 Clara Marie Nittel1*
Clara Marie Nittel1* Frederike Dobelke1Jens König2Martin Konrad2Katja Becker1
Frederike Dobelke1Jens König2Martin Konrad2Katja Becker1 Inge Kamp-Becker1Stefanie Weber3for the NEOCYST consortium
Inge Kamp-Becker1Stefanie Weber3for the NEOCYST consortium
- 1Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, Philipps University, Marburg, Germany
- 2Department of General Pediatrics, University Children’s Hospital, Münster, Germany
- 3Department of Pediatric and Adolescent Medicine, Philipps University, Marburg, Germany
This review investigates the association between neurodevelopmental disorders (NDD) and variations of the gene HNF1B. Heterozygous intragenetic mutations or heterozygous gene deletions (17q12 microdeletion syndrome) of HNF1B are the cause of a multi-system developmental disorder, termed renal cysts and diabetes syndrome (RCAD). Several studies suggest that in general, patients with genetic variation of HNF1B have an elevated risk for additional neurodevelopmental disorders, especially autism spectrum disorder (ASD) but a comprehensive assessment is yet missing. This review provides an overview including all available studies of patients with HNF1B mutation or deletion with comorbid NDD with respect to the prevalence of NDDs and in how they differ between patients with an intragenic mutation or 17q12 microdeletion. A total of 31 studies was identified, comprising 695 patients with variations in HNF1B, (17q12 microdeletion N = 416, mutation N = 279). Main results include that NDDs are present in both groups (17q12 microdeletion 25.2% vs. mutation 6.8%, respectively) but that patients with 17q12 microdeletions presented more frequently with any NDDs and especially with learning difficulties compared to patients with a mutation of HNF1B. The observed prevalence of NDDs in patients with HNF1B variations seems to be higher than in the general population, but the validity of the estimated prevalence must be deemed insufficient. This review shows that systematical research of NDDs in patients with HNF1B mutations or deletions is lacking. Further studies regarding neuropsychological characteristics of both groups are needed. NDDs might be a concomitant of HFN1B-related disease and should be considered in clinical routine and scientific reports.
Introduction
Neurodevelopmental disorders (NDDs) are characterized by an impairment in cognition, communication, behavior and/or motor skills originating from abnormal brain development (1). According to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (2), NDDs include, among others, attention deficit hyperactivity disorder (ADHD), autism spectrum disorder (ASD), intellectual disability, global developmental delay, developmental language disorder (DLD) as well as motor, learning and communication disorders (3). Although every NDD has distinct features, they share important characteristics: onset in childhood; rather steady course over lifespan; higher prevalence in males; significant overlap with other NDDs and a high heritability although multifactorial in etiology (3, 4).
One of the most common NDD is ADHD, with an estimated prevalence of 3.4% (95% CI: 2.6–4.5) in the general population (5). ADHD is characterized by inappropriate and impairing inattention, motor hyperactivity and impulsivity for the respective developmental level (6). Although much rarer, another prominent NDD is ASD (7). In 2010 the estimated global point prevalence was 1% (8). ASD is characterized by persistent deficits in communication, social interactions and restricted, repetitive patterns of behaviour, interests, or activities (2). Both, ASD and ADHD share high rates of comorbid intellectual disability (9): ADHD was diagnosed in 40%–70% of patients with ASD (10), and vice versa, ASD symptoms were found in 21% of patients with ADHD (11). Additionally, ASD and ADHD are often accompanied by other NDDs, such as intellectual disability (9, 12), language problems (13), global developmental delay (14) and motor disorder (15, 16). Although NDDs are highly heritable and various genes have been associated with them, distinct pathogenetic pathways have yet to be determined (17).
The gene HNF1B is also known as transcription factor 2 (TRF2) and early expression is seen in the kidney, liver, bile ducts, thymus, genital tract, pancreas, lung, and gut (18). Originally, variations in the hepatocyte nuclear factor-1 β (HNF-1β, OMIM 189907) have been discovered in patients with maturity-onset diabetes of the young (19), later it has been found to be one major cause of the renal cysts and diabetes syndrome [RCAD (20); OMIM 137920]. Now it is deemed a multi-system disorder, leading to end-stage kidney disease in a subset of patients, including extra-renal symptoms like pancreatic dysplasia, elevated liver enzymes and genital tract abnormalities (21–23).
Around half of the mutations of HNF1B consist of a whole-gene deletion (43%–64%) (24–26) This is caused in almost all cases by a microdeletion on chromosome 17q12, spanning a minimum of 1.4 Mb. In this review, whole gene deletions are therefore labeled 17q12 microdeletions (23, 24, 27). Both, mutations and17q12 microdeletions can arise de novo, present in around 50% of the cases (24, 25). In these cases, parents are generally genetically unaffected.
Considering the different kinds of genetic variations in HNF1B (17q12 microdeletions or intragenic mutations), no consistent genotype-phenotype correlation has so far been identified (25). Instead, phenotypes resulting from intragenic HNF1B mutations or 17q12 microdeletions are very heterogenic with a large intra- and interfamilial variability. This makes it likely, that the haploinsufficiency of HNF1B, meaning alterations of gene dosage, leads to the described multi-system disease (22, 28, 29).
From early on, a case report of patients with cognitive impairment emerged (30). Since then, there have been constantly new findings about the phenotype resulting from heterozygous mutations or whole-gene deletions of HNF1B (31–34).
Some conditions seem to be linked to 17q12 microdeletions rather than to intragenic mutations. This is the case for the Mayer-Rokitansky-Küster-Hauser syndrome (35, 36) but also NDDs, including intellectual disability (IQ < 70; ID), global developmental delay and ASD (37, 38). As there have also been reports about NDDs in patients with intragenic mutations (39), NDDs might not be exclusively linked to the 17q12 microdeletion. However, it is unknown whether the risk for NDDs differs significantly in patients with a 17q12 microdeletion from patients with an intragenic HNF1B mutation. Also, little systematical research exists so far to evaluate whether patients with variations in HNF1B have a higher risk for NDDs than the general population.
Research question
This review aims to answer the following research question: How high is the prevalence of NDDS in patients with 17q12 microdeletions and mutations of HNF1B and exceeds this prevalence the prevalence of NDDs in the general population? To answer this question, we will review findings obtained in original research. We will summarize the existing literature and investigate disease-related characteristics and the observed prevalence of NDDs in patients with a mutation of HNF1B and a 17q12 microdeletion, respectively. Further, we will compare the observed prevalence of NDDs in patients with a mutation of HNF1B and a 17q12 microdeletion with the prevalence of NDDs in the general population in the discussion section.
