 Alexandre Stambouli1
Alexandre Stambouli1 Audrey Cartault2Isabelle Oliver Petit2Solene Evrard3Eliane Mery3
Audrey Cartault2Isabelle Oliver Petit2Solene Evrard3Eliane Mery3 Frederique Savagner1,4,†*
Frederique Savagner1,4,†* Stephanie Trudel1,5,†
Stephanie Trudel1,5,†
- 1Molecular Biology Department, Federative Institute of Biology, Toulouse, France
- 2Endocrinology Department, Children's Hospital of Toulouse, Toulouse, France
- 3Pathology Department, IUCT, Institut Claudius Regaud, Toulouse, France
- 4Inserm UMR 1297, Toulouse, France
- 5Inserm UMR 1291, CHU Purpan—BP, Toulouse, France
Background: Embryonal rhabdomyosarcomas (ERMS) of the uterine cervix and corpus are rare pediatric tumors usually associated with a late age of onset and frequent somatic DICER1 mutation. It may also develop in the context of a familial predisposition such as DICER1 syndrome requiring specific medical care for children and young adults at risk for a broad range of tumors.
Case presentation: This is a case of a prepubescent 9-year-old girl who was presented to our department for metrorrhagias due to a vaginal cervical mass, initially classified as a müllerian endocervical polyp on negative myogenin immunostaining. The patient subsequently manifested growth retardation (-2DS) and learning disabilities leading to genetic explorations and the identification of a germline pathogenic DICER1 variant. The family history revealed thyroid diseases in the father, aunt and paternal grandmother before the age of 20.
Conclusion: Rare tumors such as cervical ERMS associated with a family history of thyroid disease during infancy could be related to DICER1 syndrome. Identifying at-risk relatives is challenging but necessary to detect early DICER1 spectrum tumors in young patients.
Introduction
DICER1 syndrome is a rare autosomal dominant genetic disorder which predisposes to the development of a wide range of both benign and malignant tumors. This syndrome has been linked to pathogenic germline mutations of the DICER1 gene which encodes an endoribonuclease involved in the maturation of microRNAs (miRNAs) and therefore the regulation of gene expression (1). DICER1 acts as a tumor suppressor where mutations in one gene allele lead to an increased risk of tumor development and the necessity of a somatic mutation in the second allele to produce a malignant phenotype. The majority of germline mutations are located throughout the entire gene, whereas somatic mutations have been found in the regions that encode the RNase III domains.
DICER1 syndrome is strongly suspected when typical DICER1-associated tumors such as lung cysts, pleuropulmonary blastoma (PPB), cystic nephroma, ovarian sex-cord stromal tumor and multinodular goiter develop in the early stages of childhood (2, 3). Recently a list of less commonly observed tumors including embryonal rhabdomyosarcoma (ERMS) of the bladder, uterine cervix or fallopian tubes have also been described (4–6).
Incomplete penetrance of germline pathogenic DICER1 variant and the low risk for relatives of developing a tumor before the age of 10 complicate care recommendations which have been recently discussed (7). Although DICER1 syndrome remains a rare entity it often leads to a delay in medical care for the proband with a high risk of malignant tumor development.
This is a case report of a female child incidentally diagnosed with rare DICER1-associated ERMS of the cervix after multiple investigations.
Case report
The female patient was born at term with a history of intrauterine growth restriction (IUGR; s:46 cm; w:2,550 g, hc: 34 cm) and neonatal dysmorphia, leading to neonatal urinary glycosaminoglycans (GAGs) analysis for screening of mucopolysaccharidosis with inconclusive results. She is a first child born at 39 weeks of amenorrhoea after normal pregnancy and delivery (mother's age: 24 yrs). At the age of 9 the girl was referred to our pediatric gynecology department for metrorrhagias. Clinical and physical examination revealed no sign of pubertal development but retrieved a cervically protruding vaginal mass corresponding to multiple polyps of 0.5 to 2 cm length. After surgical resection, the histopathological analysis indicated benign müllerian endocervical polyps with variable cellularity, mild atypia, squamous-like epithelium and diffuse Smooth Muscle Actin but negative Myogenin immunostaining. Serum tumor markers alpha-fetoprotein (AFP), carcinoembryonic antigen (CEA) and human chorionic gonadotropin (hCG) were negative.
