 Keith A. Sacco
Keith A. Sacco Andrea Gazzin
Andrea Gazzin Luigi D. Notarangelo
Luigi D. Notarangelo Ottavia M. Delmonte
Ottavia M. Delmonte
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 20 February 2023
Sec. Pediatric Immunology
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1110115
This article is part of the Research TopicReviews in Pediatric Primary ImmunodeficienciesView all 11 articles
Granulomas have been defined as inflammatory infiltrates formed by recruitment of macrophages and T cells. The three-dimensional spherical structure typically consists of a central core of tissue resident macrophages which may merge into multinucleated giant cells surrounded by T cells at the periphery. Granulomas may be triggered by infectious and non-infectious antigens. Cutaneous and visceral granulomas are common in inborn errors of immunity (IEI), particularly among patients with chronic granulomatous disease (CGD), combined immunodeficiency (CID), and common variable immunodeficiency (CVID). The estimated prevalence of granulomas in IEI ranges from 1%–4%. Infectious agents causing granulomas such Mycobacteria and Coccidioides presenting atypically may be ‘sentinel’ presentations for possible underlying immunodeficiency. Deep sequencing of granulomas in IEI has revealed non-classical antigens such as wild-type and RA27/3 vaccine-strain Rubella virus. Granulomas in IEI are associated with significant morbidity and mortality. The heterogeneity of granuloma presentation in IEI presents challenges for mechanistic approaches to treatment. In this review, we discuss the main infectious triggers for granulomas in IEI and the major forms of IEI presenting with ‘idiopathic’ non-infectious granulomas. We also discuss models to study granulomatous inflammation and the impact of deep-sequencing technology while searching for infectious triggers of granulomatous inflammation. We summarize the overarching goals of management and highlight the therapeutic options reported for specific granuloma presentations in IEI.
Granulomas represent the result of an immune response induced by an encounter between antigen presenting cells (predominantly monocytes and macrophages) and T cells (both CD4 + and CD8+ T cells) (1, 2). In this regard, granulomas offer an opportunity to explore an interface between the innate and adaptive immune system. However, unlike lymph nodes there is no prerequisite anatomic scaffold for granulomas to develop. Classically, granulomas in mammals have been described as having a spherical structure consisting of a central core of tissue resident macrophages which may merge into multinucleated giant cells surrounded by T cells at the periphery (3). In some cases, the central core of the granulomas shows evidence of necrosis (caseating granulomas), while other forms of granulomas do not show this feature (non-caseating granulomas). These two histological types are exemplified by granulomas occurring in the course of Mycobacterial infection and sarcoidosis, respectively (2, 4). Granulomas may be triggered by infectious and non-infectious antigens (5). Despite significant advances in imaging, histopathology, antimicrobials, and immunomodulation, granulomatous inflammation is a cause for morbidity and mortality in both adults and pediatric patients, particularly as described in common variable immunodeficiency (CVID) (6). Inborn errors of immunity (IEIs) provide a unique insight into the pathophysiology of granulomas and immune susceptibility to specific pathogens that trigger granulomatous inflammation. Tuberculous and non-tuberculous Mycobacterial infection (7) and fungal infections (8) presenting with granulomas, for example, may uncover new forms of IEI. Recently patients with disseminated Coccidioidomycosis, a dimorphic fungal infection endemic to the Southwestern United States, have helped to identify novel variants in genes affecting the immune function and inform on fungal pathogenesis (8). Rubella virus (including the attenuated live vaccine strain) is usually well controlled by the immune system in healthy individuals; however, it has been frequently identified in granulomas of patients with various forms of combined immunodeficiency (CID) (9) and has been associated with significant morbidity and mortality in IEIs (10). In a section of this review, we will highlight the current understanding of the role of infectious agents mentioned and how they interface in the development of granulomas in patients with IEIs.
Furthermore, various IEIs present with granulomas of presumed non-infectious origin involving skin and other organs. The overall prevalence of presumed non-infections granulomas in patients with IEI is estimated to be 1 to 4% with the highest prevalence being among CVID, CID, and CGD (11). In a retrospective single center pediatric study from Turkey, among 82 patients with granulomas who underwent immunological evaluation, 62 carried a diagnosis of IEI. CID was the most common diagnosis and hypogammaglobulinemia was present in 50% of the subjects (12). Granulomas have also been described in autoinflammatory diseases and primary atopic disorders (13).
In this review we will discuss updates in understanding IEIs and variants affecting host defense and predisposing to presumably non-infectious granulomas. The focus of the review will center on non-infectious granulomatous inflammation in CVID, CID, CGD and autoinflammatory disorders. We will highlight the role of infectious agents such mycobacteria and Coccidioides as ‘sentinel’ granulomatous infectious to assist with further workup of possible underlying immunodeficiency. Further, we explore the recent data pertaining to the identification of Rubella virus vaccine-strain in IEI granulomas and discuss how this discovery could shed further mechanistic insights into granuloma pathogenesis.
CVID is a clinical diagnosis characterized by severe and/or recurrent oto-sino-pulmonary infections, low serum immunoglobulins and impaired vaccine responses (14, 15). Non-infectious complications of CVID, more than infectious history are associated with decreased CVID survival (6). Granulomas in lungs, liver, spleen, lymph nodes and skin were identified in 46 CVID patients within a cohort of 473 subjects (9.7%) (6). A systematic review by van Stigt et al. (16) showed that 50% of CVID patients with granulomatous disease displays extrapulmonary granulomatous manifestation. Granulomatous and lymphocytic interstitial lung disease (GLILD) instead occurs in around 20%–30% of CVID patients (17). GLILD is a clinical, radiologic and pathologic entity characterized by lymphocytic infiltration and/or granuloma of the lung whenever other infectious causes have been excluded. Risk factors for GLILD include history of cytopenias, female gender, age between 20 and 50 years, concomitant hypersplenism and polyarthritis (17, 18). It is associated with increased risk for non-Hodgkin's lymphoma (19). Dysregulated B cells, with increased expression of B cell activating factor (BAFF), which upregulates IFN-γ signaling, have been reported in CVID with interstitial lung diseases (20). A recent study identified increased numbers of CD14 + CD16- monocytes and memory T cells, and prominent inflammation in peripheral blood of CVID patients with non-infectious complications. Further, the same patients had higher serum levels of IFN-γ, IL-6, IL-18, TNF and T-cell activation markers in peripheral blood (21). These abnormalities improved with T-cell-targeted therapy (21). While the study included various CVID patients with non-infectious complications and not only granulomas, it highlighted the role of T cell dysregulation in the pathogenesis of inflammatory manifestations associated with CVID. Screening high-resolution computerized tomogram (HRCT) is recommended for all CVID patients and should be repeated after 4–5 years if initial screening is unremarkable (22). Further, annual spirometry and 6 min-walk test are considered a cost-effective approach to identify patients who may rapidly progress to GLILD. Immunoglobulin replacement should be targeted to reach and maintain a trough IgG level >1000 mg/dl (22, 23). Increasingly, symptomatic patients may be treated with oral glucocorticoids even though the clinical response to this treatment has shown to be poor (24). Glucocorticoid-sparing agents such as weekly Rituximab 375 mg/m2 for 4 weeks repeated every 4 months for 3–4 courses (25), azathioprine 1–2 mg/kg/day or mycophenolate mofetil 250–1000 mg twice daily can be also considered (25). With such treatment, remission of extrapulmonary granulomas affecting the skin, liver and lymph nodes has been reported in 86% of patients (16). Anti-TNF-α therapy has been successful to treat extrapulmonary granulomas in CVID (16, 26).
