 Kena Wang
Kena Wang Biao Zou
Biao Zou Fan Chen
Fan Chen Jianling Zhang
Jianling Zhang Sainan Shu
Sainan Shu- Department of Pediatrics, Tongji Hospital of Tongji Medical Collage, Huazhong University of Science and Technology, Wuhan, China
Background: Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) is a common clinical phenotype of citrin deficiency in infants. Its phenotype is atypical, so genetic testing is quite necessary for the diagnosis.
Case presentation: We report 4 patients with jaundice and low body weight. Furthermore, the biochemical examination of all showed abnormal liver function and metabolic changes. DNA samples of the patients were extracted and subjected to genetic screening. All candidate pathogenic variants were validated by Sanger sequencing, and CNVs were ascertained by qPCR. The genetic screening revealed 6 variants in 4 patients, and all patients carried compound heterozygous variants of SLC25A13. Importantly, 3 variants were newly discovered: a nonsense mutation in exon17 (c.1803C > G), a frameshift mutation in exon 11(c.1141delG) and a deletion of the whole exon11. Thus, four NICCD patients were clearly caused by variants of SLC25A13. Biochemical indicators of all patients gradually returned to normal after dietary adjustment.
Conclusions: Our study clarified the genetic etiology of the four infants, expanded the variant spectrum of SLC25A13, and provided a basis for genetic counseling of the family. Early diagnosis and intervention should be given to patients with NICCD.
Introduction
Citrin deficiency (CD) is an autosomal recessive disease caused by SLC25A13 gene mutation. It includes three age-dependent clinical phenotypes: neonatal intrahepatic cholestasis caused by citrin deficiency(NICCD), adult-onset type II citrullinemia (CTLN2) and failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD) between NICCD and CTLN2 stages (1). NICCD is one of the most common genetic metabolic diseases causing infantile cholestatic hepatopathy, which is associated with persistent jaundice, abnormal coagulation function, hepatosplenomegaly, chubby face, growth retardation, metabolic abnormalities, etc (2)., but these features are not pathognomonic. So genetic analysis is regarded as a reliable method for the definite diagnosis of NICCD. With the development of genetic testing, we recognize that although NICCD is a pan-ethnic disease, it is still prevalent in East Asia. Meanwhile, China is a high-incidence area, the mutation carrier rate of SLC25A13 gene is as high as 1/65. The type of variation of SLC25A13 is also different in different regions. In China, c.852_855del4, c.1638_1660dup, IVS6 + 5G > A, and IVS16ins3kb are the four most common mutations of SLC25A13.In this study, we performed genetic screening on 4 patients with jaundice and every patient carried compound heterozygous mutations with a common variant. Unexpectedly, we also detected 3 novel variants of SLC25A13, including a nonsense mutation, a frameshift mutation, and a whole exon deletion. These findings enriched the variant spectrum of NICCD and guided clinical diagnosis and genetic counseling.
Subjects and methods
Subjects
A total of 4 patients (3 females and 1 male) from non-consanguineous families were enrolled in this study. They were all full-term, and some patients (patient 1, 2 and 4) showed low birth weight and/or low birth length. They all appeared jaundiced with normal color stool about 3 days after birth. They were given blue light irradiation, ursodeoxycholic acid, liver protection drugs and other symptomatic treatment, but there was no significant improvement in their symptoms. Patients 1, 2, and 3 were admitted to our hospital for jaundice, and patient 4 for jaundice and abnormal liver function. All of them were underweight and had no signs of chubby faces. Yellow staining of the skin and sclera was observed. Their livers were palpable 2–3 cm under the rib cage. Laboratory tests are shown in Table 1. Laboratory tests in all patients showed transaminitis, hyperbilirubinemia, elevated GGT, and hypoalbuminemia. Some patients had hypofibrinogenemia accompanied by metabolic abnormalities: hyperlactatemia, hypoglycemia, hypertriglyceridemia, citrullinemia, and elevated pyruvate. Except for patient 1, who did not check for alpha-fetoprotein (AFP), the AFP of the remaining patients was extremely elevated. Ultrasonography in Patients 1 and 2 showed the features of fatty liver. Patient 4 had no sign of fatty liver on the ultrasound, however the liver biopsy showed hepatic steatosis.