A year later, during clinical follow-up, the patient presented a decline in growth rate with growth retardation (-2 SD) (Figure 1A). A physical examination revealed prognathism, macroglossia and brachymetacarpia with no other remarkable factors. Isolated partial growth hormone (GH) deficiency was diagnosed on stimulation tests. Imaging tests, including hand and wrist x-ray, thyroid echography and pituitary magnetic resonance imaging (MRI) were all normal. Further cytogenetic and molecular testing showed normal karyotype (46XX) and no SHOX deficiency. Complementary hormonal results ([thyroid stimulating hormone (TSH), free thyroxine (FT4), parathyroid hormone (PTH), adrenocorticotropic hormone (ACTH), cortisol, prolactin, estradiol, anti-müllerian hormone (AMH), insulin-like growth factor 1 (IGF1), insulin-like growth factor binding protein-3 (IGF-BP3)]) before synthetic GH treatment (initial dose: 0.3 mg/kg/week) were all normal (details in Table 1).
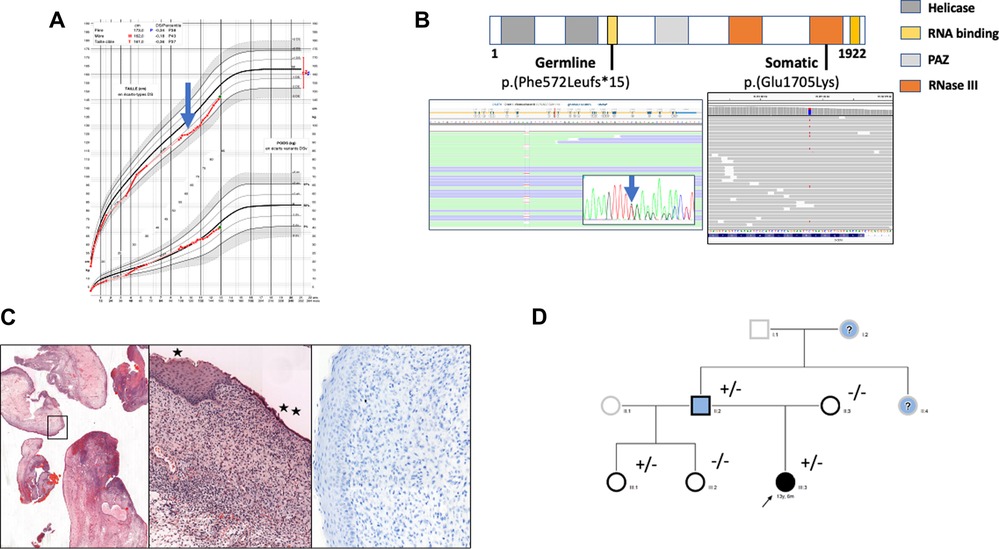
Figure 1. (A) growth curve of the female child. At 10 years old growth retardation (-2 SD) was noted (blue arrow) and synthetic growth hormone treatment was started. (B) Localization of identified somatic and germline variants. Germline heterozygous T deletion (c.1716del, p.(Phe572LeufsTer15)) and somatic heterozygous substitution c.5113G > A, p.(Glu1705Lys) were detected by next generation sequencing (Illumina, custom metabolic disorders panel) in exon 10 and 27 respectively of the DICER1 gene (LRG_492, NM_177438.3; Sequencing depth was 874 x and 756 x respectively (Integrative Genomic Viewer Software). The germline variant was confirmed by Sanger sequencing (blue arrow). (C) Histologic and immunohistochemical examination of cervical tissue samples. Edematous congestive polyp fragments, hematoxylin and eosin (HE) staining, original magnification×25 (left panel); Surface squamous (*) or mucinous (**) epithelium without atypia, the underlaying proliferative cells are monotonous without atypia or mitosis, HE×100 (middle panel); Myogenin staining showing no myogenin expression,×200 (right panel). (D) Family pedigree. Individuals screened for DICER1 germline variant are indicated by a black border. Clinical phenotypes are represented by black filling for ERMS, blue for thyroid disease and white for asymptomatic. The proband is marked with an arrow. “+” and “-” indicate the mutation status. Question marks correspond to symptomatic patients whose DICER1 mutational status is unknown.