The study of combined immunodeficiencies provides mechanistic insights into the pathophysiologic role of adaptive immune cells in granuloma formation (27, 28). Hypomorphic RAG mutations, for example, offer an interesting model to understand granulomatous inflammation. The RAG 1/2 heterotetramer is crucial for the VDJ recombination process and for the generation of a diverse repertoire of antigen-specific T-and B-cell receptors (28). The severity of the clinical phenotype correlates with residual RAG1/2 catalytic activity and may range from severe combined immunodeficiency (SCID) in null RAG 1/2 variants with absent T or B lymphocyte to a less severe phenotype with milder infections, autoimmunity, and granulomas (CID-G/AI) typically with a later onset in life (27). The latter group of patients display preservation of circulating T cells (albeit in reduced numbers and with a predominance of memory cells), and often have normal levels of immunoglobulins and variable response to immunization. High throughput sequencing analysis revealed significant abnormalities of TCR beta repertoire, especially in T regulatory cells, and of BCR repertoire, supporting the notion that relatively higher levels of RAG protein function may allow for partial preservation of the diversity of TCR and BCR repertoires, which however are enriched in self-reactive specificities that may be possibly implicated in granuloma pathogenesis (29). Granulomas in RAG deficiency infiltrate the skin, bones and/or internal organs (30, 31) and can lead to significant morbidity and physical disfiguration (32). In a cohort of 85 patients with RAG deficiency, 30 patients had CID-G/AI phenotype with 15/30 displaying granulomas. Of note, most of these patients had concomitant autoimmunity, with autoimmune cytopenias being the most prevalent autoimmune manifestation (33). In another series of 68 patients with CID-G/AI, granulomatous lesions were identified in 35% of patients with the most common location being lungs and skin but also multiple other tissues (liver, spleen, bone marrow, oropharynx, gut, testis, pancreas); usually more than one site was involved in the same individual (34). In contrast with what reported in CVID, patients with RAG deficiency showed to be refractory to glucocorticoids and biologics (i.e., anti-TNF) requiring allogeneic hematopoietic stem cell transplantation (HSCT) which appears to be the only definitive management for granulomatous inflammation in this disease (33, 35).
Skin and visceral granulomas have been described in several other IEIs with predominance of DNA repair defects including ataxia telangiectasia [AT], Artemis deficiency, Nijmegen-breakage syndrome [NBS], PRKCD deficiency (36, 37) and ligase IV deficiency (38–41) (Table 1). Cutaneous non-infectious granulomas have been extensively described in AT (Table 1) (42). Rarely, patients with AT have had granulomas detected in bones and joints (43). Granulomas are typically observed in AT patients with elevated IgM (44). In a cohort of 44 AT patients, those with granulomas had significantly decreased naïve CD8 T cells in peripheral blood (43). A skewed T cell repertoire has also been reported in AT patients with granulomas (45). IVIG, topical and systemic glucocorticoids, tacrolimus, TNF-α inhibitors have been used with variable success however a patient who underwent allogeneic hematopoietic stem cell transplantation (HCT) had complete remission from granulomas (43).
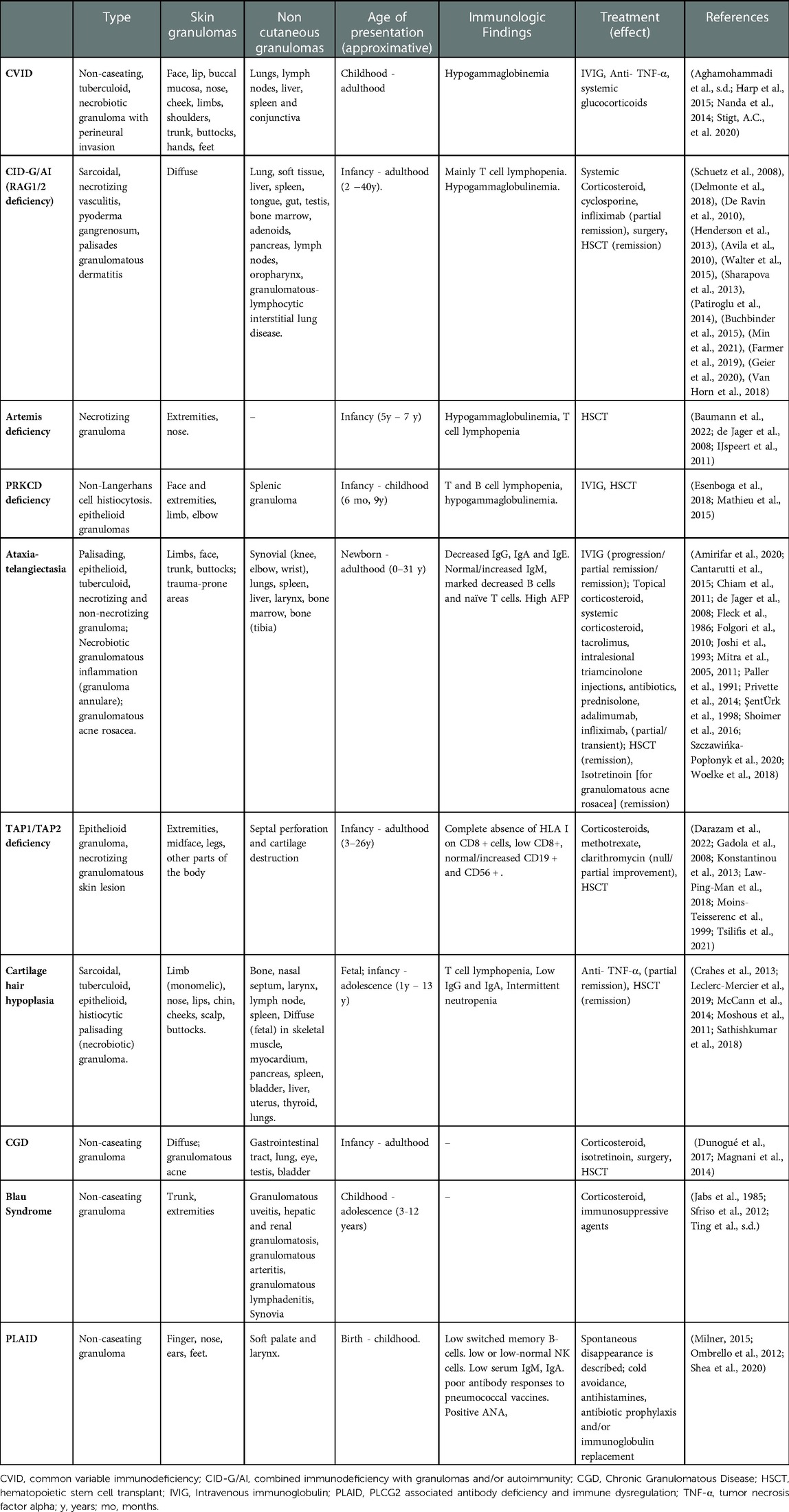
Table 1. Well-described inborn errors of immunity (IEIs) presenting with non-infectious granuloma – presentation, immunophenotype and reported therapies.
The recent identification of RA27/3 Rubella virus (RuV) vaccine strain in some patients has led to the inquiry as to whether RuV antigens may play a causative role for granuloma formation in CID (10, 38). It is unclear as to whether the virus triggers granulomatous inflammation or the impaired host defense allows viral persistence in M2 skewed macrophages and neutrophils (38, 46, 47). Infiltrative granulomas have been described also in TAP1 and TAP2 deficiency (48–52). Recently, 2 cases from Iran of TAP 2 deficiency presenting with granulomas for more than 2 decades before the genetic diagnosis were described. Three relatives of the probands that carried the same homozygous mutation had no clinical manifestation of disease supporting variable expressivity and multifactorial pathogenesis of granulomatous formations (50). Out of 17 cases of TAP2 deficiency reported in the literature, 30% manifested with skin granulomas. Immunomodulatory or immunosuppressant medications are not recommended in this disease because they may cause granuloma exacerbation (50). Data regarding HCT in MHC-I deficiency is limited; one subject displayed persistent regression of skin granulomas 15 years after HCT (53). Cartilage hair hypoplasia patients have granulomas among their clinical manifestations as well (41, 54–56). Rubella virus-associated granulomas have also been described in 21 IEI patients having cytotoxicity defects with significant frequency in MUNC13–4 and RAB27A (Griscelli syndrome type 2) deficiency (57). Of note, GLILD has also been described in Griscelli Syndrome type 2 (58). These data suggest that impaired T cell function allows persistence of macrophages to perpetuate granulomatous inflammation.
