 Mauro Cancian1*
Mauro Cancian1* Paola Triggianese2
Paola Triggianese2 Stella Modica2Francesco Arcoleo3
Stella Modica2Francesco Arcoleo3 Donatella Bignardi4
Donatella Bignardi4 Luisa Brussino5Caterina Colangelo6
Luisa Brussino5Caterina Colangelo6 Ester Di Agosta7
Ester Di Agosta7 Davide Firinu8Maria Domenica Guarino9Francesco Giardino10Marica Giliberti11
Davide Firinu8Maria Domenica Guarino9Francesco Giardino10Marica Giliberti11 Vincenzo Montinaro12
Vincenzo Montinaro12 Riccardo Senter1
Riccardo Senter1
- 1UOSD Allergologia, University Hospital of Padua, Padua, Italy
- 2Tor Vergata University Hospital, Rome, Italy
- 3UOC di Patologia Clinica e Immunologia, AOR Villa Sofia-Cervello, Palermo, Italy
- 4IRCCS Ospedale Policlinico San Martino, Genova, Italy
- 5University Hospital Ordine Mauriziano di Torino, Turin, Italy
- 6Azienda Sanitaria Locale di Pescara, Pescara, Italy
- 7Immunoallergology unit, Careggi University Hospital, Florence, Italy
- 8Department of Medical Sciences and Public Health, University of Cagliari, Cagliari, Italy
- 9Ospedale di Civitanova Marche, Civitanova Marche, Italy
- 10Azienda Ospedaliero-Universitaria Policlinico “G.Rodolico-San Marco”, Catania, Italy
- 11Azienda Ospedaliero-Universitaria Policlinico di Bari, Bari, Italy
- 12Ospedale Regionale Generale F Miulli, Acquaviva delle Fonti, Italy
Introduction: Hereditary angioedema due to C1-inhibitor deficiency is influenced by hormonal factors, with a more severe course of disease in women. Our study aims to deepen the impact of puberty on onset, frequency, location and severity of attacks.
Methods: Retrospective data were collected through a semi-structured questionnaire and shared by 10 Italian reference centers of the Italian Network for Hereditary and Acquired Angioedema (ITACA).
Results: The proportion of symptomatic patients increased significantly after puberty (98.2% vs 83.9%, p=0.002 in males; 96.3% vs 68,4%, p<0.001 in females); the monthly mean of acute attacks was significantly higher after puberty, and this occurred both in females (median (IQR) = 0.41(2) in the three years before puberty vs 2(2.17) in the three years after, p<0.001) and in males (1(1.92) vs 1.25(1.56) respectively, p<0.001). The increase was greater in females. No significant differences were detected in attack location before and after puberty.
Discussion: Overall, our study confirms previous reports on a more severe phenotype in the female gender. Puberty predisposes to increased numbers of angioedema attacks, in particular in female patients.
Introduction
The term angioedema (AE) comes from the union of the two ancient Greek words αγγɛίον (vessel) and οίδημα (edema) to emphasize how, unlike cardiovascular or renal edema that are driven by oncotic pressure and Starling's law, it is a disease that results from specific alterations in endothelial permeability (1). In most cases, especially in the pediatric population, AE is due to histamine release from mast cells and basophils, both when associated with urticaria and when it occurs in isolated manner (2, 3).
However, there are also some endotypes of AE mediated by bradykinin (BK), a vasoactive peptide produced in the circulation from high-molecular-weight kininogen (HMWK) upon activation of the contact system (4, 5). Angiotensin converting enzyme (ACE)-inhibitors (ACEI), which reduces BK catabolism by blocking ACE, the main proteases of BK, are the most frequent cause of bradykininergic AE (6, 7). Other drugs—among which gliptins, neprilysin inhibitors, and angiotensin-II-receptor antagonists—may also be involved in the pathogenesis of BK-mediated, iatrogenic angioedema that seldom affects pediatric population (8).
Infants, children and adolescents, on the other hand, represent an age cohort involved in hereditary angioedema due to C1-inhibitor (C1INH) deficiency (HAE-C1INH), which is also mediated by BK as the terminal effector (9, 10).
The history of HAE-C1INH is a long journey that started in 1882 with the first cases of angioedema in the modern era of medicine reported by Henrick Quincke, whose name later became the eponym for the disease (11). As thoroughly resumed by Reshef et al. (12), in the following decades Sir William Osler described the familiarity of the disease and Crowder the autosomal dominant transmission, but it took a century before the discovery of the pathogenetic roles of C1INH and bradykinin, respectively, by Virginia Donaldson in 1964 and Erich Nussberger in 1998. In the 2000s, six new hereditary variants of hereditary angioedema with normal C1INH have been identified to date since the mutation in the gene for factor XII described by Konrad Bork and Karen E. Binkley (13–15). Hereditary forms of angioedema of unknown origin also exist, but the most common one—although rare with an estimated prevalence of around 1:50,000—still by far remains HAE-C1INH (7, 16–21).
Although the first physiologic action of C1INH to be discovered consists in blocking the activation of the complement cascade, this protease inhibitor belonging to the serpin family exerts many actions on the contact system: in particular, it blocks the enzymatic activity of factor XII and kallikrein, thus reducing the generation of bradykinin from HMWK (22–24). Reduced or absent C1INH activity results, therefore, in increased BK production, predisposing to plasma leakage from the microcirculation (25, 26). When in HAE-C1INH, as well as in some hypertensive patients taking ACEI or in acquired forms of C1INH deficiency—even rarer than HAE (1:10) and not affecting the pediatric population as usually secondary to lymphoproliferative or autoimmune diseases (27, 28)—an additional stimulus of unknown nature locally adds, the plasma leaks from the capillary and elicits angioedema.
There is no itching or wheals, and many districts can be affected: subcutaneous tissue, with painful swelling and functional limitation that can persist several days if untreated; gastrointestinal and urogenital submucosa, with abdominal colic mimicking an acute abdomen and often leading to inappropriate surgery; upper airways, with life-threatening attacks when larynx is involved (29–32). The most common triggers of angioedema—which may be preceded by prodromes as erythema marginatum, irritability, sense of hunger, asthenia, or other signs and symptoms—are trauma, surgery, airway endoscopy, dental procedures, infections, and stressful conditions. In most cases, however, the onset of acute attacks occurs spontaneously and without any presaging sign (33–35).
C1INH deficiency may be due to impaired production of C1INH (HAE-C1INH type 1, 85% of cases) or to the synthesis of a dysfunctional protein (HAE-C1INH type 2) (1, 10). Both C1INH-HAE type 1 and type 2 have decreased C1INH activity. C1INH-HAE type 1 has a low amount of plasma C1INH caused by the lack of production from the mutated allele or production of a protein without function unrecognized in the antigen analysis. C1INH-HAE type 2 has low amount of functional C1INH in plasma and usually increased amounts of abnormal C1INH as this C1INH protein is unable to make complexes with target proteases. The phenotype and pattern of inheritance are the same, but the diagnostic approaches are different: while in HAE-C1INH type 1 it is sufficient to demonstrate an antigenic deficiency of the protein by turbidimetry, nephelometry, radial immunodiffusion, immunoblotting, or other immunologic methods, the diagnosis of HAE-C1INH type 2 requires assessment of functional C1INH activity by chromogenic or ELISAs, which need special attention even at the preanalytical stage (1, 10, 36–39). The C4 plasma concentration measurement, which is cheap, easy to perform, and worldwide available although nonspecific, can be very useful as a screening test because its level is reduced in around 95% of C1INH-deficient patients (19, 38–40). Finally, although HAE-C1INH is a genetic disease, mutational analysis of Serping1, the gene on chromosome 11 that encodes for C1INH, is usually not necessary apart from, in rare cases of doubt, prenatal (chorionic villous sampling or amniocentesis) or preimplantation diagnosis of HAE and scientific purposes (1, 10, 39–43). Moreover, gene assessment may be helpful in the first year of life because too early testing in newborns may lead to a false diagnosis of HAE-C1INH (44, 45).
