
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 10 March 2023
Sec. Pediatric Immunology
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1108207
 María Soledad Caldirola1,2*†
María Soledad Caldirola1,2*† Analía Gisela Seminario1,3
Analía Gisela Seminario1,3 Paula Carolina Luna4
Paula Carolina Luna4 Renata Curciarello5
Renata Curciarello5 Guillermo Horacio Docena5Nicolás Fernandez Escobar6Guillermo Drelichman6
Guillermo Horacio Docena5Nicolás Fernandez Escobar6Guillermo Drelichman6 Marco Gattorno7
Marco Gattorno7 Adriana A. de Jesus8
Adriana A. de Jesus8 Raphaela Goldbach-Mansky8
Raphaela Goldbach-Mansky8 María Isabel Gaillard1,9
María Isabel Gaillard1,9 Liliana Bezrodnik3
Liliana Bezrodnik3
During recent years, the identification of monogenic mutations that cause sterile inflammation has expanded the spectrum of autoinflammatory diseases, clinical disorders characterized by uncontrolled systemic and organ-specific inflammation that, in some cases, can mirror infectious conditions. Early studies support the concept of innate immune dysregulation with a predominance of myeloid effector cell dysregulation, particularly neutrophils and macrophages, in causing tissue inflammation. However, recent discoveries have shown a complex overlap of features of autoinflammation and/or immunodeficiency contributing to severe disease phenotypes. Here, we describe the first Argentine patient with a newly described frameshift mutation in SAMD9L c.2666delT/p.F889Sfs*2 presenting with a complex phenotypic overlap of CANDLE-like features and severe infection-induced cytopenia and immunodeficiency. The patient underwent a fully matched unrelated HSCT and has since been in inflammatory remission 5 years post-HSCT.
During recent years, the identification of monogenic causes of systemic autoinflammatory diseases (SAIDs) uncovered the role of innate immune sensing linked to specific proinflammatory cytokines and their signaling pathways as a cause of sterile inflammation. The availability of genetic testing has allowed physicians all over the world to make specific diagnoses in an increasing number of patients with rare severe immune dysregulation diseases (1). The clinical hallmark of SAIDs is the presence of recurrent fevers and other systemic signs of inflammation with nonspecific elevation in acute reactant markers (2, 3). Moreover, SAIDs can be misdiagnosed with systemic infections and/or immunodeficiency due to severe uncontrolled systemic and organ-specific inflammation (4, 5). The first group of monogenic SAIDs presented with dysregulation in IL-1β production, including the IL-1β activating inflammasomopathies. A recently described group of SAIDs that present with chronic upregulation of type 1 interferon (IFN)-stimulated or -regulated genes (IRGs) is referred to as autoinflammatory interferonopathies (4, 6, 7).
However, recent discoveries of additional monogenic diseases, including a disease caused by truncating mutations in SAMD9L, illustrate the existence of a complex overlap of autoinflammatory manifestations, autoimmune diseases and/or immunodeficiencies that have been included in the latest Classification of the International Union of Immunological Societies of Inborn Errors of Immunity (1, 8–10). Among these, homozygous loss-of-function mutations in PSMB8 (proteasome subunit β type 8) were first described to cause CANDLE syndrome (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature), a disease that presents with systemic inflammation, neutrophilic panniculitis, and a chronically elevated IRG signature (11). Later, mutations in different protein subunits of the proteasome–immunoproteasome system (12, 13) or proteasome assembly molecules (13, 14) were published. The prominent peripheral panniculitis in CANDLE syndrome that results in lipodystrophy is a hallmark feature that is however also seen in patients presenting with SAMD9L truncating mutations in SAMD9L and has been previously reported in patients with a wide spectrum of hematological diseases: ataxia-pancytopenia syndrome (ATXPC), aplastic anemia, cytopenia, myelodysplastic syndrome (MDS) predisposition (resembling monosomy 7 and interstitial deletion of 7q disorders), myeloid malignancies, infections, and immunodeficiency (15–19). In most instances, the disease presents later in life or even in adulthood. In 2020, de Jesus et al. published, for the first time, de novo heterozygous truncating SAMD9L mutations that cause a severe perinatal onset SAID (20). Here, we describe the first Argentine patient who presented with neutrophilic panniculitis and lipodystrophy in the context of systemic inflammation that mimicked neonatal-onset CANDLE-like autoinflammatory disease. We now report her long-term follow-up after a successful hematopoietic stem cell transplantation (HSCT). This report raises awareness of SAMD9L truncating mutations that cause severe cytopenias and autoinflammation and guides treatment decisions.