CGD is the archetypal IEI for granulomatous inflammation (59, 60). The disorder is characterized by defects in NADPH oxidase due to mutations in 6 known genes – CYBB, CYBA, NCF1, NCF2, NCF4, and CYBC1 (59). Granulomatous lesions are observed in both autosomal recessive forms (61) and in X-linked forms of CGD (62). Most granulomas in CGD are secondary to infections by organisms predisposing to granuloma formation. The infectious susceptibility and natural history of CGD is described extensively in other reviews (59, 60). However, granulomas of presumed non-infectious origin and post-infectious hyperinflammatory granulomatous inflammation are also prevalent in CGD (63). In a cohort of 71 patients with CGD around 10% suffered from post-infectious granulomas in multiple organs (64). Moreover, in a single center study on inflammatory complications of CGD in 98 patients from France, histological analysis showed presence of granulomatous formation (liver, skin, testes, and ocular) in 22 of 44 patients analyzed. This hyperinflammatory state is often associated with chronic colitis (65), granulomatous cystitis (66) and infections including Staphylococcal liver abscesses or Nocardia infection (67, 68). In these cases, systemic glucocorticoids are co-administered with empiric antimicrobials (68). It appears that hyperinflammation in CGD is triggered by an infectious antigen, however it is often perpetuated due to dysregulation in immune function – particularly defective neutrophil apoptosis (69), skewed NF-κB signaling (70), impaired leukotriene B4 and C5a degradation (71), and upregulation of pro-inflammatory cytokines TNF-α, IL-1β, IL-8, IL-17, IL-6 and G-CSF (72–75). Hyperinflammatory foci, including abscesses, lymphadenitis or granulomas often require surgical excision (76).
Granulomas caused by Mycobacterium tuberculosis and non-tuberculous mycobacteria (NTM) are significantly prevalent in IEIs due to phagocytic disorders, and T cell signaling disorders including defects in IFN-γ/Il-12 signaling (7, 77). In endemic areas where the Bacille Calmette-Guérin (BCG) vaccine, containing live-attenuated Mycobacterium bovis bacilli is administered, IEI patients may present with localized granulomatous inflammation termed BCG-itis or disseminated “BCG-osis” (77–79). This may be the first presentation of CGD or SCID and may present challenges in patient management if HSCT is considered since pre-transplant infection and/or inflammation is associated with poor outcomes. Further, granulomatous lesions may only manifest after engraftment and lead to significant morbidity (80, 81).
Very early onset IBD (VEO-IBD) includes a heterogenous group of monogenic IEIs presenting with inflammatory (non-infectious) bowel disease, occasionally with granulomas, prior to age 6 (82). Commercial targeted gene panels for VEO-IBD typically test over 65 genes (83). Readers are referred to comprehensive reviews on monogenic causes of VEO-IBD for further information (83, 84).
Autoinflammatory diseases encompass disorders of pathogenic inflammation secondary to intrinsic immune pathway hyperactivation (85). Autoinflammatory syndromes due to hyperactivation of the NF-κB signaling pathway may be associated with granuloma formation. These disorders are also typically associated with exaggerated TNF activity. The major disorder associated with granulomas in this category is Blau syndrome due autosomal dominant NOD2 pathogenic variants (86). NOD2 variants are also associated with susceptibility to Crohn's disease, characterized by non-caseating granulomas within the gastrointestinal tract (87). Patients classically present within the first decade of life with a combination of granulomatous dermatitis, erythema nodosum, uveitis and polyarticular arthritis (86). Granulomas infiltrating the liver and kidney have been described in Blau Syndrome (88). Recently, a Japanese patient with a pathogenic NOD2 variant was diagnosed with Blau syndrome following BCG vaccination suggesting that infectious triggers may play a role in granuloma formation of this disease (89). Granulomatous inflammation has also been observed in autosomal dominant RelA haploinsufficiency leading to NF-κB hyperactivation (90). Systemic glucocorticoids are used as an initial treatment however TNF-α inhibitors have shown significant therapeutic benefit in both RelA haploinsufficiency and Blau syndrome (91, 92).
Phospholipase C gamma 2–associated antibody deficiency and immune dysregulation (PLAID) is a disorder of autoinflammation, autoimmunity, immunodeficiency, and a primary atopic disorder (93). Phospholipase C gamma 2 (PLCG2) hydrolyzes phosphatidylinositol-4,5-bisphosphate into diacylglycerol and inositol trisphosphate, triggering calcium release from the endoplasmic reticulum to mediate cell activation (94). Heterozygous pathogenic deletions in the autoinhibitory domain of PLCG2 cause a PLAID phenotype since it leads to constitutive activation of the PLCG2 enzyme (95). Patients present with recurrent sinopulmonary infections, urticaria triggered by evaporative cooling, granulomatous dermatitis, hypogammaglobulinemia and various autoimmune manifestations (93, 96). Cutaneous granulomatous lesions are present in 25% of patients and among these subjects many developed skin lesions on the nose, ears and fingers within the first few days of life. These lesions may spontaneously resolve in the vast majority of patients however sometimes they may persist and lead to tissue damage and destruction of nasal and auricular cartilage (97). Furthermore, in some cases granulomatous dermatitis may have a later onset affect especially cold exposed areas (97). Histological characteristics of granulomas in PLAID are similar to CVID with a core of tissue resident macrophages including multinucleated giant cells surrounded by a lymphocytic infiltrate. In this disease the most likely pathogenetic trigger is the spontaneous activation of neutrophil and monocytes by cold exposure (98). Treatment includes cold avoidance, daily high dose nonsedating antihistamines, antibiotic prophylaxis and/or immunoglobulin replacement (96). The use of anti-inflammatory drugs or immunomodulators has not shown significant clinical efficacy in PLAID (96).
As described previously, live-attenuated RuV RA27/3 vaccine strain has been identified in cutaneous and visceral granulomas in IEI patients (99) (Table 2). This finding highlights the importance for thorough antigen screening in tissue biopsy particularly since treatment of granulomas in IEI has classically focused on use of immunosuppression. The RuV antigen and/or RNA has been identified in at least 66 IEI cases -predominantly cases of AT and CID, and defects of cytotoxicity, but much rarer in primary antibody deficiencies (10, 57). Of note, varicella zoster vaccine, mumps and RuV vaccine strain were all identified in granulomas of a patient with late onset hypomorphic RAG2 deficiency (100). The causal role for RuV in granulomas remains to be defined. Recently, both wild-type and vaccine strain RuV has been isolated from cutaneous granulomas of 4 presumed immunocompetent adults (9, 101). However, laboratory evaluation in these patients did reveal immunologic abnormalities including lower CD8+ T cells, lower T-cell mitogen responses, reversed ratio of CD8 + to CD4+ T cells, and/or low serum immunoglobulins (101).
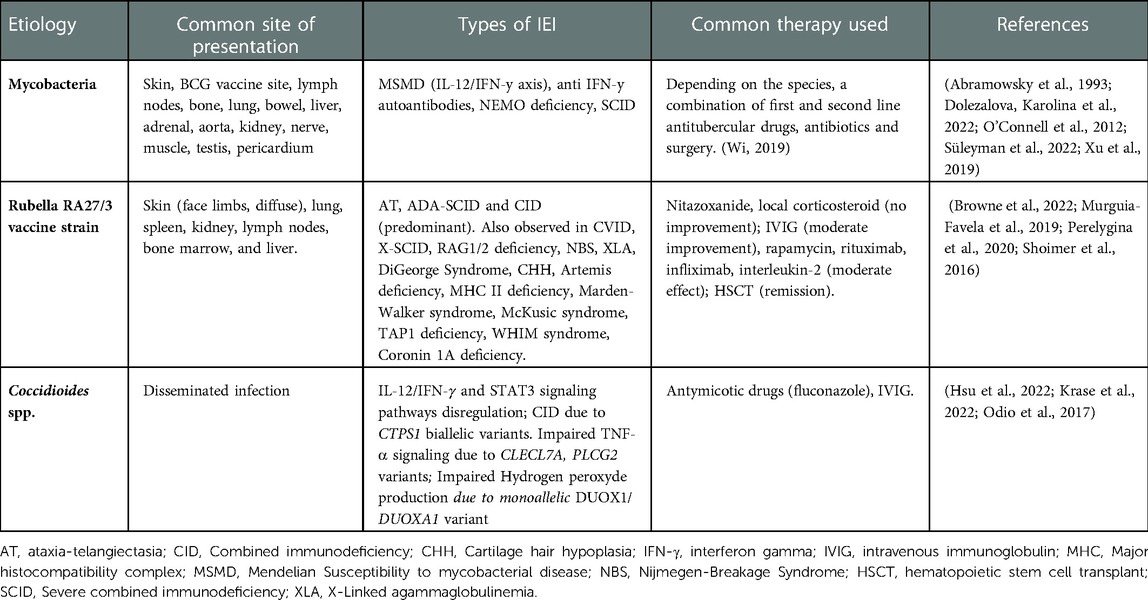
Table 2. Well-described pathogens identified in IEI granulomas.
