur during gene amplification. In this disease, ADO may cause diseased embryos to be misdiagnosed as normal embryos. To reduce ADO interference on diagnostic results, the peripheral blood gDNA of the couple and their child (proband), plus the WGA products of the embryos, were analyzed for SNP haplotypes (Figure 3). We performed linkage analysis using the SNP markers within the 2 Mb region that flanked the COL4A5 gene. Sixty informative polymorphic SNPs were selected to establish haplotypes. In this case, 15 SNPs within 1 Mb upstream and downstream of the mutation site were used for the analysis. When combined with family haplotypes (Figure 3), the results showed that embryos E1, E2, E4, E6, and E7 did not carry the maternal mutation, although E6 had the maternal haplotype. Based on pathogenic mutation detection results and linkage analysis, E1, E2, E4, and E7 did not carry the COL4A5 c.3659G>A mutation (Table 1).
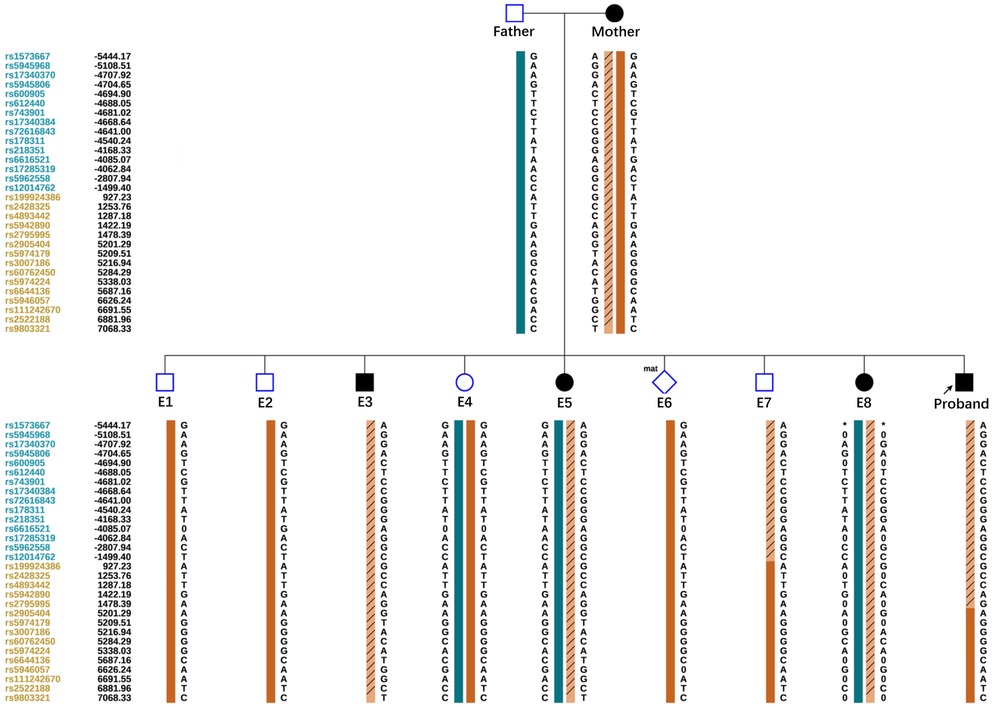
Figure 3. Schematic representation of the SNP-based haplotypes of the family members and embryos. The SNP sites are indicated on the left, and blue and orange represent SNP sites upstream or downstream, respectively, of the chromosomal position where the gene is located. Light orange left slashes to represent high-risk haplotypes, dark blue bars represent normal or low-risk haplotypes of the father, and dark orange bars represent normal or low-risk haplotypes of the mother. The results showed that E1, E2, E4, and E7 were normal embryos. E6 had the maternal haplotype.
The couple had discussions with a reproductive doctor, embryologist, and medical geneticist to determine the order of embryo transfer at our Center. Based on the developmental stage grade of the embryos, the results of the Sanger sequencing, CNV analysis, and SNP haplotype analysis, the final decision was made to transfer embryo E1 by consensus. A successful pregnancy was achieved (Table 1). At 20 weeks of gestation, the pregnant woman underwent ultrasound-guided amniocentesis, and amniotic fluid cells were cultured. Amniotic fluid cells were used for CMA, target fragment amplification, and Sanger sequencing. The results showed no abnormalities, and Sanger sequencing results were consistent with the PGT. The woman eventually gave birth to a healthy boy who did not carry the mutation.