Despite diagnostic advances, including new tools that extend the possibility of diagnosis to reference centers and physicians who eventually lack the proper facilities (46, 47), the diagnostic delay in HAE-C1INH still remains significant as typical of rare diseases (48). In patients with type II HAE-C1INH and in the de novo mutations, which represents up to 20% of cases and have no parents affected, the interval between symptom onset and diagnosis has been reported to be even greater (19, 44, 49).
Although the majority of data and studies regarding hereditary angioedema are based on adults, HAE-C1INH is a disease of pediatric interest, as typical of most congenital diseases. The very early occurrence of potential life-threatening attacks, the unpredictable nature of acute episodes, and their negative consequences on normal daily, school and social activities contribute to a significant disease burden and reduced quality of life (QoL) for both young patients and their parents, relatives, and caregivers (50–52). The onset of symptoms is reported to present in most cases in childhood, with median age of the first swelling ranging from 5 to 11 years and mean diagnostic age of diagnosis and diagnostic delay highly variable in different surveys (4, 53–60). However, these retrospective studies involve very limited populations and are so heterogeneous in their methods that they are difficult to compare with each other, and are not focused on the influence of puberty, which is expected to be relevant due to hormonal influences, psychological aspects, and the possible introduction of estroprogestin in the female population.
Our study aims to deepen our understanding of puberty on the onset, frequency, location, and severity of attacks in hereditary angioedema due to C1-inhibitor deficiency utilizing data shared by the Italian Network for Hereditary and Acquired Angioedema (ITACA, www.angioedemaitaca.org), which brings together Italian reference centers and uses an electronic disease registry where patients can directly enter data on their clinical course.
Patients and methods
This multicenter, retrospective observational study was performed on HAE patients from 10 ITACA centers throughout Italy, thus minimizing the potential bias caused by regional differences.
Inclusion criteria were age between 10 and 40 years, puberty already occurred, and enrollment in the ITACA Registry, approved by the ethics committee of the coordinating center (Comitato etico Milano area 1) on 5 May 2017. The patients were interviewed from 1 to 30 August 2022 by specialized personnel who administered a semistructured questionnaire (Supplementary Table S1) addressing patient characteristics and the effect of puberty on attack location, severity and frequency, related treatments, and overall burden of disease. Age of onset of puberty was defined as the age at menarche for females and the age at semenarche for males. In order to estimate attacks frequency, the patients were asked to report a monthly mean of attacks in the year and in the 3 years before and after puberty. All patients had previously provided written consent to be included in the registry and reiterated verbal consent at the time of the interview. For patients younger than 18 years, interviews were performed in the presence and with the consent of parents.
Statistical analysis
Continuous variables are shown as mean and SD or median and interquartile range (IQR) depending on their distribution. Shapiro–Wilk test was used to check if the variables followed a normal distribution. Categorical variables are shown as number and percentage. Student's t-test or Mann–Whitney test were used for comparisons between continuous variables, depending on their distribution; Chi-square test was used for comparisons between categorical variables (Fisher's exact test when the number in any cell was smaller than 5). Binomial logistic regression was used to investigate the ability of a set of predictor variables to predict the outcome. All the analyses performed are detailed in Supplementary Table S2. Statistical analysis was performed with Jamovi [The jamovi project (2021). jamovi Version 1.6]. p ≤ 0.05 was considered statistically significant.
Results
Population characteristics
Out of 147 eligible patients referring to the centers involved in the study, 118 (60 F, 58 M; HAE-C1INH type 1 = 113, type 2 = 5) were enrolled. As in Table 1, demographic and clinical characteristics were similar between males and females, but the age of puberty was lower in the female gender (median: 12 years in females vs. 13 years in males, p < 0.001); median age at the interview was 30 years (IQR = 13,75) in males and 29.5 years (IQR = 11,25) in females (p = 0.779); median age at first attack was 6 years (IQR = 6,5) in males and 8 years (IQR = 10) in females.
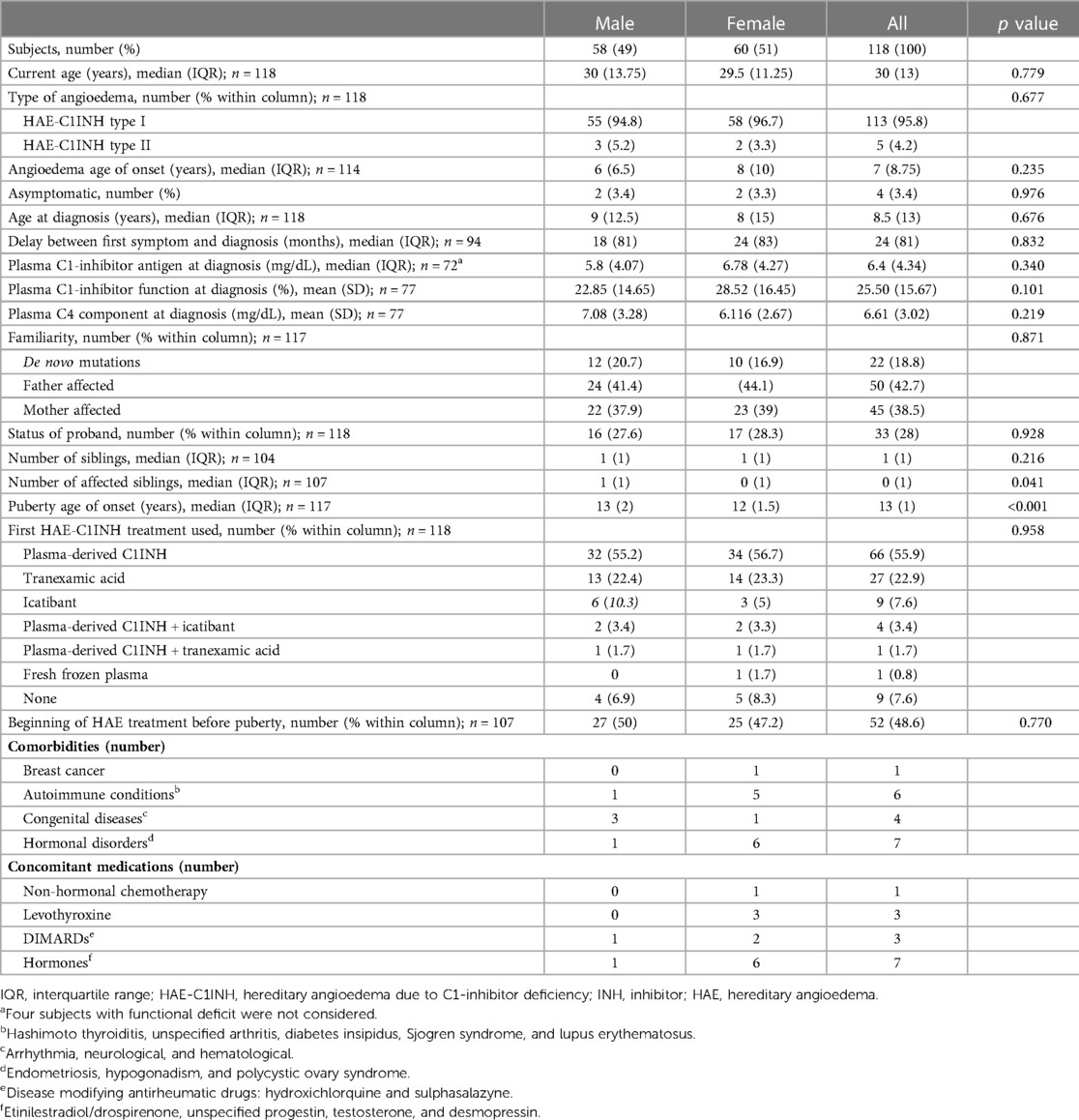
Table 1. Demographic and clinical characteristics of patients.