Viral genome sequencing has revealed multiple nucleotide and amino acid substitutions in the RA27/3 vaccine strain identified in IEI granulomas. These vaccine strains have been termed immunodeficiency-related vaccine-derived rubella viruses (iVDRVs) (47). In IEI patients iVDRVs persist within M2 macrophages, neutrophils, and epidermal keratinocytes (34). It is thought that long-term iVDRV reservoir stems from neutrophils and macrophages residing in the bone marrow (46, 47). Further natural history and mechanistic studies are required to further characterize whether iVDRV is causing granulomas in IEIs. Impaired CD8 T cell repertoire could be a plausible mechanistic insight since CD8 T cell memory is critical for Rubella virus control, and CD8 T cell repertoire decreases with age (102, 103). To date no significant clinical improvement for RuV-associated granulomas has been derived by pharmacotherapy and HSCT remains the only definitive management if indicated by the clinical severity of the underlying IEI (10).
Coccidioidomycosis, known as Valley Fever, is caused by the pathogenic fungus Coccidioides, endemic to the Southwestern United States (104, 105). Symptomatic illness occurs in around 30% of those infected with disseminated disease in <1% (104, 106). Known risk factors for disseminated disease include secondary immunodeficiency due AIDS, chemotherapy, solid organ-and hematopoietic stem cell transplantation, and immunomodulatory biologics (107). Only 14 patients with disseminated coccidioidomycosis (DCM) had been reported in the literature having mutations impairing immune function – 12 within the IL-12/IFN-γ and STAT3 signaling pathways (107–109) and a 5-year-old male with CTPS1 deficiency - a disorder of impaired lymphocyte proliferation (110). A recent publication of a largely adult DCM cohort described mutations in CLECL7A and PLCG2 which impaired TNF-α signaling, and heterozygous variants in DUOX1 and DUOXA1 which impaired hydrogen peroxide production (8). A query of the USIDNET database containing information on 5,485 IEI patients in the United States identified 10 patients with a history of Coccidioidomycosis (111). Patients with persistent presumed non-infections granulomatous inflammation can go undiagnosed for coccidioidomycosis (112). In IEI and immunocompromised patients, serology and immunofixation has poor sensitivity thus the diagnosis must be ruled out by tissue biopsy (113, 114). Identification of Coccidiodes by tissue biopsy is thus of increasing importance in IEI patients within the Southwestern United states particularly in those where the use of immunomodulators as therapeutics is being considered.
A detailed understanding of the etiology, or at least the inflammatory process of an underlying granuloma is key to direct treatment. For this, obtaining a tissue biopsy for histology and culture is imperative. Apart from acid-fast staining for Mycobacteria, and Giemsa staining for fungal etiology, cultures should be prolonged enough to detect possible fastidious organisms. In some cases where biopsy may not be obtained or cultures remain negative, cell-free DNA testing can help identify less common organisms (115) to enable targeted antimicrobial therapy. Antimicrobial target genes have been identified in granulomas of sarcoidosis patients (116). Cytokine gene expression in granulomas can be quantified using techniques such as RNAScope® (117). Complete surgical excision should be considered whenever granulomas may cause anatomic obstruction or if there is insufficient response to pharmacotherapy. Identification of an underlying pathogen warrants guideline-directed antimicrobial therapy. In the case of antimicrobial therapeutics, this is best done in liaison with infectious disease specialists and tailored to the presumed pathogen based on the underlying IEI. Recombinant interferon-gamma (IFNγ) has been used to aid pathogen clearance in IEIs mycobacterial disease, CGD and Coccidioidomycosis. In the case of immunomodulation, there is a longstanding knowledge of using systemic glucocorticoids for inflammatory granulomas in CGD (59, 118). Nitazoxanide, an antiparasitic drug with antiviral properties has been used in IEIs with RuV-associated granulomas (119). Use of oral nitazoxanide was associated with decreased Rubella virus antigen or elimination from granulomas; however, this treatment did not translate into clinically meaningful outcomes (38, 119). Several patients with RuV-associated granulomas underwent allogeneic HCT (120) Patients with lower co-morbid disorders had improved outcomes which suggests that early detection and characterization of RuV-associated granulomas has a bearing on patient outcomes (120).
The advancement of minimally invasive surgical procedures has facilitated obtaining tissue biopsy to characterize granulomas in IEI. Further, histopathologic staining and genetic sequencing can help characterize the inflammatory milieux and possible pathogenic triggers for granulomatous inflammation. This has both prognostic and therapeutic implications, particularly since empiric use of immune suppressive agents can worsen underlying latent infections. The granuloma structure has been described in the mammalian superorder Archonta (including primates) and Laurasiatheria (including carnivores and ungulates) (121). Granulomatous inflammation in preceding animals such as fish does not reflect the complex architecture seen in primates (122). Non-human primates remain a well characterized model of Tuberculosis-induced granulomas however studies are hindered by length of time needed for granuloma formation and requirement of biosafety level 3 laboratories (123). Mouse models of tuberculous granulomas further do not mirror the granuloma architecture seen in humans (124). Inoculation of iVDRV in hypomorphic models of Rag1-mutant mice (125) was unsuccessful in eliciting granulomatous inflammation (126). A heterozygous Stat4-mutant mouse model shows predisposition to disseminated Coccidioidomycosis however lung histology is characterized by lymphocyte infiltration rather than granulomatous inflammation (109, 127). In vitro human granuloma models have been developed to circumvent the difficulties of studying granulomatous inflammation in animal models (128). An in vitro human granuloma model may help characterize mechanisms in granuloma formation as has been observed in various IEI from a clinically phenotypic standpoint. IEIs provide a fascinating template to characterize the heterogeneity and kinetics of granulomatous inflammation. Further characterization of granulomas in IEI can facilitate development of diagnostics and targeted therapeutics for more common granulomatous disorders such as sarcoidosis.
KS and OD analyzed evidence from the literature and wrote the manuscript. AG elaborated the tables. LN and OD supervised the project and critically reviewed the final draft. All the authors approved the submitted manuscript and agreed to be accountable for the content of the work. All authors contributed to the article and approved the submitted version.
This work was supported by the Division of Intramural Research, National Institutes of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, United States.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AT, ataxia-telangiectasia; BCG, Bacille Calmette-Guérin; CGD, chronic granulomatous disease; CID, combined immunodeficiency; CVID, common variable immunodeficiency; GLILD, granulomatous lung interstitial lung disease; HCST, hematopoietic stem cell transplantation; IEI, inborn errors of immunity; iVDRVs, immunodeficiency-related vaccine-derived rubella viruses; NTM, non-tuberculous mycobacteria; PLAID, phospholipase C gamma 2–associated antibody deficiency and immune dysregulation; RuV, Rubella virus.