In this study, we reported an XLAS family with a mutation in the COL4A5 gene on the X chromosome. The results of genetic analyses showed that there was a single nucleotide missense mutation, c.3659G>A (p.Gly1220Asp) (NM_033380.3), in exon 41 of the COL4A5 gene. The mutant COL4A5 had a change from guanine (G) to adenine (A) at base 3,706, which resulted in amino acid 1,220 of the encoded protein being changed from glycine (Gly) to aspartic acid (Asp). The COL4A5 gene encodes the α5 chain of type IV collagen, which combines with the α3 and α4 chains of type IV collagen to form a triple helix structure (15). The main feature of the primary structure of type IV collagen was the repeated sequence of amino acids, which was represented by a glycine every three residues, and the polypeptide sequence was: -Gly-Xaa-Yaa-Gly-Xaa-Yaa-Gly-Xaa-Yaa-. Glycine is the amino acid with the lowest molecular weight of the amino acid composition of collagen. It has a single hydrogen atom in the side chain, has a larger side chain space, and allows the widest angle for the polypeptide chain to turn compared to other amino acids (16). The glycine in the Gly-Xaa-Yaa sequence is critical for forming collagen's triple helix structure. Substitution of glycine by other amino acids introduces steric hindrance and destabilizes the helical conformation, possibly leading to bulges or kinks in the linear collagen molecule. The type IV collagen α5 chain is the essential structural component of the basement membrane in the human GBM, cochlea, and lens (17). In this study, the glycine at position 1,220 was replaced by aspartic acid, which resulted in the interruption of the Gly-Xaa-Yaa repeat sequence and may have caused a weakened GBM and increased fragility of the podocyte cytoskeleton (16). Defective expression of GBM and abnormal stromal-podocyte interactions induce AS, with auditory and visual problems. The p.Gly1220Asp mutation of the COL4A5 gene, which emerged in this study, has been listed as possibly pathogenic/pathogenic in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) (variant identifier: 24666).
In our study, the wife was heterozygous, and her son was hemizygous for the COL4A5 mutation, as the COL4A5 gene was located on Xq22. The female mutation carriers in this family only showed isolated microscopic hematuria. However, the male family members who carried the mutation had more severe renal phenotypes, such as hematuria, proteinuria, renal dysfunction, and renal pathological changes. It was clear that there were significant differences in the course and severity of the disease between male and female patients. Female patients that are heterozygous for a pathogenic mutation have historically been defined as healthy carriers. However, subsequent studies have shown that female carriers display a spectrum of phenotypes that range from microscopic hematuria to ESRD and extrarenal manifestations. Approximately one in four female carriers will develop ESRD by age 60, and the risk increases with age. The mouse experiments of Rheault et al. (18) and the clinical case reports of Mastrangelo et al. (19) suggested that the extensive phenotypic characteristics of female heterozygotes may be related to digenic inheritance and skewed inactivation of the X chromosome. However, Günthner et al. (20) explored X inactivation in urine-derived cells of individual XLAS women. They found that X inactivation was not associated with age at first presentation, proteinuria, or glomerular filtration rate (eGFR). They found that the degree of X inactivation correlated with age, which may be an escape mechanism to avoid the expression of COL4A5 mutations as individuals age. Male hemizygous carriers have more obvious genotype-phenotype correlations and more severe phenotypic features than female heterozygous carriers. Nearly 90% of men with XLAS develop early chronic kidney disease (CKD) and end-stage kidney disease (ESKD) by age 40. The study found that the type of mutation was an important factor that affects the severity of XLAS in men (6, 7). Male patients with truncating mutations develop ESKD at approximately 20 years old, while those with non-truncating mutations develop ESKD at 40. It is worth noting that XLAS, caused by COL4A5 mutations, has considerable phenotypic heterogeneity and a combination of phenotypes (21–24). It may not even show typical clinical phenotypes (such as hematuria, proteinuria, and hearing and vision problems) or AS-like pathological changes, which is not conducive to early diagnosis and effective intervention (11). Therefore, in young patients with unexplained glomerular diseases, without typical manifestations, clinicians also need to consider COL4A5 mutations. Genetic testing, based on next-generation sequencing, is crucial for the diagnosis of XLAS, which is a disease with high phenotypic and genetic heterogeneity (21–23, 25).
