 Samuel Gagne
Samuel Gagne Vidya Sivaraman
Vidya Sivaraman Shoghik Akoghlanian
Shoghik Akoghlanian
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pediatr., 09 August 2023
Sec. Pediatric Immunology
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1169638
This article is part of the Research TopicImmune Dysregulation in Inborn Errors of ImmunityView all 5 articles
Type I interferonopathies are a broad category of conditions associated with increased type I interferon gene expression and include monogenic autoinflammatory diseases and non-Mendelian autoimmune diseases such as dermatomyositis and systemic lupus erythematosus. While a wide range of clinical presentations among type I interferonopathies exists, these conditions often share several clinical manifestations and implications for treatment. Presenting symptoms may mimic non-Mendelian autoimmune diseases, including vasculitis and systemic lupus erythematosus, leading to delayed or missed diagnosis. This review aims to raise awareness about the varied presentations of monogenic interferonopathies to provide early recognition and appropriate treatment to prevent irreversible damage and improve quality of life and outcomes in this unique patient population.
First discovered in the 1950 s (1), interferons (IFNs) are a large group of signaling molecules that promote and regulate innate immune system responses to viral infection. IFNs are divided into three major classes (Type I, II, and III), each with distinct roles within the immune system. Type I IFNs (IFN-1) are predominantly made up of IFN-α and IFN-β and are expressed by many cell types. IFN-1 gene expression can be induced by cytokines, IFNs themselves, and pattern recognition receptors such as toll-like receptors and cytosolic nucleic acid receptors such as MDA5 and RIG-I (2). IFN signaling proceeds via the Janus kinases (JAK)-signal transducer and activator of transcription proteins (STAT) pathway upon receptor binding, ultimately transcribing several pro-inflammatory IFN-stimulated genes (3).
Type I interferonopathies are a broad category of conditions associated with increased IFN-1 gene expression (4). Initially, interferonopathy was limited to a group of Mendelian disorders caused by a genetic defect that disrupts homeostatic control and leads to IFN-mediated responses. However, the term has been extended to non-Mendelian autoimmune diseases such as dermatomyositis, systemic sclerosis, and systemic lupus erythematosus (SLE), as well as more rare diseases such as Degos disease, where IFN signaling is important to pathogenesis (5–7). Type I interferonopathies share several clinical manifestations and treatment implications. Specifically, they frequently affect the central nervous system (CNS) and skin but are not limited to these systems (8, 9). An elevated peripheral blood IFN signature is not required but can be helpful for diagnosis (10). Unfortunately, few centers check IFN signatures, and there are no internally standardized methodologies or references.
Glucocorticoids, conventional disease-modifying anti-rheumatic drugs (DMARDs) such as methotrexate, mycophenolate mofetil (MMF), and even bone marrow transplantation (BMT) have historically been used to treat interferonopathies. More recently, biologic DMARDs to target cytokines and/or small-molecule DMARDs to target JAKs have become an increasingly common therapeutic strategy. The US Food and Drug Administration's Compassionate Use Program was initiated in 2011 to treat pediatric patients with rare Mendelian autoinflammatory diseases that have IFN-mediated pathology (3, 11). Subsequently, several case series have been published demonstrating the potential efficacy of JAK inhibitors in treating such diseases (12, 13). However, an increased risk of serious and opportunistic infections has been observed and BK viremia, nephropathy, and, rarely, progressive multifocal leukoencephalopathy have been reported with JAK inhibitor use (14, 15).
Patients with monogenic interferonopathies often present within the first week of life; however, late onset in the teenage years has been reported. Patients may share similar symptoms with more common non-Mendelian autoimmune diseases (see Table 1), leading to a delayed or missed diagnosis (Table 1). This review aims to bridge the gap between autoimmunity and monogenic interferonopathies to enable early recognition and diagnosis and improve outcomes in this unique patient population.
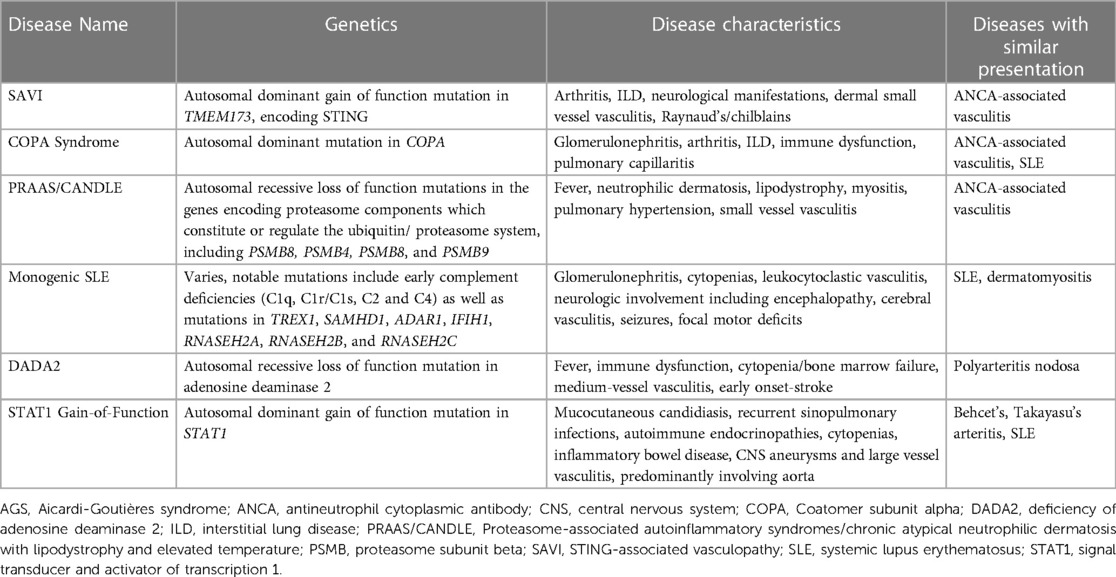
Table 1. Genetics and disease characteristics of monogenic type I interferonopathies.