1. Pagan AJ, Ramakrishnan L. The formation and function of granulomas. Annu Rev Immunol. (2018) 36:639–65. doi: 10.1146/annurev-immunol-032712-100022
2. Cronan MR, Hughes EJ, Brewer WJ, Viswanathan G, Hunt EG, Singh B, et al. A non-canonical type 2 immune response coordinates tuberculous granuloma formation and epithelialization. Cell. (2021) 184(7):1757–1774 e14. doi: 10.1016/j.cell.2021.02.046
3. Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol. (2012) 12(5):352–66. doi: 10.1038/nri3211
4. Sakthivel P, Bruder D. Mechanism of granuloma formation in sarcoidosis. Curr Opin Hematol. (2017) 24(1):59–65. doi: 10.1097/MOH.0000000000000301
5. Wang X, Liu Y. Offense and defense in granulomatous inflammation disease. Front Cell Infect Microbiol. (2022) 12:797749. doi: 10.3389/fcimb.2022.797749
6. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. (2012) 119(7):1650–7. doi: 10.1182/blood-2011-09-377945
7. Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. (2022) 42(7):1508–20. doi: 10.1007/s10875-022-01352-z
8. Hsu AP, Korzeniowska A, Aguilar CC, Gu J, Karlins E, Oler AJ, et al. Immunogenetics associated with severe coccidioidomycosis. JCI Insight. (2022) 7(22):e159491. doi: 10.1172/jci.insight.159491
9. Notarangelo LD. Rubella virus-associated granulomas in immunocompetent adults-possible implications. JAMA Dermatol. (2022) 158(6):611–3. doi: 10.1001/jamadermatol.2022.0055
10. Perelygina L, Icenogle J, Sullivan KE. Rubella virus-associated chronic inflammation in primary immunodeficiency diseases. Curr Opin Allergy Clin Immunol. (2020) 20(6):574–81. doi: 10.1097/ACI.0000000000000694
11. Leung J, Sullivan KE, Perelygina L, Icenogle JP, Fuleihan RL, Lanzieri TM. Prevalence of granulomas in patients with primary immunodeficiency disorders, United States: data from national health care claims and the US immunodeficiency network registry. J Clin Immunol. (2018) 38(6):717–26. doi: 10.1007/s10875-018-0534-7
12. Suleyman M, Serin O, Kocali B, Orhan D, Yalcin EE, Ozcelik U, et al. High inborn errors of immunity risk in patients with granuloma. J Clin Immunol. (2022) 42(8):1795–809. doi: 10.1007/s10875-022-01342-1
13. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2022) 42(7):1473–507. doi: 10.1007/s10875-022-01289-3.35748970
14. Orange JS, Glessner JT, Resnick E, Sullivan KE, Lucas M, Ferry B, et al. Genome-wide association identifies diverse causes of common variable immunodeficiency. J Allergy Clin Immunol. (2011) 127(6):1360–7.e6. doi: 10.1016/j.jaci.2011.02.039
15. de Valles-Ibanez G, Esteve-Sole A, Piquer M, Gonzalez-Navarro EA, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the genetics of common Variable immunodeficiency: monogenetic model and beyond. Front Immunol. (2018) 9:636. doi: 10.3389/fimmu.2018.00636
16. van Stigt AC, Dik WA, Kamphuis LSJ, Smits BM, van Montfrans JM, van Hagen PM, et al. What works when treating granulomatous disease in genetically undefined CVID? A systematic review. Front Immunol. (2020) 11:606389. doi: 10.3389/fimmu.2020.606389
17. Cinetto F, Scarpa R, Carrabba M, Firinu D, Lougaris V, Buso H, et al. Granulomatous lymphocytic interstitial lung disease (GLILD) in common Variable immunodeficiency (CVID): a multicenter retrospective study of patients from Italian PID referral centers. Front Immunol. (2021) 12:627423. doi: 10.3389/fimmu.2021.627423
18. Mannina A, Chung JH, Swigris JJ, Solomon JJ, Huie TJ, Yunt ZX, et al. Clinical predictors of a diagnosis of common Variable immunodeficiency-related granulomatous-lymphocytic interstitial lung disease. Ann Am Thorac Soc. (2016) 13(7):1042–9. doi: 10.1513/AnnalsATS.201511-728OC
19. Fortier JC, Haltigan E, Cavero-Chavez V, Gomez-Manjarres D, Squire JD, Reeves WH, et al. Clinical and phenotypic characterization of common Variable immunodeficiency diagnosed in younger and older adults. J Clin Immunol. (2022) 42(6):1270–9. doi: 10.1007/s10875-022-01290-w
20. Matson EM, Abyazi ML, Bell KA, Hayes KM, Maglione PJ. B cell dysregulation in common Variable immunodeficiency interstitial lung disease. Front Immunol. (2020) 11:622114. doi: 10.3389/fimmu.2020.622114
21. Abyazi ML, Bell KA, Gyimesi G, Baker TS, Byun M, Ko HM, et al. Convergence of cytokine dysregulation and antibody deficiency in common variable immunodeficiency with inflammatory complications. J Allergy Clin Immunol. (2022) 149(1):315–326.e9. doi: 10.1016/j.jaci.2021.06.008
22. Lamers OAC, Smits BM, Leavis HL, de Bree GJ, Cunningham-Rundles C, Dalm V, et al. Treatment strategies for GLILD in common Variable immunodeficiency: a systematic review. Front Immunol. (2021) 12:606099. doi: 10.3389/fimmu.2021.606099
23. Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol. (2010) 137(1):21–30. doi: 10.1016/j.clim.2010.06.012
24. Hurst JR, Verma N, Lowe D, Baxendale HE, Jolles S, Kelleher P, et al. British Lung foundation/United Kingdom primary immunodeficiency network consensus statement on the definition, diagnosis, and management of granulomatous-lymphocytic interstitial lung disease in common Variable immunodeficiency disorders. J Allergy Clin Immunol Pract. (2017) 5(4):938–45. doi: 10.1016/j.jaip.2017.01.021
25. Verbsky JW, Hintermeyer MK, Simpson PM, Feng M, Barbeau J, Rao N, et al. Rituximab and antimetabolite treatment of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency. J Allergy Clin Immunol. (2021) 147(2):704–712.e17. doi: 10.1016/j.jaci.2020.07.021
26. Harp J, Coggshall K, Ruben BS, Ramírez-Valle F, He SY, Berger TG. Cutaneous granulomas in the setting of primary immunodeficiency: a report of four cases and review of the literature. Int J Dermatol. (2015) 54(6):617–25. doi: 10.1111/ijd.12765
27. Delmonte OM, Schuetz C, Notarangelo LD. RAG Deficiency: two genes, many diseases. J Clin Immunol. (2018) 38(6):646–55. doi: 10.1007/s10875-018-0537-4
28. Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu Rev Immunol. (2000) 18:495–527. doi: 10.1146/annurev.immunol.18.1.495
29. Csomos K, Ujhazi B, Blazso P, Herrera JL, Tipton CM, Kawai T, et al. Partial RAG deficiency in humans induces dysregulated peripheral lymphocyte development and humoral tolerance defect with accumulation of T-bet(+) B cells. Nat Immunol. (2022) 23(8):1256–72. doi: 10.1038/s41590-022-01271-6
30. Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. (2008) 358(19):2030–8. doi: 10.1056/NEJMoa073966
31. Niehues T, Perez-Becker R, Schuetz C. More than just SCID–the phenotypic range of combined immunodeficiencies associated with mutations in the recombinase activating genes (RAG) 1 and 2. Clin Immunol. (2010) 135(2):183–92. doi: 10.1016/j.clim.2010.01.013
32. De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, et al. Hypomorphic rag mutations can cause destructive midline granulomatous disease. Blood. (2010) 116(8):1263–71. doi: 10.1182/blood-2010-02-267583
33. Farmer JR, Foldvari Z, Ujhazi B, De Ravin SS, Chen K, Bleesing JJH, et al. Outcomes and treatment strategies for autoimmunity and hyperinflammation in patients with RAG deficiency. J Allergy Clin Immunol Pract. (2019) 7(6):1970–1985.e4. doi: 10.1016/j.jaip.2019.02.038
34. Delmonte OM, Villa A, Notarangelo LD. Immune dysregulation in patients with RAG deficiency and other forms of combined immune deficiency. Blood. (2020) 135(9):610–9. doi: 10.1182/blood.2019000923
35. John T, Walter JE, Schuetz C, Chen K, Abraham RS, Bonfim C, et al. Unrelated hematopoietic cell transplantation in a patient with combined immunodeficiency with granulomatous disease and autoimmunity secondary to RAG deficiency. J Clin Immunol. (2016) 36(7):725–32. doi: 10.1007/s10875-016-0326-x
36. Esenboga S, Akal C, Karaatmaca B, Erman B, Dogan S, Orhan D, et al. Clin Immunol. (2018) 197:1–5. doi: 10.1016/j.clim.2018.08.002
37. Mathieu AL, Verronese E, Rice GI, Fouyssac F, Bertrand Y, Picard C, et al. PRKDC Mutations associated with immunodeficiency, granuloma, and autoimmune regulator-dependent autoimmunity. J Allergy Clin Immunol. (2015) 135(6):1578–88.e5. doi: 10.1016/j.jaci.2015.01.040
38. Perelygina L, Buchbinder D, Dorsey MJ, Eloit M, Hauck F, Hautala T, et al. Outcomes for nitazoxanide treatment in a case series of patients with primary immunodeficiencies and Rubella virus-associated granuloma. J Clin Immunol. (2019) 39(1):112–7. doi: 10.1007/s10875-019-0589-0
39. Campbell MB, Campbell WC, Rogers J, Rogers N, Rogers Z, van den Hurk AM, et al. Bloom syndrome: research and data priorities for the development of precision medicine as identified by some affected families. Cold Spring Harb Mol Case Stud. (2018) 4(2):a002816. doi: 10.1101/mcs.a002816.