Methods
Literature search
A literature research was performed on PubMed, Cochrane library, Web of Science and EBSCO host (selecting the CINAHL, APA PsycInfo, APA PsycArticles and MEDLINE databases), using the keywords autis* OR psychiatr* OR mental OR cognitive OR neurodevelopment* OR neuropsychol* AND HNF1B OR HNF1β OR 17q12 deletion OR 17q12 microdeletion OR TCF-2 OR TCF2 (=Transcription Factor 2, synonym of HNF1B), focusing on the years 1997–2022. The exact search terms were adapted to the requirements of the respective search engine. A total of 174 search results were obtained (see Figure 1). After doublets were removed a total of 98 papers remained. Included were originally published papers in English, which described patients with an HNF1B variations for which also NDD were reported. 22 papers met these criteria, nine additional studies were identified via reference lists of relevant papers, therefore a total of 31 papers are reported in this review (see Appendix A). The papers were reviewed by two researchers independently (F. D., C. M. N.) and in case of a contradictory decision, the paper was discussed with a third person (I. K.-B.).
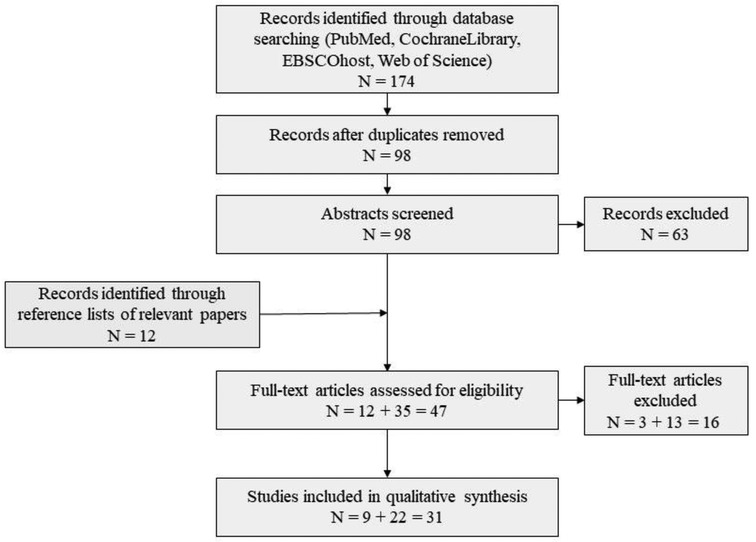
Figure 1. Flow chart of literature search.
Descriptive and statistical data analysis
The sample size, the number of cases with NDDs and percentages of NDDs in patients with a HNF1B mutation and 17q12 microdeletion were documented for every paper found (see Table 1). If obtainable, patient characteristics associated with their renal cystic disease were documented separately for patients with a 17q12 microdeletion or HNF1B mutation. To assess group differences for patients with a 17q12 microdeletion or HNF1B mutation regarding their illness-related characteristics, a χ2 test was calculated. If the expected counts in the contingency table were less than five, Fishers exact test was applied. The reported age in the studies was compared for the two groups using a Mann–Whitney-U-Test, as the requirements for a t-Test were not fulfilled. For every type of NDD, the observed prevalence was calculated. For that, the number of cases with a certain type of NDD, e.g., ASD was divided by the pooled sample size of studies investigating this type of NDD. Again, to assess group differences for patients with a 17q12 microdeletion or HNF1B mutation regarding the observed prevalence for different NDDs, a χ2 test was calculated. If the expected counts in the contingency table were less than 5, Fishers exact test was applied (see Table 2). To compare the risk for patients with 17q12 microdeletions and HNF1B mutations regarding NDDs, odd ratios for every study were calculated if the studies provided sufficient data. Odd ratios based on the pooled data for every NDD were calculated as well.
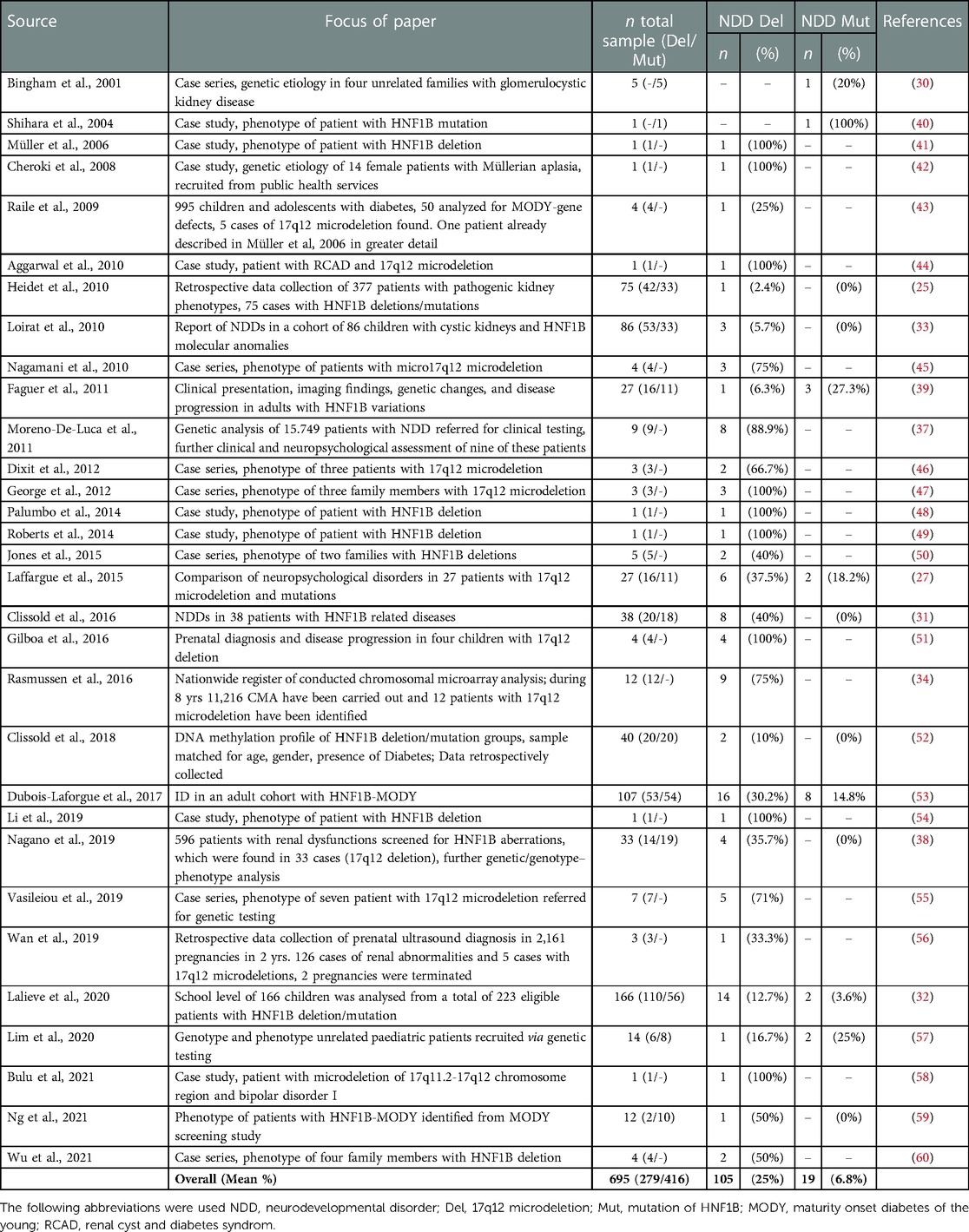
Table 1. Percentages of neurodevelopmental disorders (NDD) in studies reporting on NDDs in patients with 17q12 microdeletions and HNF1B mutations.