The impact of puberty on HAE: overall clinical picture
The proportion of symptomatic patients increased significantly after puberty in both males and females (97.3% vs. 76.1%, p < 0.001 in the whole cohort; 98.2% vs. 83.9%, p = 0.002 in males; 96.3% vs. 68.4% p < 0.001 in females) (Tables 2, 3). In the 24 patients who became symptomatic after puberty, the first attack occurred after a mean of 5.14 years and a median of 3.5 years.
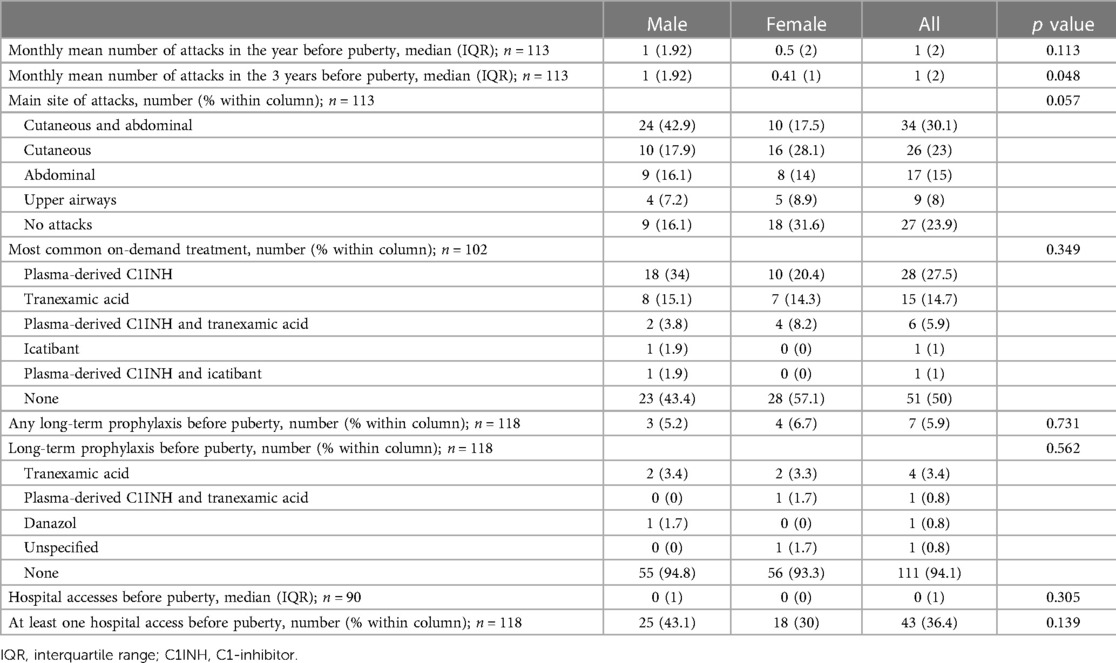
Table 2. Angioedema attacks and treatments before puberty among male and female subjects.
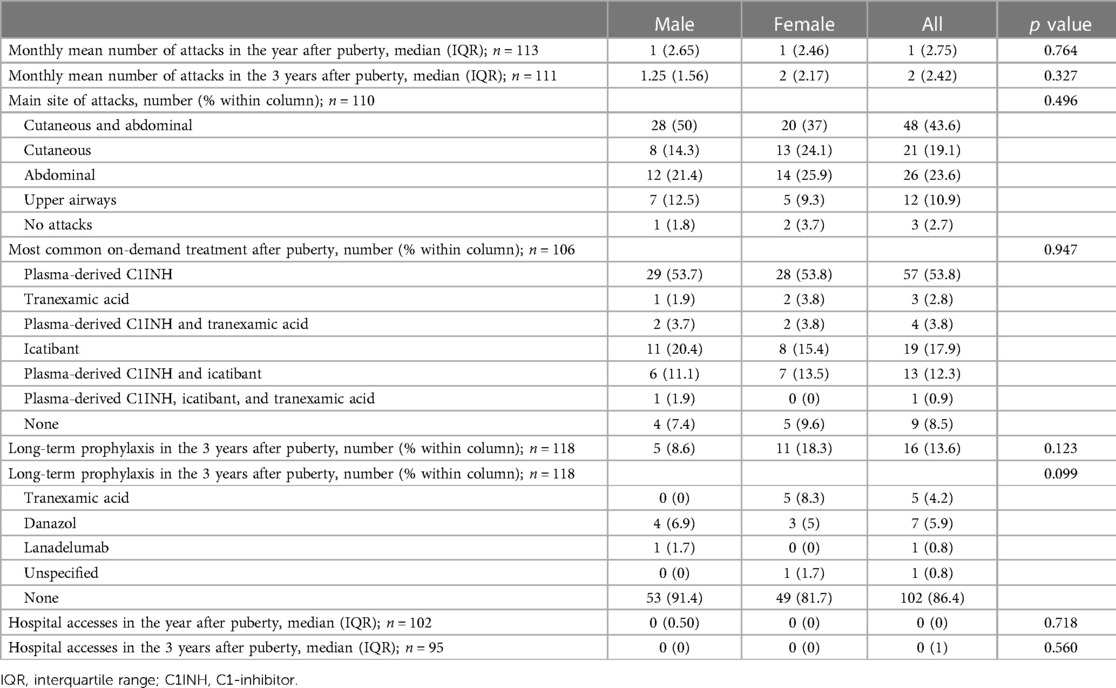
Table 3. Angioedema attacks and treatments after puberty among male and female subjects.
The numbers of acute attacks were significantly higher after puberty when comparing both 1-year and 3-year periods pre/after menarche/semenarche (p < 0.001 and p < 0.001 in the whole cohort; p = 0.035 and p < 0.001 in males; p = 0.004 and p < 0.001 in females) (Figure 1).