Stimulator of IFN genes (STING)-associated vasculopathy with onset in infancy (SAVI) is a rare autoinflammatory disease caused by gain-of-function (GOF) mutations in TMEM173 which encodes the STING transmembrane protein (16). STING is involved in the innate immune response, functioning as an adapter molecule of the cytosolic DNA danger signal-sensing machinery. In SAVI, mutations in STING lead to its sequestration from the endoplasmic reticulum Golgi intermediate compartment, leading to excessive expression of interferon stimulated genes (17). Children with SAVI typically present in early childhood with dermal small vessel vasculopathy and microangiopathic thrombosis that varies in severity from chilblain lesions and Raynaud's phenomenon to severe progressive tissue loss involving the earlobes, nose, and digits. Progressive interstitial lung disease (ILD) with or without pulmonary hypertension may be present in severe disease. Pulmonary manifestations are typically detected via chest CT scan and spirometry, although lung biopsy may be required to differentiate infectious from inflammatory disease (18, 19). Myositis and autoantibody production are common in SAVI, thus misdiagnosis of SLE or juvenile myositis is not uncommon. In SAVI, autoantibodies are not associated with disease severity but instead are likely modulated by genetic factors (19). Neurological manifestations are rare compared to other interferonopathies, and developmental delays have not been reported in children with SAVI (20, 21). Arthritis was observed in one-third of SAVI patients and can be destructive (16). SAVI can be challenging to diagnose as many patients have normal laboratory parameters, including complete blood counts and inflammatory markers. Therefore, it is crucial to consider the diagnosis of SAVI in young patients with arthritis and SLE-like symptoms. Treatment with different JAK inhibitors (ruxolitinib, baricitinib, and tofacitinib) has resulted in improvement in the lung disease, significant decrease in vasculitis flares, and prevention of progression of spontaneous amputation in patients with SAVI (13).
Coatomer subunit alpha (COPA) syndrome is a rare autosomal dominant interferonopathy caused by COPA gene mutations. COPA is part of COPI and COPII (coat member protein complex I and II) that are involved in bidirectional vesicular trafficking between the endoplasmic reticulum and the Golgi complex. COPα mutations cause dysfunctional protein binding and retrograde transport through the COPI complex (22). It has been hypothesized that this aberrant protein trafficking leads to endoplasmic reticulum stress via the unfolded protein response, leading to subsequent autoimmunity (23). However, recent research suggests that COPA mutations result in STING overexpression with chronic IFN-1 activation and autoimmunity. Specifically, defects in COPA result in an inability to traffic and degrade STING. The result is multimerization of STING which becomes spontaneously active even without the STING ligand, leading to chronic IFN-1 activation (24–26).
The presentation of COPA syndrome is clinically similar to SAVI and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis although early age of presentation assists in differentiating COPA from ANCA-associated vasculitis. COPA syndrome presents in young children with features that can include pulmonary symptoms, renal disease, arthritis, and immune dysregulation (27). Pulmonary manifestations are variable but most commonly present as progressive pulmonary hemorrhage secondary to pulmonary capillaritis (28). Chest CT scans often demonstrate diffuse ground glass opacities at the onset with eventual pulmonary nodule development and cystic changes (27, 29). Untreated, nearly all patients with COPA syndrome develop ILD, with lymphocytic interstitial pneumonia and follicular bronchitis being the most common (28, 30, 31). Renal involvement in COPA syndrome occurs in approximately 44% of patients, typically in the second decade of life (27). Renal involvement in most patients presents with glomerulonephritis with proteinuria and hematuria which can mimic ANCA-associated vasculitis or SLE (32). The histologic manifestations vary but include crescentic disease (with or without immune deposits), focal segmental glomerulosclerosis, membranous nephropathy, and IgA nephropathy. “Most (95%) patients have musculoskeletal involvement. Musculoskeletal involvement typically presents in the second decade of life as non-erosive polyarthritis although it can be the presenting symptom, leading to a misdiagnosis of juvenile idiopathic arthritis. Osteonecrosis has also been reported (27, 30). Immune dysregulation is frequently observed in 80% of patients with antinuclear antibodies, rheumatoid factor, anti-cyclic citrullinated antibodies, c-ANCA, and p-ANCA antibodies. CD4+ T-cells also frequently skew towards a Th17 phenotype. Cytopenia or hypogammaglobulinemia are not typically present (27). Less frequently, involvement of other systems, such as CNS, liver, thyroid, and skin, have been reported (30, 33). No standard treatments for COPA syndrome exist. Typically, systemic corticosteroids are given in combination with other immunosuppressants such as MMF, rituximab, and hydroxychloroquine (27, 28, 34). Successful treatment with JAK inhibitors has also been reported (35, 36).