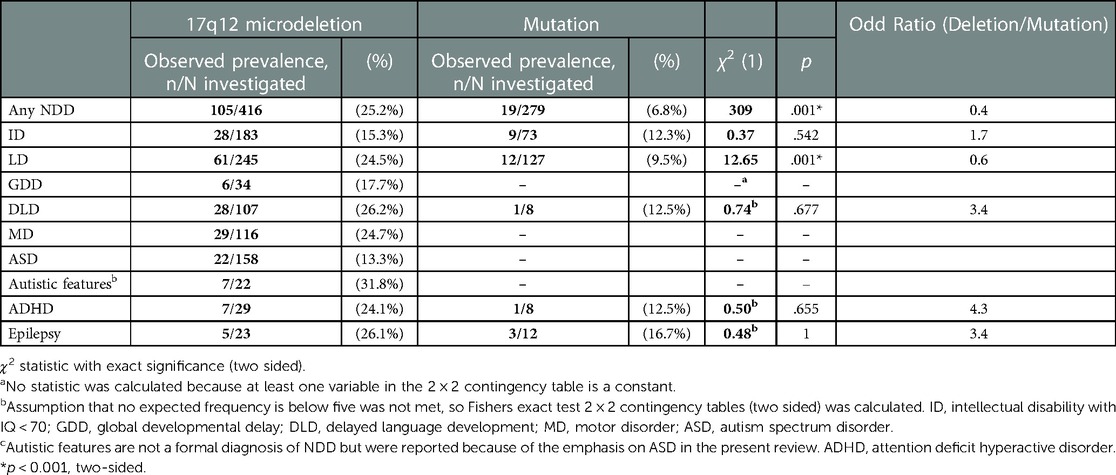
Table 2. Types of NDDs in patients with a 17q12 microdeletion or mutation of HNF1B.
Results
We will first qualitatively summarize the existing literature, then we will present the data analysis of the studies. We will summarize the 31 studies identified on NDDs in patients with variations in HNF1B and will divide this section in single case studies/case series and larger samples of patients.
Single case studies and case series
Several studies reported on patients with HNF1B variations in single case studies or case series. Considerably more case studies on patients with a 17q12 microdeletion (41, 42, 44–46, 48–50, 54, 58, 60) have been published than on patients with a mutation of HNF1B (40).
In the single case studies, all patients with a 17q12 microdeletion were reported to have some kind of NDD, including intellectual disability (41, 44, 48), ADHD (48, 58), or ASD (49). The patient in the single case study with a mutation of HNF1B was reported to have a learning disorder (40).
The case series presented either findings from sets of family members (30, 50, 60) or unrelated patients (45, 46, 55) referred for genetic testing. In the studies reporting on NDDs in family members, the observed prevalence of NDDs ranged from 20% (30) in patients with a mutation of HNF1B to 40% (50), and 100% respectively (47) in patients with a 17q12 microdeletion. In patients with a 17q12 microdeletion, cases of learning disorder, ASD, global developmental delay and motor disorder were observed (47, 50, 60), in patients with a mutation of HNF1B, a case of learning disorder was observed (30). In studies reporting on NDDs in unrelated patients, the observed prevalence of NDDs ranged from 66.7% (46) to 75% (45) in patients with a 17q12 microdeletion. In patients with a 17q12 microdeletion, cases of ASD, intellectual disability, learning disorder, developmental language disorder, motor disorder and epilepsy were observed (45, 46, 55). None of the case series in unrelated patients reported on NDDs in patients with a mutation of HNF1B.
Studies with larger sample sizes
Three studies tested for variations in HNF1B in larger samples of patients with pathogenic kidney phenotypes (25, 38) and diabetes (43). In these samples, patients with a 17q12 microdeletion mostly presented with intellectual disability, with a prevalence ranging from 2.4% to 25% (25, 38, 43). In patients with a mutation of HNF1B, no NDD was found. Two study tested for variations in HNF1B in a large sample of patients referred to genetic testing because of a NDD (34, 37). All patients had a 17q12 microdeletion and presented mostly with developmental language disorder (0.6% (34)–67% (37)), motor disorder (33% (34)–78% (37)) and learning disorder (11% (37)–58% (34)). The reported prevalence for ASD ranged from 8% (34) to 44% (37)).
Two studies followed up cases with 17q12 microdeletions which were detected prenatally (51, 56). While one study describes just one case with motor disorder and comorbid developmental language disorder (56), the other study describes NDDs in all four patients, three with ASD and one with motor disorder (51).
Two studies concentrated on the intellectual abilities of patients with variations in HNF1B (32, 53). In patients with a 17q12 microdeletion, they found a prevalence of 17% (53) for intellectual disability, the prevalence for learning disorders ranged from 13% (32) to 26% (53). In patients with a mutation of HNF1B, the prevalence for intellectual disability was 11% (53), the reported prevalence for learning disorder ranged from 4% (32) to 11% (53).
Three studies specifically focused on the occurrence of NDDs in patients with HNF1B variations and differences between patients with deletions and mutations in HNF1B (27, 31, 33). Patients with a 17q12 microdeletion presented mostly with learning disorder (23% (27)—30% (31)) and ASD (4% (27)–20% (31)). Only one study found NDDs in patients with a mutation of HNF1B (27), two patients were reported to have learning disorder (15%).
Four studies targeted somatic features of the disease and report NDDs as a secondary aspect (39, 52, 57, 59.) In patients with a 17q12 microdeletion the following NDDs were found: intellectual disability [6% (39)], ASD [5% (52)], global developmental delay (5% (52)–50% (59)), ADHD [17% (57)] and ASD [5% (52)]. Two studies reported on NDDs in patients with a mutation of HNF1B (39, 57), here, patients presented mostly with intellectual disability (13% (57)–18% (39)).
Descriptive and statistical data analysis
A total of 31 studies was identified, comprising 695 patients with variations in the HNF1B gene, among these 416 patients with a 17q12 microdeletion and 279 patients with a mutation of HNF1B. The focus of each paper and the observed prevalence of NDDs in each HNF1B deletion/mutation sample is summarized in Table 1. Of the 416 patients with a deletion, 105 (25%) were reported to have any NDD. Of the 279 patients with a mutation, 19 (7%) had any NDD.