A 5-year-old girl, the first child from nonconsanguineous parents, was born at 38 weeks with no remarkable familiar history. At 1 week of life, she developed a nonevanescent erythematous-papular generalized eruption without fever, visceromegaly, or joint involvement (Figure 1A). A skin biopsy and laboratory evaluation were performed. Her first laboratory evaluation at 24 days of life revealed an elevated white blood cell (WBC) count (32.6 × 109/L) and elevated acute phase reactants: erythrocyte sedimentation rate (ESR) of 68 mm/h (normal <15 mm/h) and C-reactive protein (CRP) level of 78.5 mg/L (normal <5.0 mg/L). Her initial immunological studies showed hypogammaglobulinemia, extremely low B cells, normal T and NK cell counts with T lymphocyte subset frequencies, and proliferative response to mitogens within range values for age (see Table 1). The skin biopsy revealed interstitial infiltrates of some atypical mononuclear cells with karyorrhexis (Figure 1B). She started on IVIG (1 g/kg/21 days) and corticosteroids (1 mg/kg). At 2 months, the severe skin lesions persisted, and she developed superinfected ulcerations on her buttocks, anemia (Hb: 90 g/L), and splenomegaly (Figure 1C). At 3 months, ADA2 enzyme activity was found to be normal at Dr. Michael S. Hershfield's Lab in Duke University Medical Center. The analysis of the IL1RN gene excluded a possible interleukin-1 receptor antagonist (DIRA). A brain MRI was normal (no calcifications), and height and weight were below the third percentile; developmentally, she lost her social smile. Due to persistent exacerbations of skin lesions, prednisone at 2 mg/kg/day and antibiotics (levofloxacin, 10 mg/kg/12 h) were started without improvement (Figure 1D). At the age of 4 months, anti-TNFα therapy with adalimumab at 0.8 mg/dose every week was started. Her WBC counts normalized. Acute phase reactants dropped, and the skin lesions improved, but she developed progressive B and NK cell cytopenia. At 6 months, after the fifth dose of adalimumab, she developed new severe skin lesions, lymphopenia, anemia, thrombocytopenia, visceromegaly, and respiratory failure requiring mechanical ventilation; biologics were discontinued (Figure 1E). She developed skin erythema and adenopathy at the BCG site, and blood culture was positive for S. aureus (Figure 1F). As the patient had a new episode of cytopenia, we added cyclosporine to previous medications with a partial response. Other molecular studies showed no mutation in CECR1/ADA2, MVK, TNFRSF1A, TMEM173/STING1, NLRP3, PSMB8, RAG1, and RAG2 genes. Due to her poor general condition, anti-IL1β treatment with anakinra at 1 mg/kg/dose was started, and cytokine studies revealed high levels of IL-6 and IL-8. The patient remained clinically stable with no major symptoms, but Mycobacterium bovis was isolated from the blood, which prompted tuberculostatic treatment with four drugs. Her lung disease improved, and she was extubated and remained clinically stable. Colleagues agreed with the diagnosis of SAID. Considering her previous and persistent clinical severity since birth, the decision to treat her with an HSCT was made.