The traditional prenatal diagnosis technology, based on amniocentesis, has been the major means to prevent the birth of children with monogenic genetic diseases in the past. However, it is traumatic and carries a risk of miscarriage. After prenatal diagnosis confirms that the fetus has AS, the pregnant woman needs to terminate the pregnancy, which causes physical and psychological harm to her and her family. Using PGT-M, the blastocyst trophoblast cells can be taken for genetic analysis, and normal embryos can be selected for transplantation into the mother's uterus. That prevents the intergenerational transmission of single-gene diseases and prevents couples from deciding to terminate the pregnancy or having affected children. Currently, single-cell expansion methods suitable for PGT include MDA and MALBAC. MDA can give higher cell concentrations after amplification, but the amplification uniformity and coverage of MALBAC are better than MDA (26). The initial concentration of DNA used for WGA in PGT is low, and the exponential amplification may cause ADO. Allele dropout refers to the failure to amplify one allele in heterozygotes, which results in the site being falsely detected as homozygous. Allele dropout is one of the characteristics of single-cell WGA and is also a key factor that leads to the underreporting of single-base mutations (26). In this case, ADO may have resulted in a false negative call. Compared with MDA, the false positive and ADO rates of MALBAC are lower (27).
In summary, MALBAC had high amplification uniformity and a low ADO rate. The WGA product from MALBAC was applied directly to CNV-seq sequencing library preparation, and it had good results in CNV analysis and SNP detection. Therefore, we used MALBAC for gene amplification of trophoblast cells. While other reports describe the application of PGT-M in XLAS (28, 29), this is the first use of the MALBAC technique for WGA. In order to reduce the impact of ADO, we used polymorphic markers from the proband and his parents and linkage analysis of mutant genes to establish haplotypes. We found that the analysis of SNP markers by NGS was beneficial in ensuring the accuracy of PGT-M (30).
Through CNV analysis, SNP haplotype analysis, and Sanger sequencing, we determined that embryos E1, E2, and E4 did not carry any pathogenic mutations or chromosomal abnormalities. We combined the results of embryo morphology and PGT and used embryo E1 (4AA) for transplantation. The pregnancy was successful. Although we performed some work to avoid misdiagnosis during PGT, we could not altogether avoid the influence of chromosomal rearrangement, mosaicism, and ADO on the diagnostic results. It was still necessary to prove the results of PGT by amniocentesis in the second trimester. In this case, the results of genetic testing on amniotic fluid cells were consistent with the results of PGT-M, and a healthy baby boy was born.
In conclusion, we reported a family with XLAS caused by a COL4A5 mutation. The family underwent PGT-M to prevent the transmission of the COL4A5 pathogenic mutation to their child. Through MALBAC-based WGA, NGS-based haplotype analysis and prenatal diagnosis in the second trimester, to reduce the risk of misdiagnosis from PGT-M, the couple finally gave birth to a healthy baby boy. Our results confirmed the feasibility of PGT-M to block the intergenerational transmission of the COL4A5 mutation, which has an important clinical application in the prevention and control of birth defects.
The raw datasets analysed during the current study are not deposited in publicly available repositories because of considerations about the security of human genetic resources and patient anonymity. Requests to access these datasets should be directed to NL,bGl1bmVuZzEwQGdtYWlsLmNvbQ==.
The studies involving human participants were reviewed and approved by Internal Ethics Committee of Zhongshan Bo'ai Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor's legal guardian for the publication of any potentially identifiable images or data included in this article.
NL and XW collected the data; and NL, XW, ZO, XF, JD, and XL reviewed the literature. NL drafted the manuscript. JD and XL provided valuable input for collecting the data and drafting the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by institutional funding from the Science and technology research projects for social welfare of Zhongshan (Project No. 2022B1013, 2022B1002 and 2022B3007), Innovation Leading talents funding project of Zhongshan (Project No. LJ2021004).