A total of 97 case descriptions of HNF1B-related kidney disease in combination with a NDD on an individual level were available in 29 papers. The remaining two papers did not offer information on an individual level (27, 32). The characteristics and clinical phenotypes of patients with deletions or mutations, respectively, are reported in Table 2. Patients with a 17q12 microdeletion differed from patients with a mutation regarding age (p = .004), kidney abnormalities (p = .018) and diabetes (p = .015). Patients with a mutation were older and suffered from kidney abnormalities/failures and diabetes more frequently.
The observed prevalence for different type of NDDs was calculated for patients with a 17q12 microdeletion or mutation separately (see Table 3). Patients with a 17q12 microdeletion differed from patients with a mutation regarding the observed prevalence of any NDD (p = .001) and learning disorder (p = .001). Compared to patients with a mutation, patients with a 17q12 microdeletion had a higher prevalence for any NDD and specifically learning disorder. We were not able to compare the observed prevalence of ADHD and ASD because of small sample sizes.
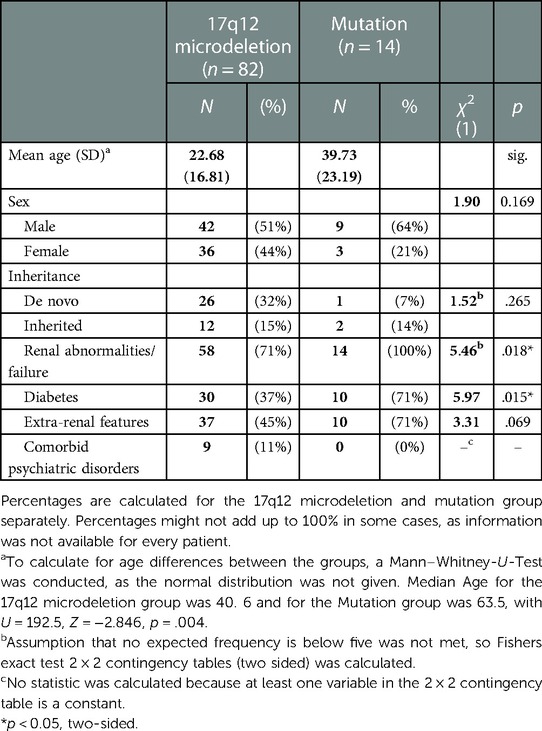
Table 3. Characteristics of patients with NDDs.
If sufficient data was presented in an original study, odd rations were computed. Despite of the descriptive differences, none of the studies showed a significant difference between patients with a 17q12 microdeletion and a mutation of HNF1B with regard to NDDs (OR = 5.6, 95% CI [0.5 63.28] (39); OR = 0.8, 95% CI [0.1 4.6] (27); OR = 0.1, 95% CI [0.03 0.2] (53); OR = 0.3, 95% CI [0.1 1.2] (32); OR = 1.7, 95% CI [0.1 24.3]).
Comparing the prevalence of NDDs in patients with HNF1B variations to the prevalence of NDDs in the general population
A recent study shows that up to 17.3% of children and adolescents between 3 and 17 years in the United States of America are affected by NDDs (61). Accordingly, the observed prevalence of NDDs is slightly higher in patients with deletions of HNF1B and lower in patience with mutations than in the general population. As stated by the DSM-5 (2), the prevalence for intellectual disability is 1%, for learning disorder 5%–15% in school children and 4% in adults, for motor disorder 5%–6% in children (5–11 years), for ASD 1%, ADHD 5% in children and 2.5% in adults. The prevalence for global developmental delay ranges from 1% to 3% in children (62), the prevalence for developmental language disorder is 7.5% (63) and for epilepsy 0.76% (64). So far, the observed prevalence of the above named NDDs in patients with deletions of HNF1B (25.2%) as well as mutations of HNF1B (6.8%) appears to exceed the reported prevalence in the general population.
Discussion
Summary of findings
This review had the purpose to summarize and compare the NDDs that have been reported for patients with a 17q12 microdeletion or intragenic mutation of HNF1B regarding the frequency and type of NDDs. The analysis of the 31 studies showed that NDDs have so far been reported considerably more often in patients with a 17q12 microdeletion compared to patients with an intragenic mutation of HNF1B (25% vs. 7%). Statistically, patients with deletions were more likely to present with NDD as well as learning disorder than patients with mutations. In contrast, patients with mutations presented with diabetes and kidney abnormalities/failure more frequently which seemed to be related to their higher age.
Patients with a 17q12 microdeletion seem to suffer from a wider variety of NDDs than patients with a mutation: patients with a 17q12 microdeletion presented with intellectual disability, learning disorder, global developmental delay, developmental language disorder, motor disorder; ASD, ADHD, epilepsy and/or autistic features (although formally not a NDD), while patients with a mutation presented only with intellectual disability, learning disorder, developmental language disorder, ADHD and/or epilepsy.
Mechanisms linking NDDs to HNF1B variations
So far, no mechanism has been determined that leads to the differences in the phenotype regarding NDDs of the two carrier groups of HNF1B alterations. A possible influence of HNF1B on the development of the hindbrain has been observed in mice and zebrafish models (65, 66), but not in humans yet. As NDDs have also been found in patients with a HNF1B mutation, the haploinsufficiency as one risk factor for NDDs can still be regarded as a possible cause. Nevertheless, other genes in the deleted region have also been suggested to play a role in the NDDs that can be observed in patients with 17q12 microdeletions. These include the gene LHX1 (37, 48, 60), which has been shown to be crucial for the migration of interneurons originating in the pre-optic area during embryonic cortical development (67). Two other genes, PIGW and PCGF2, are also hypothesized to be candidates for NDDs (55, 60) as genetic variations of those two genes have been linked with developmental delay and/or epilepsy (68–70) In addition, Laffargue et al. (27) suggested that an interaction between HNF1B and other transcription factors like homeobox protein Hox-A1 could cause NDD, as an interaction of those two transcription factors has been found during mural hindbrain development (71). Additional or synergistic effects due to “second-site” CNVs on top of a primary genomic rearrangement have been found for other genomic disorders (e.g., 16p11.2 duplication) and are also possible to play are role for 17q12 microdeletions. In a large cohort of children with syndromic ID and congenital abnormalities, 10% of the children carried a secondary CNVs (72). These effects could influence neurodevelopmental pathways and the disease outcome, e.g., the phenotype and severity of the disease, contributing to the additional NDDs.