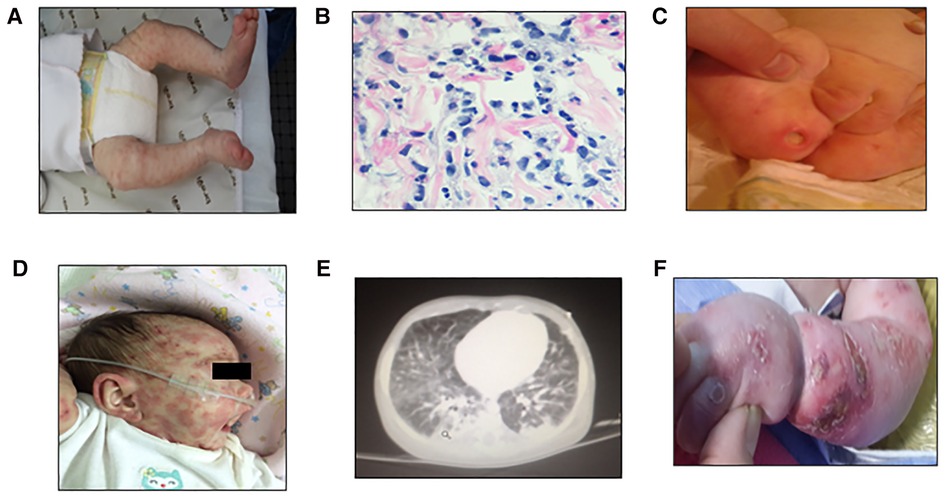
Figure 1. First skin lesions: nonevanescent erythematus papular generalized eruption at the first consult 24 days since birth (A), H&E staining showing interstitial infiltrates consisting of some atypical mononuclear cells with karyorrhexis (B), and persistent severe skin ulcers at 2 months of age (C), skin exacerbations previous to anti-TNFα therapy (D), lung MRI showing inflammatory infiltrate at 6 months after the fifth dose of anti-TNFα therapy (E), and skin erythema exacerbation in arms and legs prior to HSCT at 8 months (F).
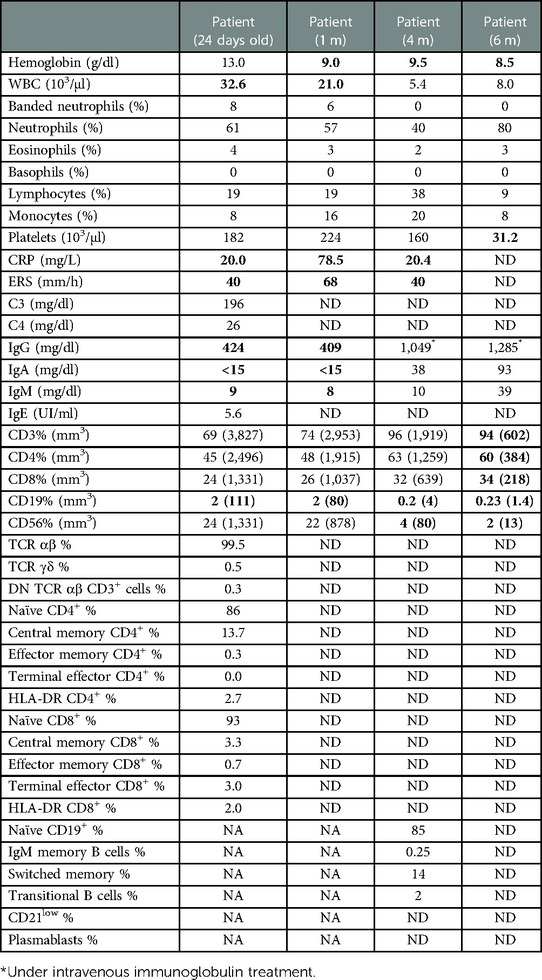
Table 1. Laboratory findings before Hematopoietic stem cell transplantation.