STAT1 Gain-of-Function (GOF) is an autosomal dominant disorder caused by several mutations that lead to STAT1 hyperphosphorylation, subsequent autoimmunity, and increased susceptibility to infection, notably chronic mucocutaneous candidiasis (37). Usually, STAT1 exists as a cytosolic transcription factor downstream of IFN and IL-27 receptors. Upon receptor binding, STAT1 is phosphorylated, forms either a heterotrimer or homodimer, and migrates to the nucleus to regulate gene expression (38). In STAT1 GOF, impaired dephosphorylation or high STAT1 levels activate pathways, decrease Th17 serum levels, and increase expression of interferon stimulated genes (37–39). These changes increase susceptibility to infections and autoimmunity; however, the exact mechanisms are unknown.
The presentation of STAT1 GOF disease is very heterogenous. However, increased susceptibility to infection is universally seen. Over 98% of patients with STAT1 GOF develop chronic mucocutaneous candidiasis which can help differentiate the diagnosis from other autoimmune diseases. Increased sinopulmonary and mucocutaneous bacterial and viral infections are also commonly observed. Autoimmune manifestations vary and occur in approximately one-third of patients. Autoimmune endocrinopathies such as thyroid disease and type I diabetes mellitus are most common. However, SLE, autoimmune skin conditions, inflammatory bowel disease, and autoimmune cytopenias can also occur (40). Vascular involvement has been well described in STAT1 GOF and predominantly presenting as cerebral aneurysms (38, 41), although large-vessel vasculitis that can mimic Takeyasu's arteritis has also been reported (42–44).
Additionally, with predominant recurrent aphthous ulcers at presentation, several patients were misdiagnosed with Behcet's disease (45, 46). Given the increased infection susceptibility and risk of autoimmunity, STAT1 GOF treatment is challenging and can include antimicrobial and immunosuppressive therapy. JAK inhibitors have emerged as a promising treatment for patients with STAT1 GOF, although many still ultimately require BMT (47, 48).
Proteasome-associated autoinflammatory syndrome (PRAAS)/chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) is a rare autosomal recessive interferonopathy caused by loss-of-function mutations in the genes encoding proteasome components that either constitute or regulate the ubiquitin/proteasome system (PSMB8, PSMA3, PSMB4, PSMB8, and PSMB9) and lead to uncontrolled IFN-1 activation. Clinically, early-onset neutrophilic dermatosis with mononuclear interstitial infiltrate appears to be pathognomonic for PRAAS/CANDLE (49). Comparable to other interferonopathies, vasculopathy, perivascular infiltrates, and neutrophilic vasculitis can be observed, and a case of ANCA-associated vasculitis has been reported (50). Self-limited attacks of myositis are frequently observed in patients with PRAAS/CANDLE. However, unlike juvenile dermatomyositis, the muscle MRIs in PRAAS/CANDLE shows patchy distribution of muscle inflammation (12). Pulmonary manifestations, such as pulmonary hypertension, can be life-threatening. Unlike SAVI and COPA, lung disease rarely progresses to ILD in PRAAS/CANDLE. Pulmonary function tests and echocardiograms are recommended for screening and monitoring. Patients with PRAAS/CANDLE may also present with neurologic features such as headaches and aseptic meningitis. Lumbar puncture typically reveals sterile cerebrospinal fluid with pleocytosis. During PRAAS/CANDLE disease flares, laboratory studies show cytopenia, nonspecific hypochromic or normocytic anemia, increased acute-phase reactant levels, elevated transaminases, and abnormal muscle enzymes (49). A child with atypical myositis and cutaneous rash who fails to respond to standard treatment regimens should raise concerns about the possibility of an underlying interferonopathy such as PRAAS/CANDLE. PRAAS/CANDLE patients are also at higher risk of developing metabolic syndrome, especially with the use of glucocorticoid therapies. The JAK inhibitor baricitinib has been used successfully with reported reduction in daily symptoms and glucocorticoid requirements (12).
SLE is a chronic multisystem inflammatory disease characterized by elevated IFN-1 activity in approximately half of patients. Monogenic or early-onset SLE is strongly linked to genetics and known SLE-associated risk alleles, whereas the more common (“classic”) SLE that presents in older children and adults is thought to be more multifactorial in etiology. Sex differences are less pronounced in monogenic SLE in comparison to classic SLE. Over 100 susceptibility loci for SLE and 35 genes for monogenic SLE and SLE-like disease have been discovered by genome-wide association studies (51, 52). Younger age at presentation of SLE predicts damaging variants in genes that cause monogenic SLE regardless of consanguinity, ancestry, sex, or the prevalence of individual clinical features (53).
Monogenic SLE was first described in the 1970s and found to be caused by early complement deficiencies (C1q, C1r/C1s, C2, and C4). While complement pathway defects continue to be the most common subcategory of monogenic SLE, the second leading cause of monogenic SLE is in mutation in genes related to IFN regulation. Autosomal recessive TREX1 gene defects have recently been linked to SLE, familial chilblain lupus, and Aicardi-Goutières syndrome (AGS) (54). TREX1 deficiency leads to the accumulation of cytosolic DNA fragments that activates the innate DNA sensing response, resulting in IFN overproduction. Mutations in genes related to IFN regulation such as SAMHD1, ADAR1, IFIH1, RNASEH2A, RNASEH2B, and RNASEH2C have also been identified as causing familial chilblain lupus and AGS (55–58).