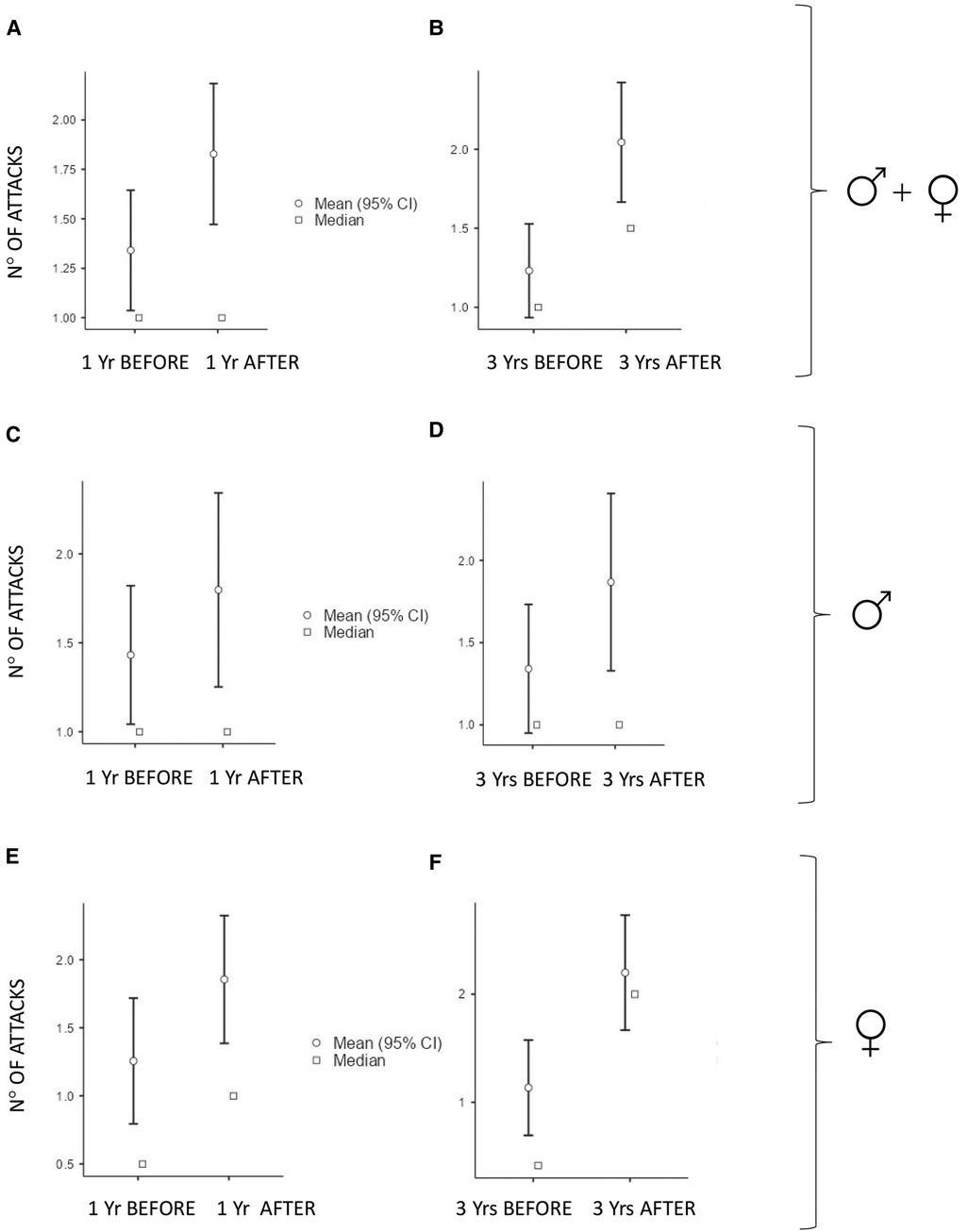
Figure 1. Number of monthly mean of attacks 1 year (Yr) before vs. 1 year after puberty and 3 years (Yrs) before vs. 3 years after puberty: In all the subjects: (A) median (IQR) = 1 (2) vs. 1 (2.75), p < 0.001 and (B) median (IQR) = 1 (2) vs. 2 (2.42), p < 0.001. In males: (C) median (IQR) = 1 (1.92) vs. 1 (2.65), p = 0.035 and (D) median (IQR) = 1 (1.92) vs. 1.25 (1.56), p < 0.001. In females: (E) median (IQR) = 0.5 (2) vs. 1 (2.46), p = 0.004 and (F) median (IQR) = 0.41 (2) vs. 2 (2.17), p < 0.001.
Characteristics of HAE attacks and related treatments before puberty are reported in Table 2, and after puberty in Table 3. No significant differences were detected in the attack location before and after puberty (Figure 2).
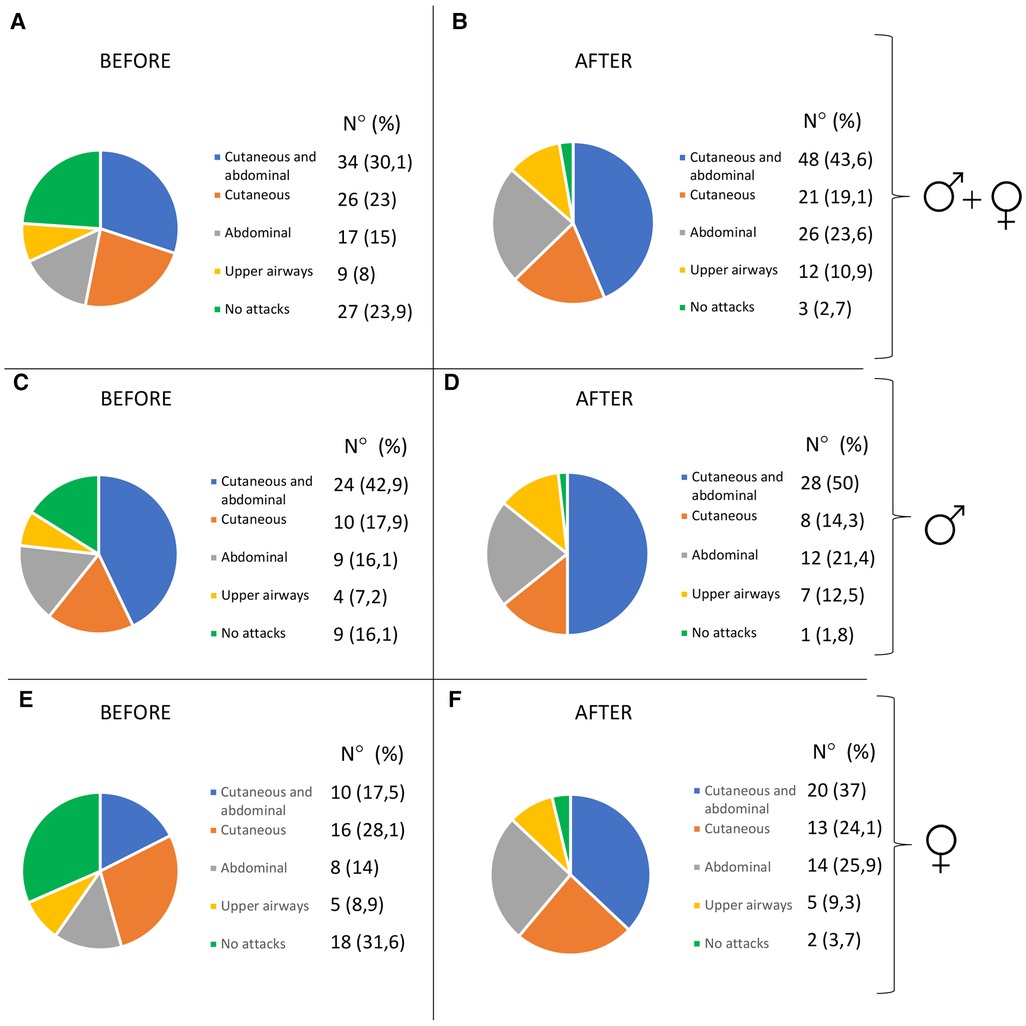
Figure 2. Pattern of attacks before and after puberty in all the patients (A,B), in males (C,D), and in females (E,F).
The impact of puberty: gender differences
Although both the genders showed an increase in the number of HAE attacks after puberty, this occurred more in females than in males as detailed in Figure 3 (differences in monthly mean of acute episodes of angioedema pre/post puberty) and Figure 4 (percentage of males and females in which the number of attacks varies pre/post puberty). Specifically, the median increase of monthly mean of attacks between 3 years before and 3 years after puberty was 1 (IQR = 2) in females and 0 (IQR = 1) in males (p = 0.02); the increase was present, even if not significant, also considering 1 year before and after puberty. Moreover, significantly more females reported an increase of attacks in both periods: in the 3 years after puberty attacks were more frequent in 63.2% of the females and in 41.2% of the males (p = 0.022).