At 10 months of age, the patient received an unrelated-donor hematopoietic stem cell transplantation, fully matched, MHC compatibility degree (10/10), with no molecular diagnosis. The conditioning regimen consisted of busulphan (4.8 mg/kg/dose IV for 4 days), fludarabine (40 mg/m2/dose IV for 4 days), and thymoglobulin (2.25 mg/kg/dose IV for 2 days). GVHD prophylaxis with tacrolimus and methotrexate (with leucovorin rescue) was administered. The source of stem cells was bone marrow (dose: 7 × 106 CD34 cells/kg/body weight). Neutrophil engraftment was achieved on day +17, and platelet engraftment was achieved on day +18. She did not present graft vs. host disease, and chimerism was evaluated by PCR, resulting in 100% donor cells. She was discharged from the clinic on day +47. A month later, Sanger sequencing trio performed at the NIAID/NIH revealed a de novo, heterozygous variant in SAMD9L c.2666delT/p.F889Sfs*2 with normal IFN-response-gene score (20).
At 6 months after the transplant, patient's WBC, CRP, and ERS were within reference values and she had recovered the B cell count (32% and 624/mm3, respectively). Between 12 and 14 months, we evaluated T and B lymphocyte compartments, which revealed beginning T cell reconstitution. We further confirmed a marked decrease in serum IL-6 and IL-8 levels in peripheral blood (Figure 2); 2 years after HSCT, the girl has fully acquired immune reconstitution, showing memory B cells and normal CD4+ and CD8+ proliferations to PHA, OKT3, and SEB (see Table 2). Her tracheotomy could not be removed on two different opportunities (2 and 4 years old) due to mucosal fibrosis and her low weight. It is worth mentioning that during an influenza episode and a second nasopharyngeal probe placement, she presented with a severe mucosal inflammation exacerbation episode requiring mechanical ventilation in the ICU that resolved with corticosteroid treatment. At present, 5 years post-HSCT, she has mild cognitive retardation, she still uses diapers, and her speech is difficult to evaluate as she still has tracheotomy. Moreover, height and weight remain below the third percentile for age. During the COVID pandemic, she had an adenovirus infection that required oxygen and steroids. Her left eye is blind due to retinal detachment. She achieved full donor chimerism without skin lesions, has recovered peripheral blood B cells with immune reconstitution, and is otherwise in good health (see Table 2).
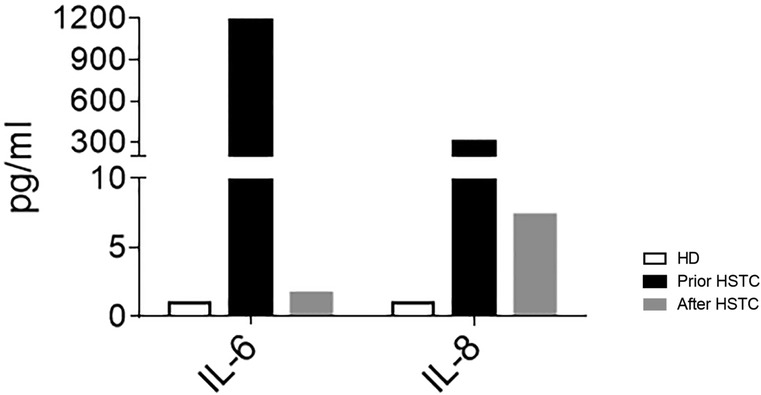
Figure 2. Plasma levels of IL-6 and IL-8 in HD (n = 5) and the patient before HSCT (black bars) and on day +420 after HSCT (gray bars).