Validity of estimations
Percentages of reported frequency of NDD in patients with HNF1B gene variations (i.e., 2.4%–100%) vary greatly; i.e., for patients with a mutation from 0% to 18.2% and for patients with a 17q12microdeletion from 2.4% to 100%. This might be due to the characteristics of the particular study. For instance, percentages tended to be higher, the smaller the samples were (e.g., n = 4, 100% in (51) or n = 5, 40% in (43)) or the more detailed the patients were described (34); n = 12; 75%). Papers who did not focus on psychological aspects tended to report a lower percentage of NDDs [e.g., (25); n = 42 patients with a 17q12microdeletion, 2.4%] as well as papers with bigger samples and reduced psychological assessment [e.g., (32); n = 110 with a deletion; 14%]. Overall, this points to the existence of a publication bias and limits the validity of the prevalence estimations.
When comparing the prevalence of NDDs in patients with HNF1B variations to the prevalence of NDDs in the general population, it strikes as odd that the prevalence of NDDs in patients with a mutation of HNF1B should be lower than in the general population. A reason for this low prevalence estimation of NDDs in patients with mutation might be a sampling bias. And the true prevalence might be higher.
The validity of the prevalence estimations are also limited by insufficient information regarding the diagnostic of NDDs in the reviewed studies. Only in three studies all participants underwent a standardized intelligence test (44, 47, 49), in six studies either only subgroups were assessed or information about the assessment was missing, e.g., name of the test (27, 37, 53–55, 58). Of 11 studies reporting on ASD cases, only three studies described the use of any diagnostic tool (27, 33, 37) and only for one patient with ASD (37) the diagnosis was assigned according to the NICE guidelines (73). Although comorbid NDDs or mental disorders were often reported, information whether comorbidities were routinely assessed were missing in the reviewed studies. However, the assessment of comorbid disorders is central for the diagnosis of ASD (73).
Further, the studies did not report the use of appropriate assessment tools for any NDDs, like standardized interviews (74, 75). Instead, information on the diagnostic process were missing or special education needs as indicators for NDDs, especially learning disorder were used (32, 34). Although, special education needs point to the existence of a NDD, they are not a valid diagnostic tool.
Overall, sample sizes were small. Only seven studies had a sample size above 30 patients (see Table 1) and only four studies investigated group differences based on inferential statistics (27, 31, 32, 53).
Overall, the small sample sizes and the quality of studies, especially regarding the diagnosis of NDDs make it impossible to draw valid conclusions about prevalence ratings in samples with HNF1B variations just yet.
Limitations
One large obstacle is the sparsity of HNF1B-associated disorders. Population based studies estimate a prevalence for 17q12 microdeletion of approximately 0.023%–0.025% (76, 77). In samples of patients with kidney abnormalities, HNF1B alterations can be found in about 5%–31% depending on the specific sample characteristics (22). The number of patients who present with an additional NDD is therefore even more limited. This reflects in the lack of good-quality studies with large, matched samples regarding age, sex and factors regarding kidney disease and diabetes.
As for a long time HNF1B-related kidney disease was regarded as primarily a physical disease, some papers report NDDs only as a “side note”. This might influence this review in several ways: In larger samples, NDDs as well as mild psychological symptoms or impairments might have easily been overlooked leading to possibly underestimated numbers. On the other hand, a publication bias might have led to an overestimation of NDDs in patients with HNF1B alterations.
Lastly, regarding the comparison of studied that focused on NDDs in particular, differences in the sensitivity of criteria for NDD (e.g., neuropsychological assessments vs. special education needs; learning disability vs. learning difficulties) and therefore differences in the percentages of NDD make it hard to compare these studies (27, 31, 32).
Conclusion
This review shows that NDDs are frequently found in patients with HNF1B variations. It seems that they are more common in patients with 17q12 microdeletion than in patients with mutation of HNF1B. The observed prevalence for NDDs, including ASD was found to be higher patients with 17q12 microdeletion than in the general population but the validity of the prevalence estimates are limited by the insufficient quality of the studies.
A first step to enhance data quality includes considering NDDs as a further possible symptom of patients with a deletion. This should include consistent reporting of existence or absence of NDDs in scientific papers as well as considering possible impairments during the clinical routine. Further studies will be needed to evaluate the neuropsychological profiles of patients with a 17q12 microdeletion or mutation of HNF1B, including matched samples and a significant sample size for statistical comparison.
If a standardized assessment of symptoms of NDD would be integrated in the clinical routine of patients with HNF1B variations, especially in patients with deletions of HNF1B, access to valuable treatment options could be facilitated and subsequently, the quality of life of patients and their families could be enhanced.
The authors are part of the NEOCYST consortium (www.neocyst.de/en/), a multicenter, interdisciplinary network of clinicians and scientists exploring early onset cystic kidney diseases and undertake a research project to assess neuropsychological symptoms in patients with HNF1B variations and other cystic kidney diseases.
Author contributions
CMN: reviewd the studies, wrote the manuscript and analysed the data. FD: reviewd the studies and summaried the studies. KB: reviwed the manuscript. IKB: reviewed the manuscript. SW: planned the study, reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was sponsored by the Federal Ministry for Education and Research, Germany (grant no 016M1903C).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. D’Souza H, Karmiloff-Smith A. Neurodevelopmental disorders. Wiley Interdiscip Rev Cogn Sci. (2017) 8:1–10. doi: 10.1002/wcs.1398
2. Autoren-Liste K. Diagnostic and statistical manual of mental disorders. DSM-5. In: keine Autoren-Liste. 5th ed. Washington, DC: American Psychiatric Assoc (2013).