We express deepest gratitude to all the peer reviewers for their suggestions. We are deeply grateful to the patients and their families for participating in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gibson J, Fieldhouse R, Chan MMY, Sadeghi-Alavijeh O, Burnett L, Izzi V, et al. Prevalence estimates of predicted pathogenic COL4A3-COL4A5 variants in a population sequencing database and their implications for alport syndrome. J Am Soc Nephrol. (2021) 32(9):2273–90. doi: 10.1681/ASN.2020071065
2. Savige J, Huang M, Croos Dabrera MS, Shukla K, Gibson J. Genotype-phenotype correlations for pathogenic COL4A3-COL4A5 variants in X-linked, autosomal recessive, and autosomal dominant alport syndrome. Front Med (Lausanne). (2022) 9:865034. doi: 10.3389/fmed.2022.865034
3. Rheault MN, Kashtan CE. Inherited glomerular diseases. J Pediatr Nephrol. (2016) 7:1918–84. doi: 10.1007/978-3-662-43596-0_79
4. Yamamura T, Horinouchi T, Aoto Y, Lennon R, Nozu K. The contribution of COL4A5 splicing variants to the pathogenesis of X-linked alport syndrome. Front Med (Lausanne). (2022) 9:841391. doi: 10.3389/fmed.2022.841391
5. Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, et al. Genotype-phenotype correlation in X-linked alport syndrome. J Am Soc Nephrol. (2010) 21(5):876–83. doi: 10.1681/asn.2009070784
6. Yamamura T, Horinouchi T, Nagano C, Omori T, Sakakibara N, Aoto Y, et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked alport syndrome. Kidney Int. (2020) 98(6):1605–14. doi: 10.1016/j.kint.2020.06.038
7. Di H, Zhang J, Gao E, Zheng C, Huang X, Wang Q, et al. Dissecting the genotype-phenotype correlation of COL4A5 gene mutation and its response to renin-angiotensin-aldosterone system blockers in Chinese male patients with alport syndrome. Nephrol Dial Transplant. (2022) 37(12):2487–95. doi: 10.1093/ndt/gfac002
8. Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. X-linked alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European community alport syndrome concerted action” study. J Am Soc Nephrol. (2003) 14(10):2603–10. doi: 10.1097/01.asn.0000090034.71205.74
9. Yamamura T, Nozu K, Fu XJ, Nozu Y, Ye MJ, Shono A, et al. Natural history and genotype-phenotype correlation in female X-linked alport syndrome. Kidney Int Rep. (2017) 2(5):850–5. doi: 10.1016/j.ekir.2017.04.011
10. Gibson JT, de Gooyer M, Huang M, Savige J. A systematic review of pathogenic COL4A5 variants and proteinuria in women and girls with X-linked alport syndrome. Kidney Int Rep. (2022) 7(11):2454–61. doi: 10.1016/j.ekir.2022.08.021
11. Kashtan CE. Alport syndrome: achieving early diagnosis and treatment. Am J Kidney Dis. (2021) 77(2):272–9. doi: 10.1053/j.ajkd.2020.03.026
12. ESHRE PGT Consortium and SIG-Embryology Biopsy Working Group, Kokkali G, Coticchio G, Bronet F, Celebi C, Cimadomo D, et al. ESHRE PGT consortium and SIG embryology good practice recommendations for polar body and embryo biopsy for PGT. Hum Reprod Open. (2020) 2020(3):hoaa020. doi: 10.1093/hropen/hoaa020
13. Zegers-Hochschild F, Adamson GD, Dyer S, Racowsky C, de Mouzon J, Sokol R, et al. The international glossary on infertility and fertility care, 2017. Hum Reprod. (2017) 32 (9):1786–801. doi: 10.1093/humrep/dex234
14. Plant KE, Green PM, Vetrie D, Flinter FA. Detection of mutations in COL4A5 in patients with alport syndrome. Hum Mutat. (1999) 13(2):124–32. doi: 10.1002/(SICI)1098-1004(1999)13:2%3C124::AID-HUMU4%3E3.0.CO;2-Z
15. Quinlan C, Rheault MN. Genetic basis of type IV collagen disorders of the kidney. Clin J Am Soc Nephrol. (2021) 16(7):1101–9. doi: 10.2215/cjn.19171220
16. Fidler AL, Vanacore RM, Chetyrkin SV, Pedchenko VK, Bhave G, Yin VP, et al. A unique covalent bond in basement membrane is a primordial innovation for tissue evolution. Proc Natl Acad Sci U S A. (2014) 111(1):331–6. doi: 10.1073/pnas.1318499111