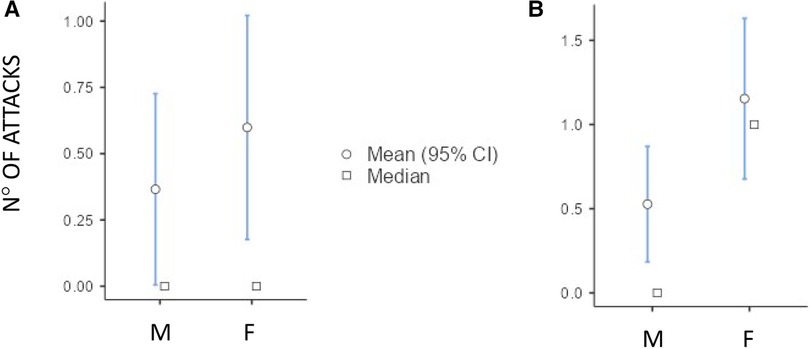
Figure 3. Increase of the number of monthly mean of attacks in male (M) and female (F) between 1 year before vs. 1 year after puberty [(A) median (IQR) = 0 (0.5) vs. 0 (1), p = 0.174] and 3 years before vs. 3 years after puberty [(B) median (IQR) = 0 (1) vs. 1 (2) p = 0.02].
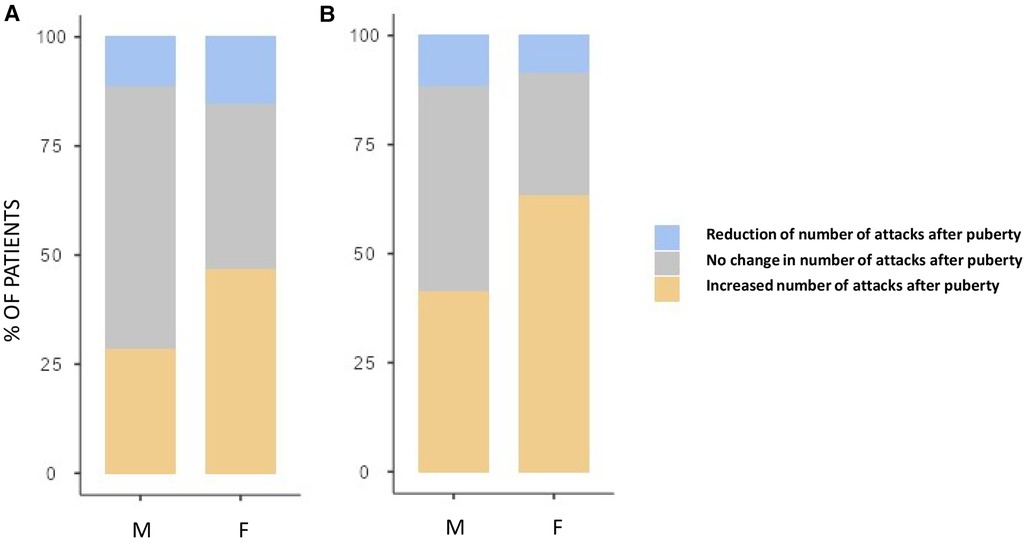
Figure 4. Proportion of subject reporting a reduction (blue), an increase (orange), or no change (gray) in the monthly mean number of attacks after puberty in male (M) and female (F) between 1 year before vs. 1 year after puberty (A) and 3 years before vs. 3 years after puberty (B) Significantly more females reported an increase of attacks in both periods: 28.3% vs. 46.6%, p = 0.048 and 41.2% vs. 63.2%, p = 0.022, respectively.
Nevertheless, no substantial differences were detected between males and females with regard to their subjective perception of illness before and after puberty (Table 4).
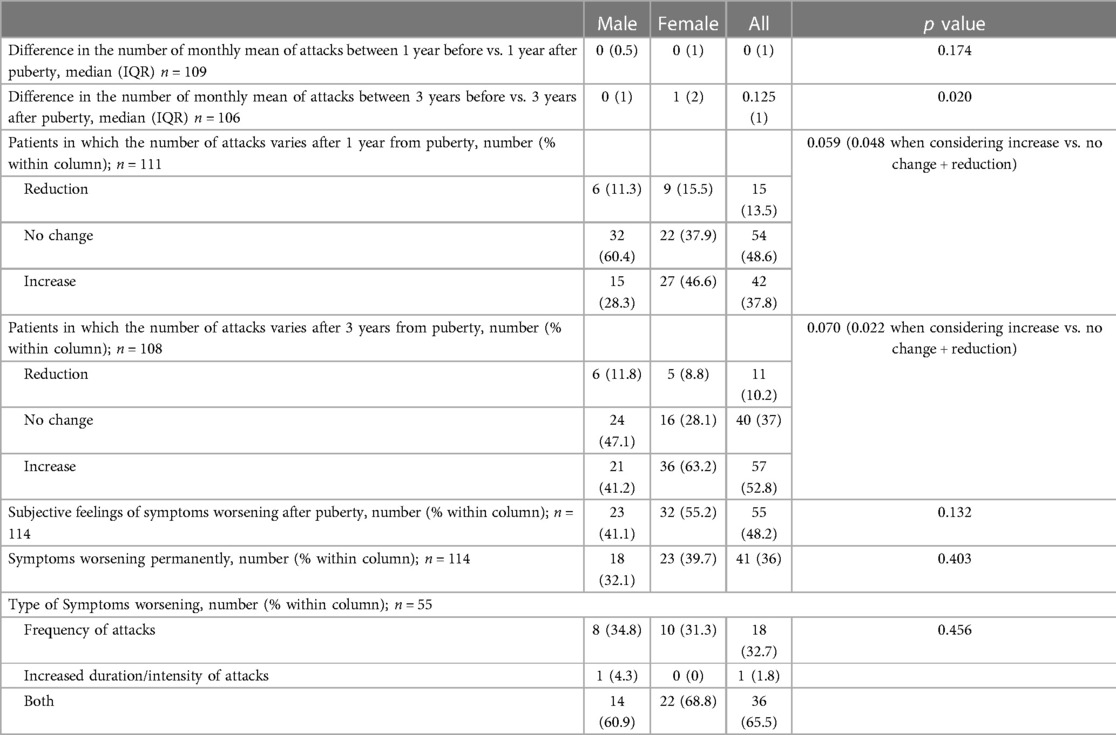
Table 4. Overall impact of puberty among male and female subjects.
The impact of puberty: predictive factors
In regression analysis, female gender predicts the worsening of symptoms for both 1-year and 3-year periods after puberty (OR 2.2, Std Err 1.47–3.3, p = 0.049 and OR 2.44, Std Err 1.65–3.63, p = 0.024, respectively). This is true for the 3-year period also when considering age of first menstruation as covariate [OR 2.43, Std Err 1.61–3.68, p = 0.031].
Discussion
Our retrospective survey in a large group of patients with HAE-C1INH from the ITACA cohort addresses the impact of puberty on the course of the disease.
Puberty is a dynamic, physical, and psychological growth process lasting several years and not a single event at a specific age (59). However, we decided to identify a significant feature of puberty easy to be recognized and remembered in order to assess the impact of puberty on patient symptoms in our survey. Thus, we chose the onset of menstruation and semenarche as the key indicator in pubertal development for their biological, psychological, and relational relevance.
The mean age indicated for these two events by our study population, 12 and 13 years for males and females (Table 1), was not different from data reported in the overall population (61–63). This finding is consistent with the empirical evidence from clinical practice of normal sexual development of children affected by HAE-C1INH.