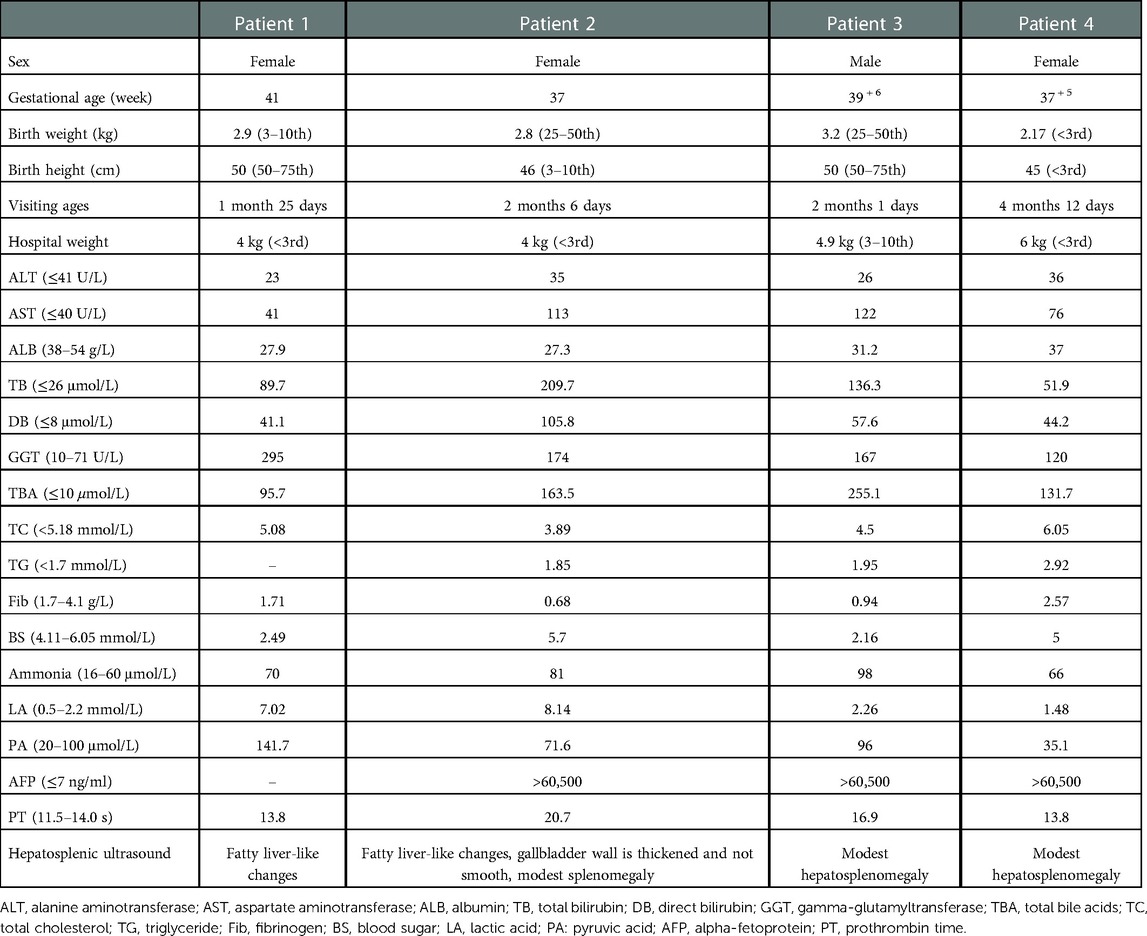
Table 1. Clinical features of the 4 patients.
Genetic testing
Genomic DNA was extracted from the peripheral blood of all probands. Genetic screening was performed for each proband: proband 1(captured by exome kit from Agilent, sequenced on Illumina platform); proband 2 (captured by metabolic liver disease panel from MyGenostics, sequenced on DNBSEQ-T7); proband 3 (captured by a panel from amcarelab, sequenced on Illumina platform); proband 4 (captured by a panel from our in-house liver disease panel, sequenced on Illumina platform). All sequencing data were mapped with human reference genome(hg19), and variants were classified according to ACMG guidelines. All candidate pathogenic variants were validated by Sanger sequencing, and CNVs were validated by quantitative real-time polymerase chain reaction (qPCR).