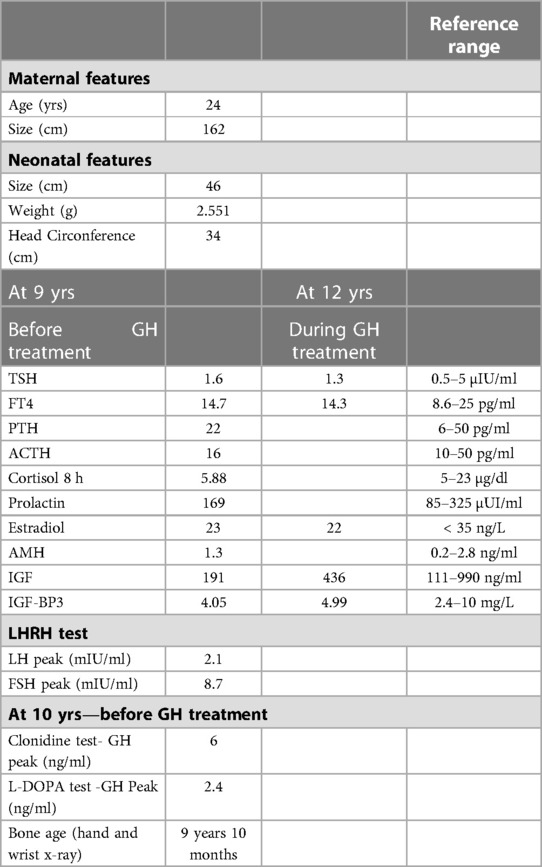
Table 1. Clinical and hormonl features for the index case and her mother.
At the age of 12, due to scholastic difficulties and learning disabilities associated with facial dysmorphism and a history of screening for mucoploysaccharidosis, a more comprehensive genetic exploration was performed using a broad gene panel for rare pediatric disorders combined with next-generation sequencing (NGS)(for complete panel see additional Supplementary Material Table S1). After obtaining written informed consent for genetic testing, no variant in mucopolysaccharidosis-related genes could be identified while an heterozygous pathogenic frameshift variant in exon 10 of the DICER1 gene (NM_177438.3) designated as c.1716del; p.(Phe572LeufsTer15) was found. This variant leads to a premature stop codon in the P-loop containing the nucleoside triphosphate hydrolase-domain of the protein. The mutation was confirmed on a second blood sample.
The identification of this DICER1 germline mutation led us to perform a molecular analysis on the endocervical mass. We found a second-hit somatic missense variation on the other DICER1 gene allele designated c.5113G > A; p.(Glu1705Lys) in exon 24 (allele frequency: 27%) corresponding to the known genetic hotspot for somatic mutations in the ribonuclease III domain (Figure 1B).
In light of DICER1 gene germline and somatic mutations, a pathological reassessment of the cervical resection was performed in a reference center, allowing reclassification from benign müllerian polyps to embryonal rhabdomyosarcoma with immunostaining focally positive for desmin and still negative for myogenin and myoD1 (Figure 1C). Together, all these results led to a diagnosis of DICER1 syndrome and the appropriate medical care for the patient and her family.
Family screening revealed that the father carried the same nucleotide deletion, which is consistent with thyroidectomy at the age of 16 for a multinodular goiter. A history of benign thyroid nodules in the father's mother and sister was also reported (Figure 1D). No other DICER1-related pathologies have been described in the family especially no PPB over three generations. Furthermore, extended genetic counseling was suggested for the father's family, in particular the proband's half-sisters. The youngest, who was 2 years old, carried the same familial deletion but presented no clinical or imaging manifestations at screening.
Discussion
DICER1 syndrome is associated with a panel of benign and malignant tumoral manifestations, which, in terms of unusual phenotypes, has not yet been completed. Our case is consistent with a double-hit tumorigenic process: the germline pathogenic variant being a frameshift caused by a deletion and the somatic variant found in the cervical tumor being a missense mutation c.5113G > A; p.(Glu1705Lys) in the catalytic domain of the protein, previously described in DICER1-related tumors (8). Concerning the germline variant c. 1716del, it was previously described in an infant with a PPB at the age of 0.8 years, corresponding to the youngest child in the series of 11 PPB-families (9).