40. Yankiwski V, Marciniak RA, Guarente L, Neff NF. Nuclear structure in normal and bloom syndrome cells. Proc Natl Acad Sci U S A. (2000) 97(10):5214–9. doi: 10.1073/pnas.090525897
41. Leclerc-Mercier S, Moshous D, Neven B, Mahlaoui N, Martin L, Pellier I, et al. Cutaneous granulomas with primary immunodeficiency in children: a report of 17 new patients and a review of the literature. J Eur Acad Dermatol Venereol. (2019) 33(7):1412–20. doi: 10.1111/jdv.15568
42. Greenberger S, Berkun Y, Ben-Zeev B, Levi YB, Barziliai A, Nissenkorn A. Dermatologic manifestations of ataxia-telangiectasia syndrome. J Am Acad Dermatol. (2013) 68(6):932–6. doi: 10.1016/j.jaad.2012.12.950
43. Woelke S, Valesky E, Bakhtiar S, Pommerening H, Pfeffermann LM, Schubert R, et al. Treatment of granulomas in patients with ataxia telangiectasia. Front Immunol. (2018) 9:2000. doi: 10.3389/fimmu.2018.02000
44. Chiam LY, Verhagen MM, Haraldsson A, Wulffraat N, Driessen GJ, Netea MG, et al. Cutaneous granulomas in ataxia telangiectasia and other primary immunodeficiencies: reflection of inappropriate immune regulation? Dermatology. (2011) 223(1):13–9. doi: 10.1159/000330335
45. Szczawinska-Poplonyk A, Olejniczak K, Tapolska-Jozwiak K, Boruczkowski M, Jonczyk-Potoczna K, Maldyk J, et al. Cutaneous and systemic granulomatosis in ataxia-telangiectasia: a clinico-pathological study. Postepy Dermatol Alergol. (2020) 37(5):760–5. doi: 10.5114/ada.2020.100485
46. Perelygina L, Plotkin S, Russo P, Hautala T, Bonilla F, Ochs HD, et al. Rubella persistence in epidermal keratinocytes and granuloma M2 macrophages in patients with primary immunodeficiencies. J Allergy Clin Immunol. (2016) 138(5):1436–1439.e11. doi: 10.1016/j.jaci.2016.06.030
47. Perelygina L, Faisthalab R, Abernathy E, Chen MH, Hao L, Bercovitch L, et al. Rubella virus infected macrophages and neutrophils define patterns of granulomatous inflammation in inborn and acquired errors of immunity. Front Immunol. (2021) 12:796065. doi: 10.3389/fimmu.2021.796065
48. Law-Ping-Man S, Toutain F, Rieux-Laucat F, Picard C, Kammerer-Jacquet S, Magerus-Chatinet A, et al. Chronic granulomatous skin lesions leading to a diagnosis of TAP1 deficiency syndrome. Pediatr Dermatol. (2018) 35(6):e375–7. doi: 10.1111/pde.13676
49. de la Salle H, Zimmer J, Fricker D, Angenieux C, Cazenave JP, Okubo M, et al. HLA Class I deficiencies due to mutations in subunit 1 of the peptide transporter TAP1. J Clin Invest. (1999) 103(5):R9–R13. doi: 10.1172/JCI5687
50. Darazam IA, Hakamifard A, Momenilandi M, Materna M, Gharehbagh FJ, Shahrooei M, et al. Delayed diagnosis of chronic necrotizing granulomatous skin lesions due to TAP2 deficiency. J Clin Immunol. (2022) 43(1):217–28. doi: 10.1007/s10875-022-01374-7.36227411
51. Konstantinou P, Kanariou M, Giliani SC, Pantelidaki A, Kokolakis A, Tosca A. Transporter associated with antigen processing deficiency syndrome: case report of an adolescent with chronic perforated granulomatous skin lesions due to TAP2 mutation. Pediatr Dermatol. (2013) 30(6):e223–5. doi: 10.1111/pde.12151
52. Caversaccio M, Bonel HM, Carter R, Williams AP, Gadola SD. TAP Deficiency syndrome: chronic rhinosinusitis and conductive hearing loss. Eur Arch Otorhinolaryngol. (2008) 265(10):1289–92. doi: 10.1007/s00405-008-0610-3
53. Tsilifis C, Moreira D, Marques L, Neves E, Slatter MA, Gennery AR. Stem cell transplantation as treatment for major histocompatibility class I deficiency. Clin Immunol. (2021) 229:108801. doi: 10.1016/j.clim.2021.108801
54. McCann LJ, McPartland J, Barge D, Strain L, Bourn D, Calonje E, et al. Phenotypic variations of cartilage hair hypoplasia: granulomatous skin inflammation and severe T cell immunodeficiency as initial clinical presentation in otherwise well child with short stature. J Clin Immunol. (2014) 34(1):42–8. doi: 10.1007/s10875-013-9962-6
55. Crahes M, Saugier-Veber P, Patrier S, Aziz M, Pirot N, Brasseur-Daudruy M, et al. Foetal presentation of cartilage hair hypoplasia with extensive granulomatous inflammation. Eur J Med Genet. (2013) 56(7):365–70. doi: 10.1016/j.ejmg.2013.04.004
56. Moshous D, Meyts I, Fraitag S, Janssen CE, Debre M, Suarez F, et al. Granulomatous inflammation in cartilage-hair hypoplasia: risks and benefits of anti-TNF-alpha mAbs. J Allergy Clin Immunol. (2011) 128(4):847–53. doi: 10.1016/j.jaci.2011.05.024
57. Gross M, Speckmann C, May A, Gajardo-Carrasco T, Wustrau K, Maier SL, et al. Rubella vaccine-induced granulomas are a novel phenotype with incomplete penetrance of genetic defects in cytotoxicity. J Allergy Clin Immunol. (2022) 149(1):388–399.e4. doi: 10.1016/j.jaci.2021.05.007
58. Szczawinska-Poplonyk A, Jonczyk-Potoczna K, Mikos M, Ossowska L, Langfort R. Granulomatous lymphocytic interstitial lung disease in a Spectrum of pediatric primary immunodeficiencies. Pediatr Dev Pathol. (2021) 24(6):504–12. doi: 10.1177/10935266211022528
59. Yu HH, Yang YH, Chiang BL. Chronic granulomatous disease: a comprehensive review. Clin Rev Allergy Immunol. (2021) 61(2):101–13. doi: 10.1007/s12016-020-08800-x
60. Holland SM. Chronic granulomatous disease. Hematol Oncol Clin North Am. (2013) 27(1):89–99., viii. doi: 10.1016/j.hoc.2012.11.002
61. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. (2010) 363(27):2600–10. doi: 10.1056/NEJMoa1007097
62. Magnani A, Brosselin P, Beaute J, de Vergnes N, Mouy R, Debre M, et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol. (2014) 134(3):655–662.e8. doi: 10.1016/j.jaci.2014.04.014
63. Hill HR. Hyperinflammatory pulmonary disease in chronic granulomatous disease. Pediatr Infect Dis J. (2011) 30(9):808–10. doi: 10.1097/INF.0b013e31822835ba
64. Dunogue B, Pilmis B, Mahlaoui N, Elie C, Coignard-Biehler H, Amazzough K, et al. Chronic granulomatous disease in patients reaching adulthood: a nationwide study in France. Clin Infect Dis. (2017) 64(6):767–75. doi: 10.1093/cid/ciw837
65. Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya-O'Brien S, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. (2004) 114(2):462–8. doi: 10.1542/peds.114.2.462
66. Kontras SB, Bodenbender JG, McClave CR, Smith JP. Interstitial cystitis in chronic granulomatous disease. J Urol. (1971) 105(4):575–8. doi: 10.1016/S0022-5347(17)61580-5
67. Freeman AF, Marciano BE, Anderson VL, Uzel G, Costas C, Holland SM. Corticosteroids in the treatment of severe nocardia pneumonia in chronic granulomatous disease. Pediatr Infect Dis J. (2011) 30(9):806–8. doi: 10.1097/INF.0b013e318218181d
68. Yamazaki-Nakashimada MA, Stiehm ER, Pietropaolo-Cienfuegos D, Hernandez-Bautista V, Espinosa-Rosales F. Corticosteroid therapy for refractory infections in chronic granulomatous disease: case reports and review of the literature. Ann Allergy Asthma Immunol. (2006) 97(2):257–61. doi: 10.1016/S1081-1206(10)60023-3