AGS presents early in infancy, often with chilblains. Neurologic manifestations are prominent in patients with AGS, such as early-onset encephalopathy, basal ganglia or white matter calcifications, focal motor findings, and seizures. Unlike the previously discussed interferonopathies, progressive neurologic impairment is common in AGS. Neuroimaging is required at baseline and follow-up to evaluate CNS vasculopathy, which is often referred to as Moyamoya disease. Cerebral vasculitis can mimic early AGS and lead to delays in diagnosis and likely worse outcomes. Similar to non-Mendelian SLE, autoantibodies can be present in monogenic SLE and AGS, along with cytopenias (e.g., anemia and thrombocytopenia) and elevated transaminases. Elevation in transaminases may be due to hepatic involvement or myositis. Cardiac involvement is rarely observed in AGS.
About 90%–95% of children with monogenic SLE generally satisfy the 2019 European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) and 2012 Systemic Lupus International Collaborating Clinics Classification criteria. Positive antinuclear antibodies with malar rash are the most common clinical features, occurring in 97% of children with monogenic SLE (53, 59). Glomerulonephritis is reported with unclear frequency and is often partially responsive to conventional immunosuppressive medications (60). IFN-1 contributes to glomerular injury through direct damage to kidney cells, induction of autoantibody generation and immune complex deposition, and inflammatory cell chemotaxis (61). The most frequent renal biopsy pattern is proliferative nephritis. However, vascular changes, such as thrombotic microangiopathy, have been reported (62). Glucocorticoids are effective. Often patients with monogenic SLE have limited response to classic SLE therapy like hydroxychloroquine, azathioprine, MMF, and cyclophosphamide. With the fundamental discoveries about pathogenesis of interferonopathies, multiple clinical trials are currently being conducted to evaluate the efficacy and safety of JAK inhibitors.
DADA2 is an autosomal recessive autoinflammatory disorder caused by mutations in the ADA2 gene (63, 64). The ADA2 enzyme is primarily found extracellularly and regulates the innate immune response. The exact mechanism by which this mutation leads to systemic inflammation is unknown. However, it has been suggested that a disruption in extracellular adenosine regulation leads to the generation of neutrophil extracellular traps (NETs). NET formation leads to macrophage activation and subsequent release of TNF-α (65, 66). Recently, patients with active DADA2 also demonstrated an IFN-1 signature that may be correlated with disease activity (67, 68). Multiple mechanisms have been proposed to explain the increased IFN-1 expression, but the significance of this increase, and its interaction with TNF-α, remains unclear (69, 70).
Clinically, DADA2 has a very similar presentation to polyarteritis nodosa (PAN) with fever, medium-vessel vasculitis, livedo racemosa, and early-onset stroke (71). Although neurologic and skin involvement is the most common, any organ system can be affected. Gastrointestinal, renal (Figure 1), cardiopulmonary, and musculoskeletal involvement can occur as well (72). Patients with DADA2 may display a degree of immune dysregulation, although this symptom is variable and strongly associated with genotype. Immune dysregulation may be limited to mild hypogammaglobulinemia and cytopenias but may be as severe as pure red cell aplasia and bone marrow failure (73, 74). While early age of onset can assist in differentiating DADA2 from PAN, some cases of infantile PAN may not have DADA2 mutations and some cases of DADA2 may not present until the second decade of life (74). Thus, it is reasonable to consider DADA2 testing in pediatric patients with symptoms consistent with PAN, especially if they have symptoms of immune dysregulation that would be more atypical in PAN. TNF inhibitors are a very effective treatment for vasculitis-predominant phenotypes (75, 76). JAK inhibitors may also be effective (77). Notably, severe hematological manifestations tend to be resistant to typical treatments and often require BMT (73).
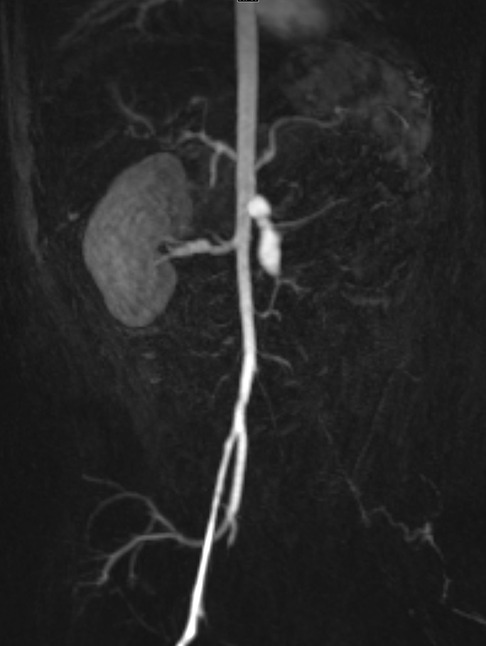
Figure 1. Renal infarct in an infant with DADA2. Severe narrowing and proximal left common iliac artery occlusion at the bifurcation. Fusiform, aneurysmal dilation of the mid to distal superior mesenteric artery segment. Renal artery and kidney are absent.
Type I interferonopathies are a group of heterogeneous diseases associated with increased IFN-1 stimulated gene expression. Many of the monogenic interferonopathies mimic more common autoimmune diseases such as SLE, juvenile myositis, PAN-, ANCA-, and CNS-vasculitis. Given the significant overlap in clinical features among these disorders, a high index of suspicion and genetic evaluation to confirm the specific disease are crucial for diagnosis and management. As testing methods such as next-generation sequencing—including targeted gene panels and whole exome or whole genome sequencing—have become more affordable and accessible over recent years, screening for pathogenic variants is now recommended by EULAR/ACR (78).