The frequency of HAE attacks before puberty was similar between males and females, with numbers of acute episodes slightly higher in males in the prepubertal 3-year period (median of monthly mean: M = 1.0; F = 0.41; p = 0.048; Table 2). However, female gender predicts a worsening of symptoms in the 1-year and 3-year period after puberty compared to the same pre-puberty time intervals (p = 0.049 and 0.024, respectively). Symptom worsening after puberty occurred to a greater extent in female subjects than in males (Figures 3, 4). On the contrary, there was no significant variation in the location of the attacks, which were reported mostly as abdominal and subcutaneous both before and after puberty by both the genders, similar to what is described for HAE-C1INH patients of any age and sex (10) (Figure 2).
Previous data suggested that adults present a higher frequency of acute episodes of angioedema than the pediatric population (64–66), although a thorough comparison between pediatric and adult patients is still lacking. Among the several biological factors involved in the process of growth that could theoretically contribute to this increase, hormonal factors certainly play a role (67–70). In fact, estrogen hormones promote a pattern of protein expression, which ultimately increases the activity of the contact system (71, 72) and female patients tend to present a more severe disease (1). In the present manuscript, we show that puberty is a watershed moment of life when the disease starts to get worse for women. Moreover, in our cohort, puberty seemed to represent a turning point also for male subjects: although on semenarche an androgen-dominated pattern of hormones is secreted (73), we might speculate that the bioactive metabolite of testosterone estradiol can act on the contact system and worsen symptoms in the male gender.
Finally, we were not able to demonstrate a parallel worsening of QoL in the period after menarche and semenarche compared to the prepubertal situation. This is because it is well recognized that the frequency of attacks is not the only factor affecting patient QoL, as the severity of single HAE episodes and the unpredictability of their onset play a crucial role in the global burden of disease (52). Moreover, the study design did not allow us to use specific tools to assess QoL variations.
Further limitations of the study derive from the retrospective approach and the approximate quantification of the number of attacks not included in the register, active since 2017, especially in patients who had been through puberty for over a decade. Several biases could influence the accuracy of attack frequency, when retrospectively reported. Among others, subjects may had been influenced by the interviewers, and attacks occurred in young age might be more easily forgotten.
Moreover, treatment strategies were highly variable, also because of the many new drugs developed in recent years. Thus, although we tried to limit these biases by including in the study only subjects under 40 years of age, we were well aware that a comparison of on-demand treatment before and after puberty was unreliable and reported only the descriptive statistics. Nevertheless, the prevalence of patients on long-term prophylaxis for HAE-C1INH had raised from 5.9% before puberty to 13.6% in the 3 years after menarche or semenarche (Tables 2, 3), thus strengthening the evidence of symptom worsening after puberty.
Overall, our study confirms previous reports on a more severe phenotype in the female gender and provides new insights on the impact of puberty on a large cohort of HAE patients from the ITACA network. Results show that puberty predisposes to increased numbers of angioedema attacks, in particular in female patients. Retrospective data collection should be confirmed in prospective assessments, which are challenging in the field of rare diseases but may be favored by patient adherence to disease registries.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by Comitato etico Milano area 1. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
MC promoted the study on behalf of ITACA. MC, PT, and RS contributed to the conceptualization and scope, and drafted the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
MC received speaker/consultancy fees from BioCryst, CSL Behring, Kalvista, Pharming, SOBI, and Takeda. PT received speaker/consultancy fees from CSL Behring and Takeda. FA received consultancy fees or research grants from CSL Behring and Takeda. DF received speaking fees or research grants from CSL-Behring and Takeda. MDG received speaker fees from CSL Behring. VM received speaker/consultancy fees from BioCryst, CSL Behring, GSK, Kyowa Kirin, Takeda, and Vifor. MG received speaker fees from Takeda, Sanofi, and Astrazeneca. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer AZ declared a past co-authorship with the authors MC, FA, VM, and RS to the handling editor.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fped.2023.1141073/full#supplementary-material.
References
1. Zuraw BL, Christiansen SC. HAE pathophysiology and underlying mechanisms. Clin Rev Allergy Immunol. (2016) 51:216–29. doi: 10.1007/s12016-016-8561-8
2. Pier J, Bingemann TA. Urticaria, angioedema, and anaphylaxis. Pediatr Rev. (2020) 41:283–92. doi: 10.1542/pir.2019-0056
3. Pattanaik D, Lieberman JA. Pediatric angioedema. Curr Allergy Asthma Rep. (2017) 17:60. doi: 10.1007/s11882-017-0729-7
4. Long AT, Kenne E, Jung R, Fuchs TA, Renné T. Contact system revised: an interface between inflammation, coagulation, and innate immunity. J Thromb Haemost. (2016) 14:427–37. doi: 10.1111/jth.13235
5. Hofman Z, de Maat S, Hack CE, Maas C. Bradykinin: inflammatory product of the coagulation system. Clin Rev Allergy Immunol. (2016) 51:152–61. doi: 10.1007/s12016-016-8540-0
6. Montinaro V, Cicardi M. ACE inhibitor-mediated angioedema. Int Immunopharmacol. (2020) 78:106081. doi: 10.1016/j.intimp.2019.106081
7. Aygören-Pürsün E, Magerl M, Maetzel A, Maurer M. Epidemiology of bradykinin-mediated angioedema: a systematic investigation of epidemiological studies. Orphanet J Rare Dis. (2018) 13:73. doi: 10.1186/s13023-018-0815-5
8. Stone C, Brown NJ. Angiotensin-converting enzyme inhibitor and other drug-associated angioedema. Immunol Allergy Clin N Am. (2017) 37:483–95. doi: 10.1016/j.iac.2017.04.006
9. Cicardi M, Zuraw BL. Angioedema due to bradykinin dysregulation. J Allergy Clin Immunol Pract. (2018) 6:1132–41. doi: 10.1016/j.jaip.2018.04.022
10. Busse PJ, Christiansen SC. Hereditary angioedema. N Engl J Med. (2020) 382:1136–48. doi: 10.1056/NEJMra1808012
12. Reshef A, Kidon M, Leibovich I. The Story of Angioedema: from Quincke to Bradykinin. Clin Rev Allerg Immunol. (2016) 51:121–39. doi: 10.1007/s12016-016-8553-8