69. Kobayashi SD, Voyich JM, Braughton KR, Whitney AR, Nauseef WM, Malech HL, et al. Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J Immunol. (2004) 172(1):636–43. doi: 10.4049/jimmunol.172.1.636
70. Bylund J, MacDonald KL, Brown KL, Mydel P, Collins LV, Hancock RE, et al. Enhanced inflammatory responses of chronic granulomatous disease leukocytes involve ROS-independent activation of NF-kappa B. Eur J Immunol. (2007) 37(4):1087–96. doi: 10.1002/eji.200636651
71. Segal BH, Kuhns DB, Ding L, Gallin JI, Holland SM. Thioglycollate peritonitis in mice lacking C5, 5-lipoxygenase, or p47(phox): complement, leukotrienes, and reactive oxidants in acute inflammation. J Leukoc Biol. (2002) 71(3):410–6. doi: 10.1189/jlb.71.3.410
72. Brown KL, Bylund J, MacDonald KL, Song-Zhao GX, Elliott MR, Falsafi R, et al. ROS-deficient monocytes have aberrant gene expression that correlates with inflammatory disorders of chronic granulomatous disease. Clin Immunol. (2008) 129(1):90–102. doi: 10.1016/j.clim.2008.06.005
73. Lekstrom-Himes JA, Kuhns DB, Alvord WG, Gallin JI. Inhibition of human neutrophil IL-8 production by hydrogen peroxide and dysregulation in chronic granulomatous disease. J Immunol. (2005) 174(1):411–7. doi: 10.4049/jimmunol.174.1.411
74. Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, et al. NADPH Oxidase limits innate immune responses in the lungs in mice. PLoS One. (2010) 5(3):e9631. doi: 10.1371/journal.pone.0009631
75. van de Veerdonk FL, Smeekens SP, Joosten LA, Kullberg BJ, Dinarello CA, van der Meer JW, et al. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci U S A. (2010) 107(7):3030–3. doi: 10.1073/pnas.0914795107
76. Feld JJ, Hussain N, Wright EC, Kleiner DE, Hoofnagle JH, Ahlawat S, et al. Hepatic involvement and portal hypertension predict mortality in chronic granulomatous disease. Gastroenterology. (2008) 134(7):1917–26. doi: 10.1053/j.gastro.2008.02.081
77. Soyak Aytekin E, Keskin A, Tan C, Yalcin E, Dogru D, Ozcelik U, et al. Differential diagnosis of primary immunodeficiency in patients with BCGitis and BCGosis: a single-centre study. Scand J Immunol. (2021) 94(4):e13084. doi: 10.1111/sji.13084
78. Chitamanni P, Anbazhagan J, Parameswaran N, Vijayakumar S. BCGosis in infants with severe combined ImmunoDeficiency (SCID). Indian J Pediatr. (2018) 85(7):585–6. doi: 10.1007/s12098-018-2624-1
79. Liu G, Xiao H, Liu L, Guo L, Guo R, Hu X, et al. Severe cases of BCGosis-susceptible primary immunodeficiency diseases identified by next-generation sequencing: implications for adjustment of BCG vaccination timing in China. J Genet Genomics. (2020) 47(4):229–32. doi: 10.1016/j.jgg.2020.03.003
80. Dorsey MJ, Wright NAM, Chaimowitz NS, Davila Saldana BJ, Miller H, Keller MD, et al. Infections in infants with SCID: isolation, infection screening, and prophylaxis in PIDTC centers. J Clin Immunol. (2021) 41(1):38–50. doi: 10.1007/s10875-020-00865-9
81. NaserEddin A, Dinur-Schejter Y, Shadur B, Zaidman I, Even-Or E, Averbuch D, et al. Bacillus calmette-guerin (BCG) vaccine-associated complications in immunodeficient patients following stem cell transplantation. J Clin Immunol. (2021) 41(1):147–62. doi: 10.1007/s10875-020-00892-6
82. Kelsen JR, Russo P, Sullivan KE. Early-Onset inflammatory bowel disease. Immunol Allergy Clin North Am. (2019) 39(1):63–79. doi: 10.1016/j.iac.2018.08.008
83. Ouahed J, Spencer E, Kotlarz D, Shouval DS, Kowalik M, Peng K, et al. Very early onset inflammatory bowel disease: a clinical approach with a focus on the role of genetics and underlying immune deficiencies. Inflamm Bowel Dis. (2020) 26(6):820–42. doi: 10.1093/ibd/izz259
84. Zheng HB, de la Morena MT, Suskind DL. The growing need to understand very early onset inflammatory bowel disease. Front Immunol. (2021) 12:675186. doi: 10.3389/fimmu.2021.675186
85. Gattorno M, Hofer M, Federici S, Vanoni F, Bovis F, Aksentijevich I, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. (2019) 78(8):1025–32. doi: 10.1136/annrheumdis-2019-215048
86. Sfriso P, Caso F, Tognon S, Galozzi P, Gava A, Punzi L. Blau syndrome, clinical and genetic aspects. Autoimmun Rev. (2012) 12(1):44–51. doi: 10.1016/j.autrev.2012.07.028
87. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's Disease. Nature. (2001) 411(6837):599–603. doi: 10.1038/35079107
88. Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Blau syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol Online J. (2014) 12:33. doi: 10.1186/1546-0096-12-33
89. Arakawa A, Kambe N, Nishikomori R, Tanabe A, Ueda M, Nishigori C, et al. NOD2 Mutation-Associated case with blau syndrome triggered by BCG vaccination. Children (Basel). (2021) 8(2):117. doi: 10.3390/children8020117.33562038
90. Steiner A, Harapas CR, Masters SL, Davidson S. An update on autoinflammatory diseases: relopathies. Curr Rheumatol Rep. (2018) 20(7):39. doi: 10.1007/s11926-018-0749-x
91. Matsuda T, Kambe N, Takimoto-Ito R, Ueki Y, Nakamizo S, Saito MK, et al. Potential benefits of TNF targeting therapy in blau syndrome, a NOD2-associated systemic autoinflammatory granulomatosis. Front Immunol. (2022) 13:895765. doi: 10.3389/fimmu.2022.895765
92. Nigrovic PA, Lee PY, Hoffman HM. Monogenic autoinflammatory disorders: conceptual overview, phenotype, and clinical approach. J Allergy Clin Immunol. (2020) 146(5):925–37. doi: 10.1016/j.jaci.2020.08.017
93. Milner JD. PLAID: a syndrome of Complex patterns of disease and unique phenotypes. J Clin Immunol. (2015) 35(6):527–30. doi: 10.1007/s10875-015-0177-x
94. Wang D, Feng J, Wen R, Marine JC, Sangster MY, Parganas E, et al. Phospholipase Cgamma2 is essential in the functions of B cell and several fc receptors. Immunity. (2000) 13(1):25–35. doi: 10.1016/S1074-7613(00)00005-4
95. Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. (2012) 366(4):330–8. doi: 10.1056/NEJMoa1102140
96. Shea J, Huynh T, Milner J, Chamlin S. PLAID Syndrome: characteristic presentation and a novel therapeutic option. Pediatr Dermatol. (2020) 37(1):147–9. doi: 10.1111/pde.13972
97. Szymanski AM, Ombrello MJ. Using genes to triangulate the pathophysiology of granulomatous autoinflammatory disease: nOD2, PLCG2 and LACC1. Int Immunol. (2018) 30(5):205–13. doi: 10.1093/intimm/dxy021
98. Aderibigbe OM, Priel DL, Lee CC, Ombrello MJ, Prajapati VH, Liang MG, et al. Distinct cutaneous manifestations and cold-induced leukocyte activation associated with PLCG2 mutations. JAMA Dermatol. (2015) 151(6):627–34. doi: 10.1001/jamadermatol.2014.5641
99. Bodemer C, Sauvage V, Mahlaoui N, Cheval J, Couderc T, Leclerc-Mercier S, et al. Live rubella virus vaccine long-term persistence as an antigenic trigger of cutaneous granulomas in patients with primary immunodeficiency. Clin Microbiol Infect. (2014) 20(10):O656–63. doi: 10.1111/1469-0691.12573
100. Dutmer CM, Asturias EJ, Smith C, Dishop MK, Schmid DS, Bellini WJ, et al. Late onset hypomorphic RAG2 deficiency presentation with fatal vaccine-strain VZV infection. J Clin Immunol. (2015) 35(8):754–60. doi: 10.1007/s10875-015-0207-8