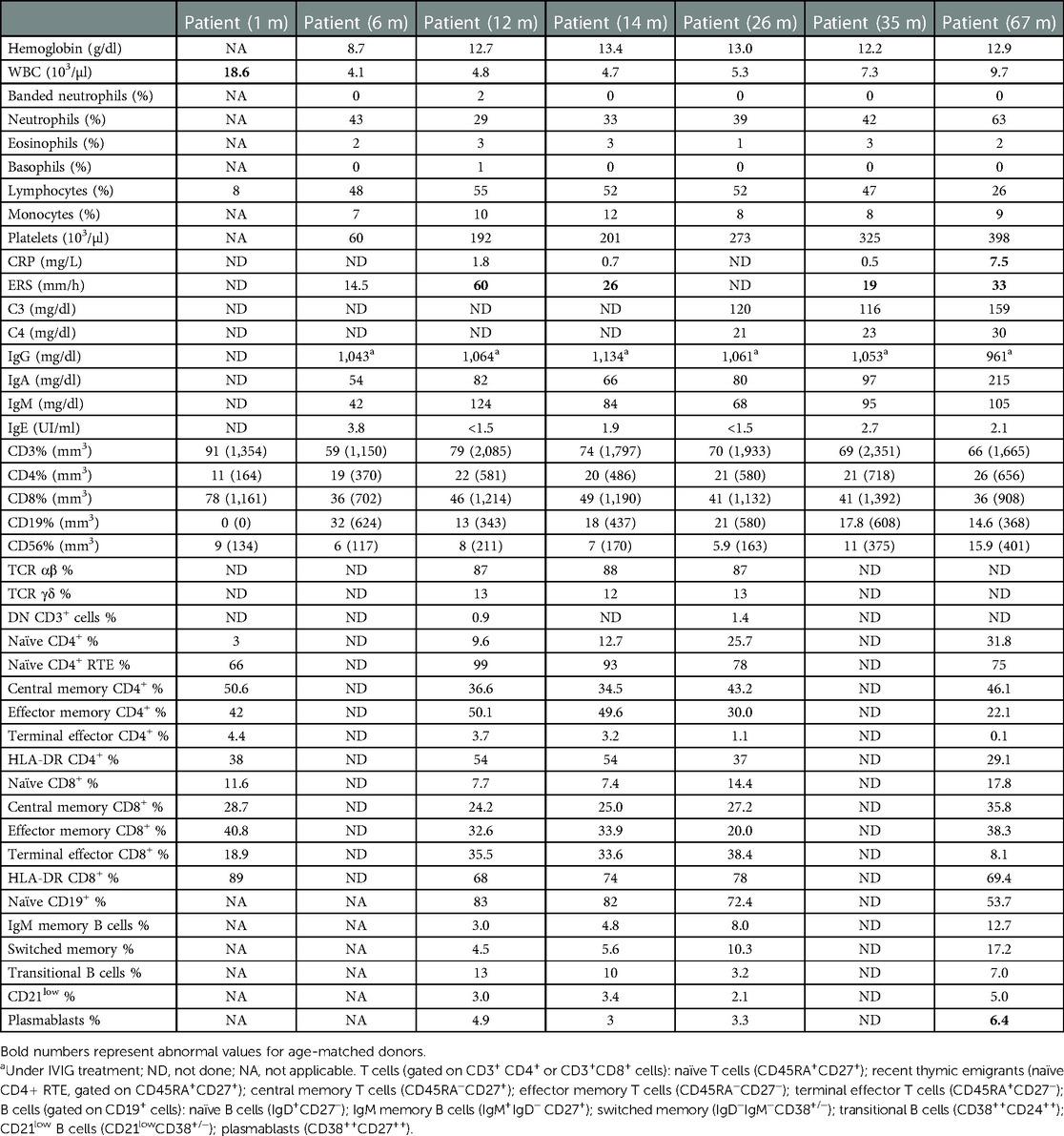
Table 2. Laboratory findings after hematopoietic stem cell transplantation.
Monogenic AD is an evolving group of complex inborn errors of immunity. In many instances, the clinical presentation of cytopenias, immunodeficiencies, and autoinflammation can be confusing, and the inability to control systemic inflammation can result in poor outcomes (21). In this report, we describe a challenging patient who presented with neonatal-onset CANDLE-like features diagnosed with a de novo truncating SAMD9L mutation and who was successfully treated with an HSCT.