17. Van Agtmael T, Bruckner-Tuderman L. Basement membranes and human disease. Cell Tissue Res. (2010) 339(1):167–88. doi: 10.1007/s00441-009-0866-y
18. Rheault MN, Kren SM, Hartich LA, Wall M, Thomas W, Mesa HA, et al. X-inactivation modifies disease severity in female carriers of murine X-linked alport syndrome. Nephrol Dial Transplant. (2010) 25(3):764–9. doi: 10.1093/ndt/gfp551
19. Mastrangelo A, Giani M, Groppali E, Castorina P, Soldà G, Robusto M, et al. X-linked alport syndrome in women: genotype and clinical course in 24 cases. Front Med. (2020) 7:580376. doi: 10.3389/fmed.2020.580376
20. Günthner R, Knipping L, Jeruschke S, Satanoskij R, Lorenz-Depiereux B, Hemmer C, et al. Renal X-inactivation in female individuals with X-linked alport syndrome primarily determined by age. Front Med (Lausanne). (2022) 9:953643. doi: 10.3389/fmed.2022.953643
21. Wuttke M, Seidl M, Malinoc A, Prischl FC, Kuehn EW, Walz G, et al. A COL4A5 mutation with glomerular disease and signs of chronic thrombotic microangiopathy. Clin Kidney J. (2015) 8(6):690–4. doi: 10.1093/ckj/sfv091
22. Li Z, Zhu P, Huang H, Pan Y, Han P, Cui H, et al. Identification of a novel COL4A5 mutation in the proband initially diagnosed as IgAN from a Chinese family with X-linked alport syndrome. Sci China Life Sci. (2019) 62(12):1572–9. doi: 10.1007/s11427-018-9545-3
23. Zhang P, Zhuo L, Zou Y, Li G, Peng K. COL4A5 Mutation causes alport syndrome with focal segmental glomerulosclerosis lesion: case report and literature review. Clin Nephrol. (2019) 92(2):98–102. doi: 10.5414/cn109737
24. Zhong L, Li Y, He X, Dey SK, Zhang Q, Banerjee S. Identification and functional characterization of a novel truncating splicing variant in COL4A5 gene causes X-linked alport syndrome with astigmatism. Chin Med J (Engl). (2023). doi: 10.1097/cm9.0000000000002417
25. Xiu X, Yuan J, Deng X, Xiao J, Xu H, Zeng Z, et al. A novel COL4A5 mutation identified in a Chinese Han family using exome sequencing. Biomed Res Int. (2014) 2014:186048. doi: 10.1155/2014/186048
26. He F, Zhou W, Cai R, Yan T, Xu X. Systematic assessment of the performance of whole-genome amplification for SNP/CNV detection and β-thalassemia genotyping. J Hum Genet. (2018) 63(4):407–16. doi: 10.1038/s10038-018-0411-5
27. Volozonoka L, Miskova A, Gailite L. Whole genome amplification in preimplantation genetic testing in the era of massively parallel sequencing. Int J Mol Sci. (2022) 23(9):4819. doi: 10.3390/ijms23094819
28. Hu X, Zhang J, Lv Y, Chen X, Feng G, Wang L, et al. Preimplantation genetic testing prevented intergenerational transmission of X-linked alport syndrome. Kidney Dis (Basel). (2021) 7(6):514–20. doi: 10.1159/000517796
29. Shi WH, Ye MJ, Chen SC, Zhang JY, Chen YY, Zhou ZY, et al. Case report: preimplantation genetic testing and pregnancy outcomes in women with alport syndrome. Front Genet. (2021) 12:633003. doi: 10.3389/fgene.2021.633003
Keywords: X-Linked alport syndrome, COL4A5, next-generation sequencing, type IV collagen, preimplantation genetic testing
Citation: Liu N, Wen X, Ou Z, Fang X, Du J and Lin X (2023) Case report: Preimplantation genetic testing for X-linked alport syndrome caused by variation in the COL4A5 gene. Front. Pediatr. 11:1177019. doi: 10.3389/fped.2023.1177019
Received: 30 April 2023; Accepted: 28 July 2023;
Published: 10 August 2023.
Edited by:
Mohiuddin Mohammed Taher, Umm al-Qura University, Saudi ArabiaReviewed by:
Santasree Banerjee, Beijing Genomics Institute (BGI), China© 2023 Liu, Wen, Ou, Fang, Du and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Du Ym9haWR1amluZ0AxNjMuY29t Xiufeng Lin enNib2FpMjAxNEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.