3. Thapar A, Cooper M, Rutter M. Neurodevelopmental disorders. Lancet Psychiatry. (2017) 4:339–46. doi: 10.1016/S2215-0366(16)30376-5
4. May T, Adesina I, McGillivray J, Rinehart NJ. Sex differences in neurodevelopmental disorders. Curr Opin Neurol. (2019) 32:622–6. doi: 10.1097/WCO.0000000000000714
5. Polanczyk GV, Salum GA, Sugaya LS, Caye A, Rohde LA. Annual research review: a meta-analysis of the worldwide prevalence of mental disorders in children and adolescents. J Child Psychol Psychiatry. (2015) 56:345–65. doi: 10.1111/jcpp.12381
6. Thapar A, Cooper M. Attention deficit hyperactivity disorder. Lancet. (2016) 387:1240–50. doi: 10.1016/S0140-6736(15)00238-X
7. Ismail FY, Shapiro BK. What are neurodevelopmental disorders? Curr Opin Neurol. (2019) 32:611–6. doi: 10.1097/WCO.0000000000000710
8. Zeidan J, Fombonne E, Scorah J, Ibrahim A, Durkin MS, Saxena S, et al. Global prevalence of autism: a systematic review update. Autism Res. (2022) 15:778–90. doi: 10.1002/aur.2696
9. Zeidan J, Fombonne E, Scorah J, Ibrahim A, Durkin MS, Saxena S, et al. Prevalence of autism spectrum disorder and co-morbidities in children and adolescents: a systematic literature review. Front Psychiatry. (2021) 12:744709. doi: 10.3389/fpsyt.2021.744709
10. Joshi G, Faraone SV, Wozniak J, Petty C, Fried R, Galdo M, Furtak SL, et al. Examining the clinical correlates of autism spectrum disorder in youth by ascertainment source. J Autism Dev Disord. (2014) 44:2117–26. doi: 10.1007/s10803-014-2063-4
11. Hollingdale J, Woodhouse E, Young S, Fridman A, Mandy W. Autistic spectrum disorder symptoms in children and adolescents with attention-deficit/hyperactivity disorder: a meta-analytical review. Psychol Med. (2020) 50:2240–53. doi: 10.1017/S0033291719002368
12. McClain MB, Hasty Mills AM, Murphy LE. Inattention and hyperactivity/impulsivity among children with attention-deficit/hyperactivity-disorder, autism spectrum disorder, and intellectual disability. Res Dev Disabil. (2017) 70:175–84. doi: 10.1016/j.ridd.2017.09.009
13. Korrel H, Mueller KL, Silk T, Anderson V, Sciberras E. Research review: language problems in children with attention-deficit hyperactivity disorder - a systematic meta-analytic review. J Child Psychol Psychiatry. (2017) 58:640–54. doi: 10.1111/jcpp.12688
14. Shan L, Feng J-Y, Wang T-T, Xu Z-D, Jia F-Y. Prevalence and developmental profiles of autism spectrum disorders in children with global developmental delay. Front Psychiatry. (2021) 12:794238. doi: 10.3389/fpsyt.2021.794238
15. Mohd Nordin A, Ismail J, Kamal Nor N. Motor development in children with autism spectrum disorder. Front Pediatr. (2021) 9:598276. doi: 10.3389/fped.2021.598276
16. Licari MK, Alvares GA, Varcin K, Evans KL, Cleary D, Reid SL, et al. Prevalence of motor difficulties in autism spectrum disorder: analysis of a population-based cohort. Autism Res. (2020) 13:298–306. doi: 10.1002/aur.2230
17. Homberg JR, Kyzar EJ, Scattoni ML, Norton WH, Pittman J, Gaikwad S, et al. Genetic and environmental modulation of neurodevelopmental disorders: translational insights from labs to beds. Brain Res Bull. (2016) 125:79–91. doi: 10.1016/j.brainresbull.2016.04.015
18. Edghill EL, Bingham C, Ellard S, Hattersley AT. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet. (2006) 43:84–90. doi: 10.1136/jmg.2005.032854
19. Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. (1997) 17:384–5. doi: 10.1038/ng1297-384
20. Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. (2006) 17:2864–70. doi: 10.1681/ASN.2006030277
21. Bockenhauer D, Jaureguiberry G. HNF1B-associated clinical phenotypes: the kidney and beyond. Pediatr Nephrol. (2016) 31:707–14. doi: 10.1007/s00467-015-3142-2
22. Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat Rev Nephrol. (2015) 11:102–12. doi: 10.1038/nrneph.2014.232
23. Okorn C, Goertz A, Vester U, Beck BB, Bergmann C, Habbig S, et al. HNF1B nephropathy has a slow-progressive phenotype in childhood-with the exception of very early onset cases: results of the German multicenter HNF1B childhood registry. Pediatr Nephrol. (2019) 34:1065–75. doi: 10.1007/s00467-018-4188-8
24. Ulinski T, Lescure S, Beaufils S, Guigonis V, Decramer S, Morin D, et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol. (2006) 17:497–503. doi: 10.1681/ASN.2005101040
25. Heidet L, Decramer S, Pawtowski A, Morinière V, Bandin F, Knebelmann B, et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol. (2010) 5:1079–90. doi: 10.2215/CJN.06810909
26. Chen YZ, Gao Q, Zhao XZ, Chen YZ, Bennett CL, Xiong XS, et al. Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chin Med J. (2010) 123:3326–33. PMID: 21163139
27. Laffargue F, Bourthoumieu S, Llanas B, Baudouin V, Lahoche A, Morin D, et al. Towards a new point of view on the phenotype of patients with a 17q12 microdeletion syndrome. Arch Dis Child. (2015) 100:259–64. doi: 10.1136/archdischild-2014-306810
28. Edghill EL, Oram RA, Owens M, Stals KL, Harries LW, Hattersley AT, et al. Hepatocyte nuclear factor-1beta gene deletions–a common cause of renal disease. Nephrol Dial Transplant. (2008) 23:627–35. doi: 10.1093/ndt/gfm603
29. El-Khairi R, Vallier L. The role of hepatocyte nuclear factor 1β in disease and development. Diabetes Obes Metab. (2016) 18(Suppl 1):23–32. doi: 10.1111/dom.12715
30. Bingham C, Bulman MP, Ellard S, Allen LI, Lipkin GW, Hoff WG, et al. Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet. (2001) 68:219–24. doi: 10.1086/316945
31. Clissold RL, Shaw-Smith C, Turnpenny P, Bunce B, Bockenhauer D, Kerecuk L, et al. Chromosome 17q12 microdeletions but not intragenic HNF1B mutations link developmental kidney disease and psychiatric disorder. Kidney Int. (2016) 90:203–11. doi: 10.1016/j.kint.2016.03.027
32. Laliève F, Decramer S, Heidet L, Baudouin V, Lahoche A, Llanas B, et al. School level of children carrying a HNF1B variant or a deletion. Eur J Hum Genet. (2020) 28:56–63. doi: 10.1038/s41431-019-0490-6
33. Loirat C, Bellanné-Chantelot C, Husson I, Deschênes G, Guigonis V, Chabane N. Autism in three patients with cystic or hyperechogenic kidneys and chromosome 17q12 deletion. Nephrol Dial Transplant. (2010) 25:3430–3. doi: 10.1093/ndt/gfq380