ERMS is the most common subtype of rhabdomyosarcoma, with a high prevalence in the head and neck region as well as in the genitourinary tract, usually characterized by a positive immunohistochemistry for desmin, myogenin and MyoD1 (10, 11). The “botryoid” variant is a particular form of ERMS found in mucosa, typically presenting as a polyp. However, ERMS of the uterine cervix and corpus are particularly rare affections and, in our case the primary analysis showed no such typical immunostaining profile, which led to an inaccurate diagnosis of benign müllerian polyps. Moreover, DICER1-associated ERMS occurs most frequently in pubertal and post-pubertal adolescent girls and young women (mean 10–20 years). A recent study including five cervical and four uterine ERMS showed DICER1 pathogenic variants in all the uterine lesions whereas these variants were absent in the urinary tract ERMS. The authors suggest that DICER1-associated ERMS could be qualified as a distinct subtype in future classifications, especially when related to a specific DNA methylation profile (12).
Growth retardation was an important feature in this case, but is not usually related to DICER1 syndrome. In their exploration of pituitary development in families with the DICER1-truncating variant Zhang et al. showed that changes in miRNAs level could be the cause of clinical pituitary disorders but no evidence of a direct relation with growth hormone deficiency was noted (13). However, in a recent study on unusual symptoms of the DICER1 syndrome, Venger et al. described developmental delay and facial dysmorphism in a family carrying a DICER1 frameshift variant (6). Considering the higher risk of cancer development for pediatric patients receiving GH treatment, this young girl carrying a germline DICER1 pathogenic variant should benefit from reinforced sureveillance relative to current guidelines (14).
Family history is an important aspect in evaluating the risk of DICER1 syndrome. In this case, the association of a childhood tumor, although initially considered to be benign, and a familial history of multinodular goiter should alert the clinician to initiate genetic testing for DICER1 (ideally included in a NGS gene panel). In addition, screening and surveillance recommendations differ based on age and gender. Therefore, chest imaging is recommended from birth until 12 years of age since PPB is the main lung expression of DICER1 syndrome, whereas thyroid and pelvic ultrasound (US) are only recommended as of 8 years of age throughout life (7, 15). For this young girl who became pubertal in accordance with the standard timelines, follow-up was initiated and included regular physical exams, yearly pelvic and abdominal US until the age of 40, and thyroid US every 3 years. Fine needle aspiration biopsy (FNAB) could be used when focal changes in the thyroid gland appeared but due to the 16 to 18-fold increased risk of differentiated thyroid cancer especially in female DICER1 carrier, thyroidiectomy should be rapidly considered (16, 17). This was also the case for all the relatives' female children who were carriers of DICER1 deletion. The initial treatment was surgical resection alone (no additional treatments), and four years later any local or distant recurrences were monitored.
This case report highlights that DICER1 syndrome should be suspected in the presence of early typical DICER1-related tumors as well as in case of unusual tumors during childhood, especially when there is a family history of thyroid diseases during infancy or young adulthood.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
AS: physician manuscript writing AC and IOP: physicians, diagnosis, follow up, genetic counselling and taking care SE and EM: pathologists, expertise on ERMs and somatic molecular analysis. FS and ST: germinal molecular analysis and manuscript design. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1150418/full#supplementary-material.
References
1. Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. (2014) 14:662–72. doi: 10.1038/nrc3802
2. Réguerre Y, Golmard L, Brisse HJ, Oliver Petit I, Savagner F, Boudjemaa S, et al. Syndrome associé aux variants pathogènes constitutionnels de DICER1: où en sommes-nous en 2019? Bull Cancer. (2019) 106:1177–89. doi: 10.1016/j.bulcan.2019.08.016
3. Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. Germline DICER1 mutations in familial pleuropulmonary blastoma. Science. (2009) 325:965. doi: 10.1126/science.1174334