Results
SLC25A13 mutations
Compound heterozygous variants from SLAC25A13 were detected in all four probands; all variants were classified as likely pathogenic or pathogenic according to ACMG guidelines (3) (Table 2): Patient 1 [NM001160210: c.1803C > G, p.Y601X (Figure 1A) and IVS16ins3kb, p.A584fs]; Patient 2 [NM001160210: c.1803C > G, p.Y601X (Figure 1B) and c.1663_1664insGAGATTACAGGTGGCTGCCCGGG, p.A555fs] (Figure 1C) ]; Patient 3 [NM_014251: c.1141delG, p.V381Cfs*27 (Figure 1D) and c.1750_1751 ins3Kb (Figure 1E), patient 4 [NM_014251: c.852_855del, p.M285Pfs*2 (Figure 1F) and exon11 del (Figure 1G)].

Figure 1. (A) Patient 1 carried the variant of c.1803C > G, the results of sanger sequencing indicated that it was inherited from her mother. (B)The novel variant c.1803C > G was detected in patient 2, which is derived from her mother. (C) c.1663_1664insGAGATTACAGGTGGCTGCCCGGG in patient 2 was confirmed from her father by Sanger sequencing. (D)The deletion variant c.1141delG in patient 3 was from his father. (E) Patient 3 and his mother is heterozygous nonsense mutation of c.1750_1751 ins3Kb, his father is normal. (F) Patient 4 was a carrier of c.852_855del, which is derived from her mother. (G) The another variant exon11 del in patient 4 were validated by qPCR, the image from left to right is her mother, her father, herself and the sample, so that the mutation originated from her father.
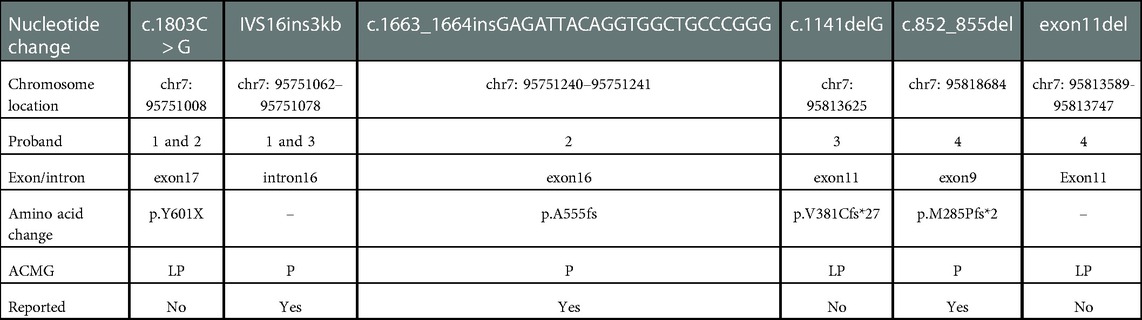
Table 2. Variants detected summary.
Clinical outcome
All four patients adjusted their diets: breastfeeding was stopped and it was replaced by a lactose-free and MCT-rich formula. The clinical manifestations improved significantly and none of them suffered long-term complications (We called the family of the patient 2, who informed us that the patient's liver function and AFP levels had returned to normal, but the report was lost. The follow-up results of patients 1, 3, and 4 are showed in Table 3). Most of the patients revealed specific preference for high-protein food but not with an obvious aversion to foods high in carbohydrate.