In recent years, germline heterozygous missense mutations in SAMD9L have been reported as the monogenic causes for sporadic or autosomal dominant cases of ATXPC and MDS (15, 17, 19, 22), but these patients do not present with early-onset disease and their inflammatory markers are normal. These clinical presentations differed from the early onset severe manifestations described by de Jesus et al., who reported a cohort of SAID patients in which some had monogenic truncating mutations in SAMD9L that presented with IFN and NFκB-mediated dysregulation during flares (20). In total, eight patients with SAMD9L-SAAD have been described so far: three are deceased (one in the context of a BMT), and two have been successfully bone marrow-transplanted including this patient. Severe and progressive B and NK cell cytopenias without alterations of myeloid subpopulations have also been described (22).
Patients with missense SAMD9L mutations who developed MDS have received HSCT, resulting in high donor engraftment (23). We now report that HSCT also leads to the resolution of the systemic and skin manifestations 5 years post-transplantion and provides a positive outcome on an HSCT that was conducted in a life-threatening disease with no good treatment options. However, questions regarding the long-term management of transplanted patients remain unanswered, including the safety of vaccinations, continued benefit to skin inflammation and viral infections, and the prevention of the development of cerebellar atrophy, a feature seen in adults with missense mutations.
In summary, this report raises awareness of SAMD9L-SAAD as a differential diagnosis of a CANDLE-like syndrome with immunodeficiency, and our data suggest HSCT as a treatment option.
The original contributions presented in the study are included in the article/supplementary materials; further inquiries can be directed to the corresponding author.
Ethical review and approval were not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participant’s legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
MC wrote the manuscript, collected the data and performed some laboratory analysis. AS, PL, NE, and GD gently contributed with medical record data. RC and GD kindly helped with some patient sample assays. AJ and RG-M made the molecular diagnosis and provided gene scores. MG, AJ, and RG-M made critical revisions and provided feedback. MG contributed to data analysis and interpretation. LB contributed to critical discussions and supervised patient management. All authors contributed to the article and approved the submitted version.
We especially thank our patient's parents for trusting in physicians, biochemists, and technicians of the immunology service of our institution and our colleagues around the world for their generous collaboration in this complex case.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ADA2, adenosine deaminase; ATXPC, ataxia-pancytopenia syndrome; CANDLE, chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; FC, flow cytometry; GVHD, graft vs. host disease; IFN, interferon; IVIG, intravenous immunoglobulin; MRI, magnetic resonance imaging; MDS, myelodysplastic syndrome; PID, primary immunodeficiencies; SAMD9L, sterile alpha motif domain containing 9 like; URD, unrelated donor; WBC, white blood cell.
1. Martinon F, Aksentijevich I. New players driving inflammation in monogenic autoinflammatory diseases. Nat Rev Rheumatol. (2015) 11:11–20. doi: 10.1038/nrrheum.2014.158
3. Lachmann HJ. Autoinflammatory syndromes as causes of fever of unknown origin. Clin Med (Lond). (2015) 15:295–8. doi: 10.7861/clinmedicine.15-3-295
4. Notarangelo LD, Bacchetta R, Casanova JL, Su HC. Human inborn errors of immunity: an expanding universe. Sci Immunol. (2020) 5(49):eabb1662. doi: 10.1126/sciimmunol.abb1662
5. Beck DB, Aksentijevich I. Biochemistry of autoinflammatory diseases: catalyzing monogenic disease. Front Immunol. (2019) 10:101. doi: 10.3389/fimmu.2019.00101
6. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol. (2017) 18:832–42. doi: 10.1038/ni.3777