4. Abbo O, Pinnagoda K, Brouchet L, Leobon B, Savagner F, Oliver I, et al. Wilms tumor, pleuropulmonary blastoma, and DICER1: case report and literature review. World J Surg Oncol. (2018) 16:164. doi: 10.1186/s12957-018-1469-4
5. Robertson JC, Jorcyk CL, Oxford JT. DICER1 Syndrome: dICER1 mutations in rare cancers. Cancers (Basel). (2018) 10:E143. doi: 10.3390/cancers10050143
6. Venger K, Elbracht M, Carlens J, Deutz P, Zeppernick F, Lassay L, et al. Unusual phenotypes in patients with a pathogenic germline variant in DICER1. Fam Cancer. (2021. doi: 10.1007/s10689-021-00271-z. [Epub ahead of print]34331184
7. Schultz KAP, Williams GM, Kamihara J, Stewart DR, Harris AK, Bauer AJ, et al. DICER1 And associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res. (2018) 24:2251–61. doi: 10.1158/1078-0432.CCR-17-3089
8. Kim J, Schultz KAP, Hill DA, Stewart DR. The prevalence of germline DICER1 pathogenic variation in cancer populations. Mol Genet Genomic Med. (2019) 7:e555. doi: 10.1002/mgg3.555
9. Slade I, Bacchelli C, Davies H, Murray A, Abbaszadeh F, Hanks S, et al. DICER1 Syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet. (2011) 48:273–8. doi: 10.1136/jmg.2010.083790
10. Leiner J, Le Loarer F. The current landscape of rhabdomyosarcomas: an update. Virchows Arch. (2020) 476:97–108. doi: 10.1007/s00428-019-02676-9
11. Ibrahim U, Saqib A, Mohammad F, Ding J, Salman B, Collado FK, et al. Embryonal rhabdomyosarcoma of the cervix: a rare disease at an uncommon age. Cureus. (2017) 9:e1864. doi: 10.7759/cureus.1864
12. Kommoss FKF, Stichel D, Mora J, Esteller M, Jones DTW, Pfister SM, et al. Clinicopathologic and molecular analysis of embryonal rhabdomyosarcoma of the genitourinary tract: evidence for a distinct DICER1-associated subgroup. Mod Pathol. (2021) 34:1558–69. doi: 10.1038/s41379-021-00804-y
13. Zhang Z, Florez S, Gutierrez-Hartmann A, Martin JF, Amendt BA. MicroRNAs regulate pituitary development, and MicroRNA 26b specifically targets lymphoid enhancer factor 1 (lef-1), which modulates pituitary transcription factor 1 (pit-1) expression. J Bio Chem. (2010) 285:34718–28. doi: 10.1074/jbc.M110.126441
14. Raman S, Grimberg A, Waguespack SG, Miller BS, Sklar CA, Meacham LR, et al. Risk of neoplasia in pediatric patients receiving growth hormone therapy–A report from the pediatric endocrine society drug and therapeutics committee. J Clin Endocrinol Metab. (2015) 100:2192–203. doi: 10.1210/jc.2015-1002
15. Rossi S, Barresi S, Stracuzzi A, Lopez-Nunez O, Chiaravalli S, Ferrari A, et al. DICER1-associated Malignancies mimicking germ cell neoplasms: report of two cases and review of the literature. Pathol—Res and Pract. (2021) 225:153553. doi: 10.1016/j.prp.2021.153553
16. Khan NE, Bauer AJ, Schultz KAP, Doros L, Decastro RM, Ling A, et al. Quantification of thyroid cancer and multinodular goiter risk in the DICER1 syndrome: a family-based cohort study. J Clin Endocrinol Metab. (2017) 102:1614–22. doi: 10.1210/jc.2016-2954
17. Bakhuizen JJ, Hanson H, van der Tuin K, Lalloo F, Tischkowitz M, Wadt K, et al. SIOPE Host genome working group, CanGene-CanVar clinical guideline working group, expert network members. Surveillance recommendations for DICER1 pathogenic variant carriers: a report from the SIOPE host genome working group and CanGene-CanVar clinical guideline working group. Fam Cancer. (2021) 20:337–48. doi: 10.1007/s10689-021-00264-y
Keywords: DICER1 syndrome, cervical embryonal rhabdomyosarcoma, thyroid pathologies, surveillance recommendations, genetic testing
Citation: Stambouli A, Cartault A, Petit IO, Evrard S, Mery E, Savagner F and Trudel S (2023) DICER1 syndrome and embryonal rhabdomyosarcoma of the cervix: a case report and literature review. Front. Pediatr. 11:1150418. doi: 10.3389/fped.2023.1150418
Received: 24 January 2023; Accepted: 18 April 2023;
Published: 5 May 2023.
Edited by:
Malgorzata Gabriela Wasniewska, University of Messina, ItalyReviewed by:
Artur Bossowski, Medical University of Bialystok, PolandValeria Calcaterra, University of Pavia, Italy
Maria Cristina Vigone, San Raffaele Hospital (IRCCS), Italy
© 2023 Stambouli, Cartault, Petit, Evrard, Mery, Savagner and Trudel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frederique Savagner c2F2YWduZXIuZkBjaHUtdG91bG91c2UuZnI=
†These authors have contributed equally to this work