101. Wanat KA, Perelygina L, Chen MH, Hao L, Abernathy E, Bender NR, et al. Association of persistent Rubella virus with idiopathic skin granulomas in clinically immunocompetent adults. JAMA Dermatol. (2022) 158(6):626–33. doi: 10.1001/jamadermatol.2022.0828
102. Lovett AE, Hahn CS, Rice CM, Frey TK, Wolinsky JS. Rubella virus-specific cytotoxic T-lymphocyte responses: identification of the capsid as a target of major histocompatibility complex class I-restricted lysis and definition of two epitopes. J Virol. (1993) 67(10):5849–58. doi: 10.1128/jvi.67.10.5849-5858.1993
103. Blackman MA, Woodland DL. The narrowing of the CD8 T cell repertoire in old age. Curr Opin Immunol. (2011) 23(4):537–42. doi: 10.1016/j.coi.2011.05.005
104. Brown J, Benedict K, Park BJ, Thompson GR. Coccidioidomycosis: epidemiology. Clin Epidemiol. (2013) 5:185–97.23843703
105. Huang JY, Bristow B, Shafir S, Sorvillo F. Coccidioidomycosis-associated deaths, United States, 1990-2008. Emerg Infect Dis. (2012) 18(11):1723–8. doi: 10.3201/eid1811.120752
106. Galgiani JN, Ampel NM, Blair JE, Catanzaro A, Geertsma F, Hoover SE, et al. Infectious diseases society of America (IDSA) clinical practice guideline for the treatment of coccidioidomycosis. Clin Infect Dis. (2016) 63(6):e112–46. doi: 10.1093/cid/ciw360
107. Odio CD, Marciano BE, Galgiani JN, Holland SM. Risk factors for disseminated coccidioidomycosis, United States. Emerg Infect Dis. (2017) 23(2):308–11. doi: 10.3201/eid2302.160505.28098554
108. Hung CY, Hsu AP, Holland SM, Fierer J. A review of innate and adaptive immunity to coccidioidomycosis. Med Mycol. (2019) 57(Supplement_1):S85–92. doi: 10.1093/mmy/myy146
109. Powell DA, Hsu AP, Shubitz LF, Butkiewicz CD, Moale H, Trinh HT, et al. Mouse model of a human STAT4 point mutation that predisposes to disseminated coccidiomycosis. Immunohorizons. (2022) 6(2):130–43. doi: 10.4049/immunohorizons.2200007
110. Krase IZ, Woodward J, Bauer CS, Miller H, Sacco K. Seronegative mediastinal coccidioidomycosis as a novel presentation of CTPS1 combined immunodeficiency. Open Forum Infect Dis. (2022) 9(8):ofac403. doi: 10.1093/ofid/ofac403
111. Krase IZ, Garabedian E, Fuleihan R, Consortium U, Sacco K. Prevalence of coccidioidomycosis in primary immunodeficiency: data from the USIDNET registry. Clin Immunol. (2022) 245:109135. doi: 10.1016/j.clim.2022.109135
112. Mangold AR, DiCaudo DJ, Blair JE, Sekulic A. Chronic interstitial granulomatous dermatitis in coccidioidomycosis. Br J Dermatol. (2016) 174(4):881–4. doi: 10.1111/bjd.14295
113. Crum NF. Coccidioidomycosis: a contemporary review. Infect Dis Ther. (2022) 11(2):713–42. doi: 10.1007/s40121-022-00606-y
114. Bilal J, Kollampare S, Bode B, Lisse JR, Hoover SE, Sudano D, et al. Management of asymptomatic coccidioidomycosis in patients with rheumatic diseases. Rheumatol Int. (2019) 39(7):1257–62. doi: 10.1007/s00296-019-04307-7
115. Lehman HK, Yu KOA, Towe CT, Risma KA. Respiratory infections in patients with primary immunodeficiency. J Allergy Clin Immunol Pract. (2022) 10(3):683–691.e1. doi: 10.1016/j.jaip.2021.10.073
116. Rotsinger JE, Celada LJ, Polosukhin VV, Atkinson JB, Drake WP. Molecular analysis of sarcoidosis granulomas reveals antimicrobial targets. Am J Respir Cell Mol Biol. (2016) 55(1):128–34. doi: 10.1165/rcmb.2015-0212OC
117. Palmer MV, Thacker TC, Waters WR. Differential cytokine gene expression in granulomas from lungs and lymph nodes of cattle experimentally infected with aerosolized Mycobacterium bovis. PLoS One. (2016) 11(11):e0167471.27902779
118. Chiriaco M, Salfa I, Di Matteo G, Rossi P, Finocchi A. Chronic granulomatous disease: clinical, molecular, and therapeutic aspects. Pediatr Allergy Immunol. (2016) 27(3):242–53. doi: 10.1111/pai.12527
119. Perelygina L, Hautala T, Seppanen M, Adebayo A, Sullivan KE, Icenogle J. Inhibition of rubella virus replication by the broad-spectrum drug nitazoxanide in cell culture and in a patient with a primary immune deficiency. Antiviral Res. (2017) 147:58–66. doi: 10.1016/j.antiviral.2017.09.019
120. Buchbinder D, Hauck F, Albert MH, Rack A, Bakhtiar S, Shcherbina A, et al. Rubella virus-associated cutaneous granulomatous disease: a unique complication in immune-deficient patients, not limited to DNA repair disorders. J Clin Immunol. (2019) 39(1):81–9. doi: 10.1007/s10875-018-0581-0
121. Wilson JL, Mayr HK, Weichhart T. Metabolic programming of macrophages: implications in the pathogenesis of granulomatous disease. Front Immunol. (2019) 10:2265. doi: 10.3389/fimmu.2019.02265
122. Polinas M, Padros F, Merella P, Prearo M, Sanna MA, Marino F, et al. Stages of granulomatous response against histozoic metazoan parasites in mullets (osteichthyes: mugilidae). Animals (Basel). (2021) 11(6):1501. doi: 10.3390/ani11061501
123. Kauffman KD, Sakai S, Lora NE, Namasivayam S, Baker PJ, Kamenyeva O, et al. PD-1 blockade exacerbates Mycobacterium tuberculosis infection in rhesus macaques. Sci Immunol. (2021) 6(55):eabf3861. doi: 10.1126/sciimmunol.abf3861
124. Bohrer AC, Castro E, Hu Z, Queiroz ATL, Tocheny CE, Assmann M, et al. Eosinophils are part of the granulocyte response in tuberculosis and promote host resistance in mice. J Exp Med. (2021) 218(10):e20210469. doi: 10.1084/jem.20210469
125. Ott de Bruin LM, Bosticardo M, Barbieri A, Lin SG, Rowe JH, Poliani PL, et al. Hypomorphic Rag1 mutations alter the preimmune repertoire at early stages of lymphoid development. Blood. (2018) 132(3):281–92. doi: 10.1182/blood-2017-12-820985
126. Sacco K, Bosticardo M, Pala F, Pittaluga S, Corsino C, Hao L, et al. Developing a Rag1-mutant murine model of Rubella vaccine-associated granulomatous inflammation. in JOURNAL OF CLINICAL IMMUNOLOGY. (2021). SPRINGER/PLENUM PUBLISHERS 233 SPRING ST, NEW YORK, NY 10013 USA.
127. Powell DA, Hsu AP, Butkiewicz CD, Trinh HT, Frelinger JA, Holland SM, et al. Vaccine protection of mice with primary immunodeficiencies against disseminated coccidioidomycosis. Front Cell Infect Microbiol. (2021) 11:790488. doi: 10.3389/fcimb.2021.790488
Keywords: granuloma - etiology, combined immune deficiency, GLILD, rubella (MMR) vaccine, coccidiodomycosis, autoinflammatory and autoimmunological diseases
Citation: Sacco KA, Gazzin A, Notarangelo LD and Delmonte OM (2023) Granulomatous inflammation in inborn errors of immunity. Front. Pediatr. 11:1110115. doi: 10.3389/fped.2023.1110115
Received: 28 November 2022; Accepted: 23 January 2023;
Published: 20 February 2023.
Edited by:
Nina Kathrin Serwas, Arcus Biosciences, United StatesReviewed by:
Hanna IJspeert, Erasmus University Rotterdam, Netherlands© 2023 Sacco, Gazzin, Notarangelo and Delmonte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ottavia M. Delmonte b3R0YXZpYS5kZWxtb250ZUBuaWguZ292
Specialty Section: This article was submitted to Pediatric Immunology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.