34. Rasmussen M, Vestergaard EM, Graakjaer J, Petkov Y, Bache I, Fagerberg C, et al. 17q12 Deletion and duplication syndrome in Denmark-A clinical cohort of 38 patients and review of the literature. Am J Med Genet A. (2016) 170:2934–42. doi: 10.1002/ajmg.a.37848
35. Williams LS, Demir Eksi D, Shen Y, Lossie AC, Chorich LP, Sullivan ME, et al. Genetic analysis of Mayer-Rokitansky-Kuster-Hauser syndrome in a large cohort of families. Fertil Steril. (2017) 108:145–151.e2. doi: 10.1016/j.fertnstert.2017.05.017
36. Fontana L, Gentilin B, Fedele L, Gervasini C, Miozzo M. Genetics of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Clin Genet. (2017) 91:233–46. doi: 10.1111/cge.12883
37. Moreno-De-Luca D, Mulle JG, Kaminsky EB, Sanders SJ, Myers SM, Adam MP, et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am J Hum Genet. (2010) 87:618–30. doi: 10.1016/j.ajhg.2010.10.004
38. Nagano C, Morisada N, Nozu K, Kamei K, Tanaka R, Kanda S, et al. Clinical characteristics of HNF1B-related disorders in a Japanese population. Clin Exp Nephrol. (2019) 23:1119–29. doi: 10.1007/s10157-019-01747-0
39. Faguer S, Decramer S, Chassaing N, Bellanné-Chantelot C, Calvas P, Beaufils S, et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int. (2011) 80:768–76. doi: 10.1038/ki.2011.225
40. Shihara N, Horikawa Y, Onishi T, Ono M, Kashimada K, Takeda J. Identification of a new case of hepatocyte nuclear factor-1beta mutation with highly varied phenotypes. Diabetologia. (2004) 47:1128–9. doi: 10.1007/s00125-004-1402-y
41. Müller D, Klopocki E, Neumann LM, Mundlos S, Taupitz M, Schulze I, et al. A complex phenotype with cystic renal disease. Kidney Int. (2006) 70:1656–60. doi: 10.1038/sj.ki.5001746
42. Cheroki C, Krepischi-Santos ACV, Szuhai K, Brenner V, Kim CAE, Otto PA, et al. Genomic imbalances associated with mullerian aplasia. J Med Genet. (2008) 45:228–32. doi: 10.1136/jmg.2007.051839
43. Raile K, Klopocki E, Holder M, Wessel T, Galler A, Deiss D, et al. Expanded clinical spectrum in hepatocyte nuclear factor 1b-maturity-onset diabetes of the young. J Clin Endocrinol Metab. (2009) 94:2658–64. doi: 10.1210/jc.2008-2189
44. Aggarwal V, Krishnamurthy S, Seth A, Bingham C, Ellard S, Mukherjee SB, et al. The renal cysts and diabetes (RCAD) syndrome in a child with deletion of the hepatocyte nuclear factor-1β gene. Indian J Pediatr. (2010) 77:1429–31. doi: 10.1007/s12098-010-0215-x
45. Nagamani SCS, Erez A, Shen J, Li C, Roeder E, Cox S, et al. Clinical spectrum associated with recurrent genomic rearrangements in chromosome 17q12. Eur J Hum Genet. (2010) 18:278–84. doi: 10.1038/ejhg.2009.174
46. Dixit A, Patel C, Harrison R, Jarvis J, Hulton S, Smith N, et al. 17q12 Microdeletion syndrome: three patients illustrating the phenotypic spectrum. Am J Med Genet A. (2012) 158A:2317–21. doi: 10.1002/ajmg.a.35520
47. George AM, Love DR, Hayes I, Tsang B. Recurrent transmission of a 17q12 microdeletion and a variable clinical spectrum. Mol Syndromol. (2012) 2:72–5. doi: 10.1159/000335344
48. Palumbo P, Antona V, Palumbo O, Piccione M, Nardello R, Fontana A, et al. Variable phenotype in 17q12 microdeletions: clinical and molecular characterization of a new case. Gene. (2014) 538:373–8. doi: 10.1016/j.gene.2014.01.050
49. Roberts JL, Gandomi SK, Parra M, Lu I, Gau CL, Dasouki M, et al. Clinical report of a 17q12 microdeletion with additionally unreported clinical features. Case Rep Genet. (2014) 2014:264947. doi: 10.1155/2014/264947
50. Jones GE, Mousa HA, Rowley H, Houtman P, Vasudevan PC. Should we offer prenatal testing for 17q12 microdeletion syndrome to all cases with prenatally diagnosed echogenic kidneys? Prenatal findings in two families with 17q12 microdeletion syndrome and review of the literature. Prenat Diagn. (2015) 35:1336–41. doi: 10.1002/pd.4701
51. Gilboa Y, Perlman S, Pode-Shakked N, Pode-Shakked B, Shrim A, Azaria-Lahav E, et al. Prenatal diagnosis of 17q12 deletion syndrome: from fetal hyperechogenic kidneys to high risk for autism. Prenat Diagn. (2016) 36:1027–32. doi: 10.1002/pd.4926
52. Clissold RL, Ashfield B, Burrage J, Hannon E, Bingham C, Mill J, et al. Genome-wide methylomic analysis in individuals with HNF1B intragenic mutation and 17q12 microdeletion. Clin Epigenetics. (2018) 10:97. doi: 10.1186/s13148-018-0530-z
53. Dubois-Laforgue D, Bellanné-Chantelot C, Charles P, Jacquette A, Larger E, Ciangura C, et al. Intellectual disability in patients with MODY due to hepatocyte nuclear factor 1B (HNF1B) molecular defects. Diabetes Metab. (2017) 43:89–92. doi: 10.1016/j.diabet.2016.10.003
54. Li HJ, Groden C, Hoenig MP, Ray EC, Ferreira CR, Gahl W, et al. Case report: extreme coronary calcifications and hypomagnesemia in a patient with a 17q12 deletion involving HNF1B. BMC Nephrol. (2019) 20. doi: 10.1186/s12882-019-1533-5
55. Vasileiou G, Hoyer J, Thiel CT, Schaefer J, Zapke M, Krumbiegel M, et al. Prenatal diagnosis of HNF1B-associated renal cysts: is there a need to differentiate intragenic variants from 17q12 microdeletion syndrome? Prenatal Diagn. (2019) 39:1136–47. doi: 10.1002/pd.5556
56. Wan S, Zheng Y, Dang Y, Song T, Chen B, Zhang J. Prenatal diagnosis of 17q12 microdeletion and microduplication syndrome in fetuses with congenital renal abnormalities. Mol Cytogenet. (2019) 12:19. doi: 10.1186/s13039-019-0431-7
57. Lim SH, Kim JH, Han KH, Ahn YH, Kang HG, Ha IS, et al. Genotype and phenotype analyses in pediatric patients with HNF1B mutations. J Clin Med. (2020) 9:1–11. doi: 10.3390/jcm9072320
58. Bulu E, Kalayci YA, Agirbasli D, Demirel OF, Aksoy Poyraz C, Seven M. Co-existing bipolar disease and 17q12 deletion: a rare case report. Psychiatr Genet. (2021) 1:30–3. doi: 10.1097/YPG.0000000000000302
59. Ng N, Mijares Zamuner M, Siddique N, Kim J, Burke M, Byrne MM. Genotype-phenotype correlations and response to glucose lowering therapy in subjects with HNF1β associated diabetes. Acta Diabetol. (2021) 1:83–93. doi: 10.1007/s00592-021-01794-8
60. Wu HX, Li L, Zhang H, Tang J, Zhang MB, Tang HN, et al. Accurate diagnosis and heterogeneity analysis of a 17q12 deletion syndrome family with adulthood diabetes onset and complex clinical phenotypes. Endocrine. (2021) 73:37–46. doi: 10.1007/s12020-021-02682-5