Including such diverse diseases under type I interferonopathies also has important treatment implications. Historically these diseases have required multiple immunosuppressive treatments and sometimes even BMT. However, the development of JAK inhibitors has led to significant advances in treatment. New recommendations from EULAR/ACR now include using JAK inhibitors to treat PRAAS/CANDLE, SAVI, and AGS, and growing evidence suggests additional utility in other type I interferonopathies (78). In addition, several ongoing clinical trials with JAK inhibitors are being conducted in patients with SLE.
Despite the significant advances in the last decade, it remains unclear why some interferonopathies lead to systemic autoinflammatory and autoimmune disease while others present with prominent neurologic or vascular features. Additionally, JAK inhibitors have demonstrated promise in treating type I interferonopathies. However, they may be ineffective in some cases. Therefore, further work is necessary to identify novel mutations and subsequently elucidate pathophysiology to develop new therapeutic options that will improve quality of life and decrease complications of these rare diseases.
SG and SA contributed to the conception and design of the article. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. (1957) 147(927):258–67. doi: 10.1098/rspb.1957.0048
2. Kopitar-Jerala N. The role of interferons in inflammation and inflammasome activation. Front Immunol. (2017) 8:873. doi: 10.3389/fimmu.2017.00873
3. de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol. (2015) 33:823–74. doi: 10.1146/annurev-immunol-032414-112227
4. Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. (2011) 1238:91–8. doi: 10.1111/j.1749-6632.2011.06220.x
5. Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med. (2016) 213(12):2527–38. doi: 10.1084/jem.20161596
6. Magro CM, Poe JC, Kim C, Shapiro L, Nuovo G, Crow MK, et al. Degos disease: a C5b-9/interferon-alpha-mediated endotheliopathy syndrome. Am J Clin Pathol. (2011) 135(4):599–610. doi: 10.1309/AJCP66QIMFARLZKI
7. Muskardin TLW, Niewold TB. Type I interferon in rheumatic diseases. Nat Rev Rheumatol. (2018) 14(4):214–28. doi: 10.1038/nrrheum.2018.31
8. d’Angelo DM, Di Filippo P, Breda L, Chiarelli F. Type I interferonopathies in children: an overview. Front Pediatr. (2021) 9:631329. doi: 10.3389/fped.2021.631329
9. Crow YJ, Stetson DB. The type I interferonopathies: 10 years on. Nat Rev Immunol. (2022) 22(8):471–83. doi: 10.1038/s41577-021-00633-9
10. Demers-Mathieu V. Optimal selection of Ifn-alpha-inducible genes to determine type I interferon signature improves the diagnosis of systemic lupus erythematosus. Biomedicines. (2023) 11(3):864. doi: 10.3390/biomedicines11030864
11. Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. (2015) 15(7):429–40. doi: 10.1038/nri3850
12. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. Jak1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. (2018) 128(7):3041–52. doi: 10.1172/JCI98814
13. Wang Y, Wang F, Zhang X. Sting-Associated vasculopathy with onset in infancy: a familial case series report and literature review. Ann Transl Med. (2021) 9(2):176. doi: 10.21037/atm-20-6198
14. Winthrop KL. The emerging safety profile of jak inhibitors in rheumatic disease. Nat Rev Rheumatol. (2017) 13(4):234–43. doi: 10.1038/nrrheum.2017.23
15. Wathes R, Moule S, Milojkovic D. Progressive multifocal leukoencephalopathy associated with ruxolitinib. N Engl J Med. (2013) 369(2):197–8. doi: 10.1056/NEJMc1302135
16. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated sting in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371(6):507–18. doi: 10.1056/NEJMoa1312625
17. Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of sting requires palmitoylation at the Golgi. Nat Commun. (2016) 7:11932. doi: 10.1038/ncomms11932
18. Lin B, Berard R, Al Rasheed A, Aladba B, Kranzusch PJ, Henderlight M, et al. A novel sting1 variant causes a recessive form of sting-associated vasculopathy with onset in infancy (savi). J Allergy Clin Immunol. (2020) 146(5):1204–8 e6. doi: 10.1016/j.jaci.2020.06.032
19. Lu W, Zhang Y, McDonald DO, Jing H, Carroll B, Robertson N, et al. Dual proteolytic pathways govern glycolysis and immune competence. Cell. (2014) 159(7):1578–90. doi: 10.1016/j.cell.2014.12.001
20. Fremond ML, Hadchouel A, Berteloot L, Melki I, Bresson V, Barnabei L, et al. Overview of sting-associated vasculopathy with onset in infancy (savi) among 21 patients. J Allergy Clin Immunol Pract. (2021) 9(2):803–18 e11. doi: 10.1016/j.jaip.2020.11.007