7. Hoffman HM, Broderick L. The role of the inflammasome in patients with autoinflammatory diseases. J Allergy Clin Immunol. (2016) 138:3–14. doi: 10.1016/j.jaci.2016.05.001
8. Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. (2010) 140:784–90. doi: 10.1016/j.cell.2010.03.002
9. Almeida de Jesus A, Goldbach-Mansky R. Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin Immunol. (2013) 147:155–74. doi: 10.1016/j.clim.2013.03.016
10. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol.. (2022) 42:1473–507. doi: 10.1007/s10875-022-01289-3.35748970
11. Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity: mutations in PSMB8 cause candle syndrome. Arthritis Rheum. (2012) 64:895–907. doi: 10.1002/art.33368
12. Torrelo A. CANDLE syndrome as a paradigm of proteasome-related autoinflammation. Front Immunol. (2017) 8:927. doi: 10.3389/fimmu.2017.00927
13. de Jesus AA, Brehm A, VanTries R, Pillet P, Parentelli A-S, Montealegre Sanchez GA, et al. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J Allergy Clin Immunol. (2019) 143:1939–43.e8. doi: 10.1016/j.jaci.2018.12.1012
14. Poli MC, Ebstein F, Nicholas SK, de Guzman MM, Forbes LR, Chinn IK, et al. Heterozygous truncating variants in POMP escape nonsense-mediated decay and cause a unique immune dysregulatory syndrome. Am J Hum Genet. (2018) 102:1126–42. doi: 10.1016/j.ajhg.2018.04.010
15. Wong JC, Bryant V, Lamprecht T, Ma J, Walsh M, Schwartz J, et al. Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight. (2018) 3(14):e121086. doi: 10.1172/jci.insight.121086
16. Kennedy AL, Shimamura A. Genetic predisposition to MDS: clinical features and clonal evolution. Blood. (2019) 133:1071–85. doi: 10.1182/blood-2018-10-844662
17. Bluteau O, Sebert M, Leblanc T, Peffault de Latour R, Quentin S, Lainey E, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood. (2018) 131:717–32. doi: 10.1182/blood-2017-09-806489
18. Tesi B, Davidsson J, Voss M, Rahikkala E, Holmes TD, Chiang SCC, et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood. (2017) 129:2266–79. doi: 10.1182/blood-2016-10-743302
19. Allenspach EJ, Soveg F, Finn LS, So L, Gorman JA, Rosen ABI, et al. Germline SAMD9L truncation variants trigger global translational repression. J Exp Med. (2021) 218:e20201195. doi: 10.1084/jem.20201195
20. de Jesus AA, Hou Y, Brooks S, Malle L, Biancotto A, Huang Y, et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest. (2020) 130:1669–82. doi: 10.1172/JCI129301
21. Nigrovic PA, Lee PY, Hoffman HM. Monogenic autoinflammatory disorders: conceptual overview, phenotype, and clinical approach. J Allergy Clin Immunol. (2020) 146:925–37. doi: 10.1016/j.jaci.2020.08.017
22. Gorcenco S, Komulainen-Ebrahim J, Nordborg K, Suo-Palosaari M, Andréasson S, Krüger J, et al. Ataxia-pancytopenia syndrome with SAMD9L mutations. Neurol Genet. (2017) 3:e183. doi: 10.1212/NXG.0000000000000183
Keywords: autoinflammatory syndromes, CANDLE-like syndrome, primary immunodeficiencies, SAMD9L, sterile alpha motif domain containing 9 like, case report
Citation: Caldirola MS, Seminario AG, Luna PC, Curciarello R, Docena GH, Fernandez Escobar N, Drelichman G, Gattorno M, de Jesus AA, Goldbach-Mansky R, Gaillard MI and Bezrodnik L (2023) Case report: De novo SAMD9L truncation causes neonatal-onset autoinflammatory syndrome which was successfully treated with hematopoietic stem cell transplantation. Front. Pediatr. 11:1108207. doi: 10.3389/fped.2023.1108207
Received: 25 November 2022; Accepted: 16 February 2023;
Published: 10 March 2023.
Edited by:
Jean Donadieu, Hôpital Armand Trousseau, FranceReviewed by:
Leonardo Oliveira Mendonça, University of São Paulo, Brazil© 2023 Caldirola, Seminario, Luna, Curciarello, Docena, Fernandez Escobar, Drelichman, Gattorno, de Jesus, Goldbach-Mansky, Gaillard and Bezrodnik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Soledad Caldirola bWFyaWFzb2xlZGFkY2FsZGlyb2xhQGdtYWlsLmNvbQ==
†ORCID María Soledad Caldirola orcid.org/0000-0001-7899-5878
Specialty Section: This article was submitted to Pediatric Immunology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.