61. Zablotsky B, Black LI, Maenner MJ, Schieve LA, Danielson ML, Bitsko RH, et al. Prevalence and trends of developmental disabilities among children in the United States: 2009–2017. Pediatrics. (2019) 144:1–21. doi: 10.1542/peds.2019-0811
62. Majnemer A, Shevell MI. Diagnostic yield of the neurologic assessment of the developmentally delayed child. J Pediatr. (1995) 127:193–9. doi: 10.1016/S0022-3476(95)70294-6
63. Norbury CF, Gooch D, Wray C, Baird G, Charman T, Simonoff E, et al. The impact of nonverbal ability on prevalence and clinical presentation of language disorder: evidence from a population study. J Child Psychol Psychiatry. (2016) 57:1247–57. doi: 10.1111/jcpp.12573
64. Fiest KM, Sauro KM, Wiebe S, Patten SB, Kwon CS, Dykeman J, et al. Prevalence and incidence of epilepsy: a systematic review and meta-analysis of international studies. Neurology. (2017) 88:296–303. doi: 10.1212/WNL.0000000000003509
65. Sun Z, Hopkins N. vHnf1, the MODY5 and familial GCKD-associated gene, regulates regional specification of the zebrafish gut, pronephros, and hindbrain. Genes Dev. (2001) 15:3217–29. doi: 10.1101/gad946701
66. Pouilhe M, Gilardi-Hebenstreit P, Desmarquet-Trin Dinh C, Charnay P. Direct regulation of vHnf1 by retinoic acid signaling and MAF-related factors in the neural tube. Dev Biol. (2007) 309:344–57. doi: 10.1016/j.ydbio.2007.07.003
67. Symmank J, Gölling V, Gerstmann K, Zimmer G. The transcription factor LHX1 regulates the survival and directed migration of POA-derived cortical interneurons. Cereb Cortex. (2019) 29:1644–58. doi: 10.1093/cercor/bhy063
68. Hogrebe M, Murakami Y, Wild M, Ahlmann M, Biskup S, Hörtnagel K, et al. A novel mutation in PIGW causes glycosylphosphatidylinositol deficiency without hyperphosphatasia. Am J Med Genet A. (2016) 170:3319–22. doi: 10.1002/ajmg.a.37950
69. Turnpenny PD, Wright MJ, Sloman M, Caswell R, van Essen AJ, Gerkes E, et al. Missense mutations of the Pro65 residue of PCGF2 cause a recognizable syndrome associated with craniofacial, neurological, cardiovascular, and skeletal features. Am J Hum Genet. (2018) 103:786–93. doi: 10.1016/j.ajhg.2018.09.012.
70. Chiyonobu T, Inoue N, Morimoto M, Kinoshita T, Murakami Y. Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with West syndrome and hyperphosphatasia with mental retardation syndrome. J Med Genet. (2014) 51:203–7. doi: 10.1136/jmedgenet-2013-102156
71. Makki N, Capecchi MR. Identification of novel Hoxa1 downstream targets regulating hindbrain, neural crest and inner ear development. Dev Biol. (2011) 357:295–304. doi: 10.1016/j.ydbio.2011.06.042
72. Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. (2012) 367:1321–31. doi: 10.1056/NEJMoa1200395
73. National Institute for Health and Care Excellence (NICE). Autism spectrum disorder in adults: diagnosis and management. London: keine Autoren-Liste (2021).
74. Shaffer D, Fisher P, Lucas CP, Dulcan MK, Schwab-Stone ME. NIMH Diagnostic interview schedule for children version IV (NIMH DISC-IV): description, differences from previous versions, and reliability of some common diagnoses. J Am Acad Child Adolesc Psychiatry. (2000) 39:28–38. doi: 10.1097/00004583-200001000-00014
75. Weller EB, Weller RA, Fristad MA, Rooney MT, Schecter J. Children’s interview for psychiatric syndromes (ChIPS). J Am Acad Child Adolesc Psychiatry. (2000) 39:76–84. doi: 10.1097/00004583-200001000-00019
76. Smajlagić D, Lavrichenko K, Berland S, Helgeland Ø, Knudsen GP, Vaudel M, et al. Population prevalence and inheritance pattern of recurrent CNVs associated with neurodevelopmental disorders in 12,252 newborns and their parents. Eur J Hum Genet. (2021) 29:205–15. doi: 10.1038/s41431-020-00707-7
77. Calle Sánchez X, Helenius D, Bybjerg-Grauholm J, Pedersen C, Hougaard DM, Børglum AD, et al. Comparing copy number variations in a Danish case cohort of individuals with psychiatric disorders. JAMA psychiatry. (2022) 79:59–69. doi: 10.1001/jamapsychiatry.2021.3392
CrossRef Full Text | Google Scholar
Appendix A
A literature research was performed on PubMed, Cochrane library, Web of Science and EBSCO host (selecting the CINAHL, APA PsycInfo, APA PsycArticles and MEDLINE databases), using the keywords autis* OR psychiatr* OR mental OR cognitive OR neurodevelopment* OR neuropsychol* AND HNF1B OR HNF1β OR 17q12 deletion OR 17q12 microdeletion OR TCF-2 OR TCF2 (= Transcription Factor 2, synonym of HNF1B), focusing on the years 1997-2022. The exact search terms were adapted to the requirements of the respective search engine. A total of 174 search results were obtained. After doublets were removed a total of 98 papers remained. Included were originally published papers in English, which described patients with an HNF1B variations for which also NDD were reported. 22 papers met these criteria, nine additional studies were identified via reference lists of relevant papers therefore a total of 31 papers are reported in this review (see Figure 1). The papers were reviewed by two research independently (F. D., C. M. N.) and in case of a contradictory decision, the paper was discussed with a third person (I. K.-B.).
Keywords: renal cyst and diabetes syndrome, HNF1B, neurodevelopmental disorder (NDD), autism spectrum disorder, review
Citation: Nittel CM, Dobelke F, König J, Konrad M, Becker K, Kamp-Becker I, Weber S and for the NEOCYST consortium (2023) Review of neurodevelopmental disorders in patients with HNF1B gene variations. Front. Pediatr. 11:1149875. doi: 10.3389/fped.2023.1149875
Received: 23 January 2023; Accepted: 20 February 2023;
Published: 9 March 2023.
Edited by:
Lovro Lamot, University of Zagreb, CroatiaReviewed by:
Danijela Petković Ramadža, University Hospital Centre Zagreb, CroatiaTanja Kersnik Levart, University Medical Centre Ljubljana, Slovenia
© 2023 Nittel, Dobelke, König, Konrad, Becker, Kamp-Becker, Weber and and for the NEOCYST consortium. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clara Marie Nittel Y2xhcmEubml0dGVsQHVuaS1tYXJidXJnLmRl
Specialty Section: This article was submitted to Pediatric Nephrology, a section of the journal Frontiers in Pediatrics