21. Livingston JH, Stivaros S, van der Knaap MS, Crow YJ. Recognizable phenotypes associated with intracranial calcification. Dev Med Child Neurol. (2013) 55(1):46–57. doi: 10.1111/j.1469-8749.2012.04437.x
22. Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, et al. Copa mutations impair Er-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. (2015) 47(6):654–60. doi: 10.1038/ng.3279
23. Volpi S, Tsui J, Mariani M, Pastorino C, Caorsi R, Sacco O, et al. Type I interferon pathway activation in Copa syndrome. Clin Immunol. (2018) 187:33–6. doi: 10.1016/j.clim.2017.10.001
24. Deng Z, Chong Z, Law CS, Mukai K, Ho FO, Martinu T, et al. A defect in copi-mediated transport of sting causes immune dysregulation in Copa syndrome. J Exp Med. (2020) 217(11):e20201045. doi: 10.1084/jem.20201045
25. Lepelley A, Martin-Niclos MJ, Le Bihan M, Marsh JA, Uggenti C, Rice GI, et al. Mutations in Copa lead to abnormal trafficking of sting to the Golgi and interferon signaling. J Exp Med. (2020) 217(11):e20200600. doi: 10.1084/jem.20200600
26. Fremond ML, Crow YJ. Sting-Mediated lung inflammation and beyond. J Clin Immunol (2021) 41(3):501–14. doi: 10.1007/s10875-021-00974-z
27. Vece TJ, Watkin LB, Nicholas S, Canter D, Braun MC, Guillerman RP, et al. Copa syndrome: a novel autosomal dominant immune dysregulatory disease. J Clin Immunol. (2016) 36(4):377–87. doi: 10.1007/s10875-016-0271-8
28. Tsui JL, Estrada OA, Deng Z, Wang KM, Law CS, Elicker BM, et al. Analysis of pulmonary features and treatment approaches in the Copa syndrome. ERJ Open Res. (2018) 4(2):00017-2018. doi: 10.1183/23120541.00017-2018
29. Noorelahi R, Perez G, Otero HJ. Imaging findings of Copa syndrome in a 12-year-old boy. Pediatr Radiol. (2018) 48(2):279–82. doi: 10.1007/s00247-017-3961-3
30. Taveira-DaSilva AM, Markello TC, Kleiner DE, Jones AM, Groden C, Macnamara E, et al. Expanding the phenotype of Copa syndrome: a kindred with typical and atypical features. J Med Genet. (2019) 56(11):778–82. doi: 10.1136/jmedgenet-2018-105560
31. Prenzel F, Harfst J, Schwerk N, Ahrens F, Rietschel E, Schmitt-Grohe S, et al. Lymphocytic interstitial pneumonia and follicular bronchiolitis in children: a registry-based case series. Pediatr Pulmonol. (2020) 55(4):909–17. doi: 10.1002/ppul.24680
32. Lodi L, Mastrolia MV, Bello F, Rossi GM, Angelotti ML, Crow YJ, et al. Type I interferon-related kidney disorders. Kidney Int. (2022) 101(6):1142–59. doi: 10.1016/j.kint.2022.02.031
33. Thaivalappil SS, Garrod AS, Borowitz SM, Watkin LB, Lawrence MG. Persistent unexplained transaminitis in Copa syndrome. J Clin Immunol. (2021) 41(1):205–8. doi: 10.1007/s10875-020-00832-4
34. Guan Y, Liu H, Tang X, Xu H, Peng Y, Zhou C, et al. Effective sirolimus treatment of 2 Copa syndrome patients. J Allergy Clin Immunol Pract. (2021) 9(2):999–1001 e1. doi: 10.1016/j.jaip.2020.10.019
35. Krutzke S, Rietschel C, Horneff G. Baricitinib in therapy of Copa syndrome in a 15-year-old girl. Eur J Rheumatol. (2019) 7(Suppl 1):1–4. doi: 10.5152/eurjrheum.2019.18177
36. Fremond ML, Legendre M, Fayon M, Clement A, Filhol-Blin E, Richard N, et al. Use of ruxolitinib in Copa syndrome manifesting as life-threatening alveolar haemorrhage. Thorax. (2020) 75(1):92–5. doi: 10.1136/thoraxjnl-2019-213892
37. Scott O, Lindsay K, Erwood S, Mollica A, Roifman CM, Cohn RD, et al. Stat1 gain-of-function heterozygous cell models reveal diverse interferon-signature gene transcriptional responses. NPJ Genom Med. (2021) 6(1):34. doi: 10.1038/s41525-021-00196-7
38. Okada S, Asano T, Moriya K, Boisson-Dupuis S, Kobayashi M, Casanova JL, et al. Human Stat1 gain-of-function heterozygous mutations: chronic mucocutaneous candidiasis and type I interferonopathy. J Clin Immunol. (2020) 40(8):1065–81. doi: 10.1007/s10875-020-00847-x
39. Kaleviste E, Saare M, Leahy TR, Bondet V, Duffy D, Mogensen TH, et al. Interferon signature in patients with Stat1 gain-of-function mutation is epigenetically determined. Eur J Immunol. (2019) 49(5):790–800. doi: 10.1002/eji.201847955
40. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous Stat1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. (2016) 127(25):3154–64. doi: 10.1182/blood-2015-11-679902
41. Bierman-Chow S, Freeman AF, Holland SM, Lynch J, Cho HJ. Cerebral aneurysm in three pediatric patients with Stat1 gain-of-function mutations. J Neurol. (2022) 269(10):5638–42. doi: 10.1007/s00415-022-11131-w