13. Binkley KE, Davis A. Clinical, biochemical, and genetic characterization of a novel estrogen-dependent inherited form of angioedema. J Allergy Clin Immunol. (2000) 106:546–50. doi: 10.1067/mai.2000.108106
14. Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. (2000) 356:213–7. doi: 10.1016/S0140-6736(00)02483-1
15. Bork K, Wulff K, Möhl BS, Steinmüller-Magin L, Witzke G, Hardt J, et al. Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J Allergy Clin Immunol. (2021) 148:1041–8. doi: 10.1016/j.jaci.2021.01.011
16. Stray-Pedersen A, Abrahamsen TG, Frøland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol. (2000) 20:477–85. doi: 10.1023/A:1026416017763
17. Roche O, Blanch A, Caballero T, Sastre N, Callejo D, López-Trascasa M. Hereditary angioedema due to C1 inhibitor deficiency: patient registry and approach to the prevalence in Spain. Ann Allergy Asthma Immunol. (2005) 94:498–503. doi: 10.1016/S1081-1206(10)61121-0
18. Bygum A. Hereditary angio-oedema in Denmark: a nationwide survey. Br J Dermatol. (2009) 161:1153–8. doi: 10.1111/j.1365-2133.2009.09366.x
19. Zanichelli A, Arcoleo F, Barca MP, Borrelli P, Bova M, Cancian M, et al. A nationwide survey of hereditary angioedema due to C1 inhibitor deficiency in Italky. Orphanet J Rare Dis. (2015) 6(10):11. doi: 10.1186/s13023-015-0233-x
20. Schöffl C, Wiednig M, Koch L, Blagojevic D, Duschet P, Hawranek T, et al. Hereditary angioedema in Austria: prevalence and regional peculiarities. Dtsch Dermatol Ges. (2019) 17:416–23. doi: 10.1111/ddg.13815
21. Lumry WR, Settipane RA. Hereditary angioedema: epidemiology and burden of disease. Allergy Asthma Proc. (2020) 41(Suppl 1):S08–13. doi: 10.2500/aap.2020.41.200050
22. Davis AE, Mejia P, Lu F. Biological activities of C1 inhibitor. Mol Immunol. (2008) 45: 4057–63. doi: 10.1016/j.molimm.2008.06.028
23. Schoenfeld AK, Lahrsen E, Alban S. Regulation of complement and contact system activation via C1 inhibitor potentiation and factor XIIa activity modulation by sulfated glycans—structure-activity relationships. PLoS One. (2016) 11:e0165493. doi: 10.1371/journal.pone.0165493
24. Karknaukhova E. C1-inhibitor: structure, functional diversity and therapeutic development. Curr Med Chem. (2022) 29:467–88. doi: 10.2174/0929867328666210804085636
25. Davis AE. The pathopisiology of hereditary angioedema. Clin Immunol. (2005) 114:3–9. doi: 10.1016/j.clim.2004.05.007
26. Cugno M, Zanichelli A, Foieni F, Caccia S, Cicardi M. C1-inhibitor deficiency and angioedema: molecular mechanisms and clinical progress. Trends Mol Med. (2009) 15:69–78. doi: 10.1016/j.molmed.2008.12.001
27. Cugno M, Castelli R, Cicardi M. Angioedema due to acquired C1-inhibitor deficiency: a bridging condition between autoimmunity and lymphoproliferation. Autoimmun Rev. (2008) 8:156–9. doi: 10.1016/j.aurev.2008.05.003
28. Cicardi M, Zanichelli A. Acquired angioedema. Allergy Asthma Clin Immunol. (2010) 6:14. doi: 10.1186/1710-1492-6-14
29. Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. (2008) 359:1027–36. doi: 10.1056/NEJMcp0803977
30. Rubinstein E, Stolz LE, Sheffer AL, Stevens C, Bousvaros A. Abdominal attacks and treatment in hereditary angioedema with C1-inhibitor deficiency. BMC Gastroenterol. (2014) 14:71. doi: 10.1186/1471-230X-14-71
31. Bork K, Siedlecki K, Bosch S, Schopf RE, Kreuz W. Asphyxiation by laryngeal edema in patients with hereditary angioedema. Mayo Clin Proc. (2000) 75:349–54. doi: 10.4065/75.4.349
32. Bork K. Recurrent angioedema and the threat of asphyxation. Dtsch Arztebl Int. (2010) 107: 408–14. doi: 10.3238/arztebl.2010.0408
33. Craig T. Triggers and short-term prophylaxis in patients with hereditary angioedema. Allergy Asthma Proc. (2020) 41(Suppl 1):S30–4. doi: 10.2500/aap.2020.41.200058
34. Kemp JG, Craig TJ. Variability of prodromal signs and symptoms associated with hereditary angioedema attacks: a literature review. Allergy Asthma Proc. (2009) 30:493–9. doi: 10.2500/aap.2009.30.3278
35. Leibovich-Nassi I, Golander H, Somech R, Har-Even D, Reshef A. New instruments for the evaluation of prodromes and attacks of hereditary angioedema (HAE-EPA). Clin Rev Alklergy Immunol. (2021) 61:29–39. doi: 10.1007/s12016-021-08843-8
36. Gompels MM, Lock RJ, Morgan JE, Osborne J, Brown A, Virgo PF. A multicentre evaluation of the diagnostic efficiency of serological investigations for C1 inhibitor deficiency. J Clin Pathol. (2002) 55: 145–7. doi: 10.1136/jcp.55.2.145
37. Wagenaar-Bos IG, Drouet C, Aygören-Pursun E, Bork K, Bucher C, Bygum A, et al. Functional C1-inhibitor diagnostics in hereditary angioedema: assay evaluation and recommendations. J Immunol Methods. (2008) 338: 14–20. doi: 10.1016/j.jim.2008.06.004
38. Porebski G, Kwitniewski M, Reshef A. Biomarkers in hereditary angioedema. Clin Rev Allergy Immunol. (2021) 60:404–15. doi: 10.1007/s12016-021-08845-6
39. Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui IJ, Aygören-Pürsün E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema—the 2021 revision and update. Allergy. (2022) 77:1961–90. doi: 10.1111/all.15214
40. Cancian M. Diagnostic and therapeutic management of hereditary angioedema due to C1-inhibitor deficiency: the Italian experience. Curr Opin Allergy Clin Immunol. (2015) 15: 383–91. doi: 10.1097/ACI.0000000000000186
41. Loules G, Zamanakou M, Parsopoulou F, Vatsiou S, Psarros F, Csuka D, et al. Targeted next-generation sequencing for the molecular diagnosis of hereditary angioedema due to C1-inhibitor deficiency. Gene. (2018) 667:76–82. doi: 10.1016/j.gene.2018.05.029
42. Germenis AE, Margaglione M, Pesquero JB, Farkas H, Cichon S, Csuka D, et al. International consensus on the use of genetics in the management of hereditary angioedema. J Allergy Clin Immunol Pract. (2020) 8:901–11. doi: 10.1016/j.jaip.2019.10.004
43. Drouet C, López-Lera A, Ghannam A, López-Trascasa M, Cichon S, Ponard D, et al. SERPING1 variants and C1-INH biological function: a close relationship with C1-INH-HAE. Front Allergy. (2022) 3:835503. doi: 10.3389/falgy.2022.835503
44. Farkas H, Martinez-Saguer I, Bork K, Bowen T, Craig T, Frank M, et al. International consensus on the diagnosis and management of pediatric patients with hereditary angioedema with C1 inhibitor deficiency. Allergy. (2017) 72:300–13. doi: 10.1111/all.13001
45. Pedrosa M, Phillips-Angels E, López -Lera A, López-Trascasa M, Caballero T. Complement study versus CINH gene testing for the diagnosis of type I hereditary angioedema in children. J Clin Immunol. (2016) 36:16–8. doi: 10.1007/s10875-015-0222-9
46. Lai Y, Zhang G, Inhaber N, Bernstein JA, Cwik M, Zhou Z, et al. A robust multiplexed assay to quantify C1-inhibitor, C1q, and C4 proteins for in vitro diagnosis of hereditary angioedema from dried blood spot. J Pharm Biomed Anal. (2021) 195:113844. doi: 10.1016/j.jpba.2020.113844
47. Förster TM, Magerl M, Maurer M, Zülbahar S, Zielke S, Inhaber N, et al. HAE patient self-sampling for biomarker establishment. Orphanet J Rare Dis. (2021) 16:399. doi: 10.1186/s13023-021-02021-x