42. Tanimura M, Dohi K, Hirayama M, Sato Y, Sugiura E, Nakajima H, et al. Recurrent inflammatory aortic aneurysms in chronic mucocutaneous candidiasis with a gain-of-function Stat1 mutation. Int J Cardiol. (2015) 196:88–90. doi: 10.1016/j.ijcard.2015.05.183
43. Sener S, Basaran O, Batu ED, Atalay E, Esenboga S, Cagdas D, et al. Childhood-onset Takayasu arteritis and immunodeficiency: case-based review. Clin Rheumatol. (2022) 41(9):2883–92. doi: 10.1007/s10067-022-06295-9
44. Maeshima K, Ishii K, Shibata H. An adult fatal case with a Stat1 gain-of-function mutation associated with multiple autoimmune diseases. J Rheumatol. (2019) 46(3):325–7. doi: 10.3899/jrheum.180210
45. Papadopoulou C, Omoyinmi E, Standing A, Pain CE, Booth C, D’Arco F, et al. Monogenic mimics of Behcet's disease in the young. Rheumatology (Oxford). (2019) 58(7):1227–38. doi: 10.1093/rheumatology/key445
46. Girardelli M, Valencic E, Moressa V, Margagliotta R, Tesser A, Pastore S, et al. Genetic and immunologic findings in children with recurrent aphthous stomatitis with systemic inflammation. Pediatr Rheumatol Online J. (2021) 19(1):70. doi: 10.1186/s12969-021-00552-y
47. Deya-Martinez A, Riviere JG, Roxo-Junior P, Ramakers J, Bloomfield M, Guisado Hernandez P, et al. Impact of jak inhibitors in pediatric patients with Stat1 gain of function (Gof) mutations-10 children and review of the literature. J Clin Immunol. (2022) 42(5):1071–82. doi: 10.1007/s10875-022-01257-x
48. Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (Stat1) or Stat3 mutations. J Allergy Clin Immunol. (2018) 142(5):1665–9. doi: 10.1016/j.jaci.2018.07.020
49. Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, et al. Mutations in proteasome subunit Beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. (2012) 64(3):895–907. doi: 10.1002/art.33368
50. Yamazaki-Nakashimada MA, Santos-Chavez EE, de Jesus AA, Rivas-Larrauri F, Guzman-Martinez MN, Goldbach-Mansky R, et al. Systemic autoimmunity in a patient with candle syndrome. J Investig Allergol Clin Immunol. (2019) 29(1):75–6. doi: 10.18176/jiaci.0338
51. Yin X, Kim K, Suetsugu H, Bang SY, Wen L, Koido M, et al. Meta-analysis of 208370 east asians identifies 113 susceptibility loci for systemic lupus erythematosus. Ann Rheum Dis. (2021) 80(5):632–40. doi: 10.1136/annrheumdis-2020-219209
52. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in Trex1, Rnaseh2a, Rnaseh2b, Rnaseh2c, Samhd1, Adar, and Ifih1. Am J Med Genet A. (2015) 167A(2):296–312. doi: 10.1002/ajmg.a.36887
53. Misztal MC, Liao F, Couse M, Cao J, Dominguez D, Lau L, et al. Genome-wide sequencing identified rare genetic variants for childhood-onset monogenic lupus. J Rheumatol. (2023) 50(5):671–5. doi: 10.3899/jrheum.220513
54. Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, et al. Heterozygous mutations in Trex1 cause familial chilblain lupus and dominant aicardi-goutieres syndrome. Am J Hum Genet. (2007) 80(4):811–5. doi: 10.1086/513443
55. Ramantani G, Hausler M, Niggemann P, Wessling B, Guttmann H, Mull M, et al. Aicardi-Goutieres syndrome and systemic lupus erythematosus (Sle) in a 12-year-old boy with Samhd1 mutations. J Child Neurol. (2011) 26(11):1425–8. doi: 10.1177/0883073811408310
56. Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, et al. Gain-of-function mutations in Ifih1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. (2014) 46(5):503–9. doi: 10.1038/ng.2933
57. Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, et al. Mutations in Adar1 cause aicardi-goutieres syndrome associated with a type I interferon signature. Nat Genet. (2012) 44(11):1243–8. doi: 10.1038/ng.2414
58. Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, et al. Mutations involved in aicardi-goutieres syndrome implicate Samhd1 as regulator of the innate immune response. Nat Genet. (2009) 41(7):829–32. doi: 10.1038/ng.373
59. Batu ED, Kosukcu C, Taskiran E, Sahin S, Akman S, Sozeri B, et al. Whole exome sequencing in early-onset systemic lupus erythematosus. J Rheumatol. (2018) 45(12):1671–9. doi: 10.3899/jrheum.171358
60. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3'-5’ DNA exonuclease Trex1 are associated with systemic lupus erythematosus. Nat Genet. (2007) 39(9):1065–7. doi: 10.1038/ng2091
61. Ding X, Ren Y, He X. Ifn-I mediates lupus nephritis from the beginning to renal fibrosis. Front Immunol. (2021) 12:676082. doi: 10.3389/fimmu.2021.676082
62. Kavanagh D, McGlasson S, Jury A, Williams J, Scolding N, Bellamy C, et al. Type I interferon causes thrombotic microangiopathy by a dose-dependent toxic effect on the microvasculature. Blood. (2016) 128(24):2824–33. doi: 10.1182/blood-2016-05-715987
63. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-Onset stroke and vasculopathy associated with mutations in Ada2. N Engl J Med. (2014) 370(10):911–20. doi: 10.1056/NEJMoa1307361
64. Elkan PN, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis Nodosa vasculopathy. N Engl J Med. (2014) 370(10):921–31. doi: 10.1056/NEJMoa1307362
65. Kendall JL, Springer JM. The many faces of a monogenic autoinflammatory disease: adenosine deaminase 2 deficiency. Curr Rheumatol Rep. (2020) 22(10):64. doi: 10.1007/s11926-020-00944-1
66. Carmona-Rivera C, Khaznadar SS, Shwin KW, Irizarry-Caro JA, O'Neil LJ, Liu Y, et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated netosis and tnf production in patients with Dada2. Blood. (2019) 134(4):395–406. doi: 10.1182/blood.2018892752
67. Trivioli G, Gelain E, Angelotti ML, Ravaglia F, Allinovi M, Lodi L, et al. A report of 2 cases of kidney involvement in Ada2 deficiency: different disease phenotypes and the tissue response to type I interferon. Am J Kidney Dis. (2022) 80(5):677–82. doi: 10.1053/j.ajkd.2022.05.008
68. Insalaco A, Moneta GM, Pardeo M, Caiello I, Messia V, Bracaglia C, et al. Variable clinical phenotypes and relation of interferon signature with disease activity in Ada2 deficiency. J Rheumatol. (2019) 46(5):523–6. doi: 10.3899/jrheum.180045
69. Greiner-Tollersrud OK, Boehler V, Bartok E, Krausz M, Polyzou A, Schepp J, et al. Ada2 is a lysosomal dnase regulating the type-I interferon response. bioRxiv. (2020):2020.06.21.162990. doi: 10.1101/2020.06.21.162990
70. Dhanwani R, Takahashi M, Mathews IT, Lenzi C, Romanov A, Watrous JD, et al. Cellular sensing of extracellular purine nucleosides triggers an innate Ifn-Beta response. Sci Adv. (2020) 6(30):eaba3688. doi: 10.1126/sciadv.aba3688
71. Meyts I, Aksentijevich I. Deficiency of adenosine deaminase 2 (Dada2): updates on the phenotype, genetics, pathogenesis, and treatment. J Clin Immunol. (2018) 38(5):569–78. doi: 10.1007/s10875-018-0525-8
72. Lee PY, Aksentijevich I, Zhou Q. Mechanisms of vascular inflammation in deficiency of adenosine deaminase 2 (Dada2). Semin Immunopathol. (2022) 40:269–80. doi: 10.1007/s00281-022-00918-8
73. Lee PY, Kellner ES, Huang Y, Furutani E, Huang Z, Bainter W, et al. Genotype and functional correlates of disease phenotype in deficiency of adenosine deaminase 2 (Dada2). J Allergy Clin Immunol. (2020) 145(6):1664–72 e10. doi: 10.1016/j.jaci.2019.12.908
74. Gibson KM, Morishita KA, Dancey P, Moorehead P, Drogemoller B, Han X, et al. Identification of novel adenosine deaminase 2 gene variants and varied clinical phenotype in pediatric vasculitis. Arthritis Rheumatol. (2019) 71(10):1747–55. doi: 10.1002/art.40913
75. Deuitch NT, Yang D, Lee PY, Yu X, Moura NS, Schnappauf O, et al. Tnf inhibition in vasculitis management in adenosine deaminase 2 deficiency (Dada2). J Allergy Clin Immunol. (2022) 149(5):1812–6.e6. doi: 10.1016/j.jaci.2021.10.030
76. Ombrello AK, Qin J, Hoffmann PM, Kumar P, Stone D, Jones A, et al. Treatment strategies for deficiency of adenosine deaminase 2. N Engl J Med. (2019) 380(16):1582–4. doi: 10.1056/NEJMc1801927
77. Du Y, Liu M, Nigrovic P, Dedeoglu F, Lee PY. Biologics and jak inhibitors for the treatment of monogenic systemic autoinflammatory diseases in children. J Allergy Clin Immunol (2023) 151(3):607–18. doi: 10.1016/j.jaci.2022.12.816
78. Cetin Gedik K, Lamot L, Romano M, Demirkaya E, Piskin D, Torreggiani S, et al. The 2021 European alliance of associations for rheumatology/American college of rheumatology points to consider for diagnosis and management of autoinflammatory type I interferonopathies: Candle/Praas, Savi, and Ags. Arthritis Rheumatol. (2022) 74(5):735–51. doi: 10.1002/art.42087
Keywords: early-onset, vasculitis, vasculopathy, interferonopathy, autoinflammatory, monogenic lupus
Citation: Gagne S, Sivaraman V and Akoghlanian S (2023) Interferonopathies masquerading as non-Mendelian autoimmune diseases: pattern recognition for early diagnosis. Front. Pediatr. 11:1169638. doi: 10.3389/fped.2023.1169638
Received: 19 February 2023; Accepted: 5 July 2023;
Published: 9 August 2023.
Edited by:
Eveline Wu, University of North Carolina at Chapel Hill, United StatesReviewed by:
Theresa Lynn Wampler Muskardin, Hospital for Special Surgery, United States© 2023 Gagne, Sivaraman and Akoghlanian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samuel Gagne c2FtdWVsamdhZ25lQGdtYWlsLmNvbQ== Shoghik Akoghlanian U2hvZ2hpay5ha29naGxhbmlhbkBuYXRpb253aWRlY2hpbGRyZW5zLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.