48. Isono M, Kokado M, Kato K. Why does it take so long for rare disease patients to get an accurate diagnosis?—a qualitative investigation of patient experiences of hereditary angioedema. PLoS One. (2022) 17:e0265847. doi: 10.1371/journal.pone.0265847
49. Zanichelli A, Magerl M, Longhurst H, Fabien V, Maurer M. Hereditary angioedema with C1 inhibitor deficiency: delay in diagnosis in Europe. Allergy Asthma Clin Immunol. (2013) 9(1):29 doi: 10.1186/1710-1492-9-29
50. Savarese L, Bova M, De Falco C, Guarino MD, De Luca Picione R, Petraroli A, et al. Emotional processes and stress in children affected by hereditary angioedema with C1-inhibitor deficiency: a multicenter, prospective study. Orphanet J Rare Dis. (2018) 13:115. doi: 10.1186/s13023-018-0871-x
51. Savarese L, Freda MF, De Luca Picione R, Dolce P, De Falco R, Alessio M, et al. The experience of living with a chronic disease in pediatrics from the mothers’ narrative: the clinical interview on parental sense of grip on the disease. Health Psychol Open. (2020):1–13. doi: 10.1177/2055102920971496
52. Bork K, Anderson JT, Caballero T, Craig T, Johnston DT, Li HH, et al. Assessment and management of disease burden and quality of life in patients with hereditary angioedema: a consensus report. Allergy Asthma Clin Immunol. (2021) 17:40. doi: 10.1186/s13223-021-00537-2
53. Farkas H. Pediatric hereditary angioedema due to C1-inhibitor deficiency. Allergy Asthma Clin Immunol. (2010) 6:18. doi: 10.1186/1710-1492-6-18
54. Nanda MK, Elenburg S, Bernstein J, Assa’ad AH. Clinical features of pediatric angioerdema. J Allergy Clin Immunol Pract. (2015) 3:392–5. doi: 10.1016/j.jaip.2014.11.012
55. Christiansen SC, Davis DK, Castaldo AJ, Zuraw BL. Pediatric hereditary angioedema: onset, diagnostic delay, and disease severity. Clin Pediatr. (2016) 55:935–42. doi: 10.1177/0009922815616886
56. Karadža-Lapić L, Barešić M, Vrsalović R, Ivković-Jureković I, Sršen S, Prkačin I, et al. Hereditary angioedema due to C1-inhibitor deficiency in pediatric patients in Croatia—first national study, diagnostic and prophylactic challenges. Acta Clin Croat. (2019) 58: 139–46. doi: 10.20471/acc.2019.58.01.18
57. Cancian M, Perego F, Senter R, Arcoleo F, De Pasquale T, Zoli A, et al. Pediatric angioedema: essential features and preliminary results from the hereditary angioedema global registry in Italy. Pediatr Allergy Immunol. (2020) 31(Suppl 24):22–4. doi: 10.1111/pai.13170
58. Andrási N, Balla Z, Szilági A, Csuka D, Varga L, Farkas H. Diagnosing pediatric patients with hereditary C1-inhibitor deficiency—experience from the Hungarian center of reference and excellence. Front Allergy. (2022) 3:860355. doi: 10.3389/falgy.2022.860355
59. Patton GC, Viner R. Pubertal transitions in health. Lancet (2007) 369(9567):1130–9. doi: 10.1016/S0140-6736(07)60366-3
60. Zanichelli A, Magerl M, Longhurst HJ, Aberer W, Caballero T, Bouillet L, et al. Improvement in diagnostic delays over time in patients with hereditary angioedema: findings from the Icatibant Outcome Survey. Clin Transl Allergy. (2018) 8:42. doi: 10.1186/s13601-018-0229-4
61. Walker IV, Smith CR, Davies JH, Inskip HM, Baird J. Methods for determining pubertal status in research studies: literature review and opinions of experts and adolescents. J Dev Orig Health Dis. (2020) 11(2):168–87. doi: 10.1017/S2040174419000254
62. Kim T, Yun JW, Son M, Kim CB, Choe SA. Age at menarche of adolescent girls and the neighbourhood socioeconomic status of their school area. Eur J Contracept Reprod Health Care. (2022):1–7. doi: 10.1080/13625187.2022.2104834
63. Suutela M, Miettinen PJ, Kosola S, Rahkonen O, Varimo T, Tarkkanen A, et al. Timing of puberty and school performance: a population-based study. Front Endocrinol (Lausanne). (2022) 13:936005. doi: 10.3389/fendo.2022.936005
64. Aabom A, Andersen KE, Fagerberg C, Fisker N, Jakobsen MA, Bygum A. Clinical characteristics and real-life diagnostic approaches in all Danish children with hereditary angioedema. Orphanet J Rare Dis. (2017) 12(1):55. doi: 10.1186/s13023-017-0604-6.63
65. Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. (2006) 119(3):267–74. doi: 10.1016/j.amjmed.2005.09.064.b
66. Piotrowicz-Wójcik K, Bulanda M, Juchacz A, Jamróz-Brzeska J, Gocki J, Kuziemski K, et al. Clinical characteristics and management of angioedema attacks in Polish adult patients with hereditary angioedema due to C1-inhibitor deficiency. J Clin Med. (2021) 10(23):5609. doi: 10.3390/jcm10235609
67. Bouillet L, Gompel A. Hereditary angioedema in women: specific challenges. Immunol Allergy Clin North Am. (2013) 33(4):505–11. doi: 10.1016/j.iac.2013.07.006
68. Caballero T, Farkas H, Bouillet L, Bowen T, Gompel A, Fagerberg C, et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol. (2012) 129(2):308–20. doi: 10.1016/j.jaci.2011.11.025
69. Agostoni A, Cicardi M. Hereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patients. Medicine (Baltimore). (1992) 71(4):206–15. doi: 10.1097/00005792-199207000-00003
70. Triggianese P, Senter R, Petraroli A, Zoli A, Lo Pizzo M, Bignardi D, et al. Pregnancy in women with hereditary angioedema due to C1-inhibitor deficiency: results from the ITACA cohort study on outcome of mothers and children with in utero exposure to plasma-derived C1-inhibitor. Front Med (Lausanne). (2022) 9:930403. doi: 10.3389/fmed.2022.930403
71. Kulkarni M, Travers JB, Rohan C. High estrogen states in hereditary angioedema: a spectrum. Clin Rev Allergy Immunol. (2021) 60(3):396–403. doi: 10.1007/s12016-021-08863-4
72. Gompel A, Fain O, Boccon-Gibod I, Gobert D, Bouillet L. Exogenous hormones and hereditary angioedema. Int Immunopharmacol. (2020) 78:106080. doi: 10.1016/j.intimp.2019.106080
Keywords: C1-inhibitor (C1INH), contact system, rare disease, hereditary angioedema (HAE), pediatric, puberty, ITACA
Citation: Cancian M, Triggianese P, Modica S, Arcoleo F, Bignardi D, Brussino L, Colangelo C, Di Agosta E, Firinu D, Guarino MD, Giardino F, Giliberti M, Montinaro V and Senter R (2023) The impact of puberty on the onset, frequency, location, and severity of attacks in hereditary angioedema due to C1-inhibitor deficiency: A survey from the Italian Network for Hereditary and Acquired Angioedema (ITACA). Front. Pediatr. 11:1141073. doi: 10.3389/fped.2023.1141073
Received: 9 January 2023; Accepted: 8 March 2023;
Published: 18 April 2023.
Edited by:
Sezgin Sahin, Istanbul University-Cerrahpasa, TürkiyeReviewed by:
Erik Waage Nielsen, Nord University, NorwayAndrea Zanichelli, University of Milan, Italy
© 2023 Cancian, Triggianese, Modica, Arcoleo, Bignardi, Brussino, Colangelo, Di Agosta, Firinu, Guarino, Giardino, Giliberti, Montinaro and Senter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauro Cancian bWNhbmNpYW5AdW5pcGQuaXQ=
Specialty Section: This article was submitted to Pediatric Rheumatology, a section of the journal Frontiers in Pediatrics