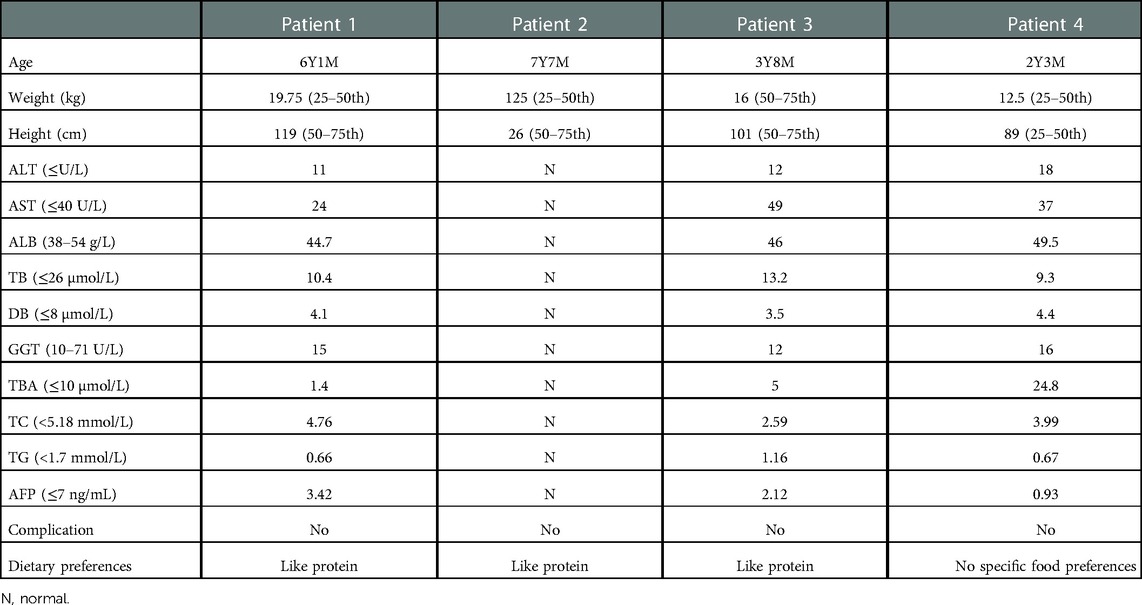
Table 3. Follow-up of the patients.
Discussion
SLC25A13 is the pathogenic gene of NICCD, which encodes the citrin protein. Citrin plays significant roles in the urea cycle, malic acid-aspartate shuttle, and gluconeogenesis (3). As a result, variants of SLC25A13 can lead to metabolic abnormalities such as citrullinemia, hyperammonemia, hypoglycemia, and hyperlipidemia. Our study results were similar to those of the previous studies (4), some patients with NICCD showed low birth weight and low birth height according to sex and gestational age. At the same time, the patients with NICCD can also be accompanied by elevated liver enzymes, hyperbilirubinemia, significantly elevated alpha-fetoprotein, hypoalbuminemia, prolonged prothrombin time and fatty liver. Predictably, four patients in our study also had these characteristics. The sign of hepatic steatosis is common in patients with NICCD. While both the ultrasonography and liver biopsy of patient 3 did not indicate evidence of hepatic steatosis. Therefore, consistent with Miyamoto's report (5), NICCD cannot be ruled out even if hepatic steatosis is not seen in patients with cholestasis. None of the above changes are specific. A variety of neonatal intrahepatic cholestasis (such as cytomegalovirus hepatitis, Alagille syndrome, etc.) can manifest as jaundice, elevated liver enzyme, hepatosplenomegaly, and growth retardation. Meanwhile, other inherited metabolic diseases(such as galactosemia (6), hepatocerebral mitochondrial DNA depletion syndrome (7–9), arginylsuccinuria (10), etc.) can also have symptoms similar to NICCD. It can be seen that it is a big challenge to distinguish NICCD from other diseases. Therefore, we should actively conduct genetic testing for patients with unexplained infantile cholestasis to avoid misdiagnosis and missed diagnosis.
So far, more than 300 variants of SLC25A13 have been reported worldwide, including 292 single nucleotide variations, 28 deletion mutations, 8 duplation mutations, 4 microsatellite mutations, 3 insertion mutations, and 1 insertional deletion variant. In our study, four patients all harbored compound heterozygous variants of SLC25A13, and a total of 6 variant types were detected, among which three have not been reported: c.1803C > G, c.1141delG and exon 11 del. The common mutation c.1803C > G identified in patient1 and 2, which is located on exon 17 and is predicted to cause the early appearance of amino acid termination code (tyrosine > termination) in protein synthesis, and according to ACMG guidelines, pathogenicity analysis is PVS1 + PM2 + PP4, it is classified as likely pathogenic, so far as we know, which has not been reported previously. While in our study, it was found in two patients and two family members. After a detailed medical history we found no consanguinity between the two families; however, they all came from Hubei Province, China, which spans the Yangtze River and belongs to the central city in China. The previous large-scale studies on NICCD were generally in the south and north of China, with few central cities. As we all know, there is a significant difference in the mutation carrier rate of SLC25A13 gene between North and South populations along the boundary of 30° North latitude in China. The results of our study suggested that the carrier rates of c.1803C > G may be frequent in the central cities of China, and there are differences in the distribution of the mutation spectrum in different geographical areas. As for the relationship between genotype and phenotype, although some studies declared that there were likely to be phenotype–genotype correlations in the specific genotypes (11), in our study, no obvious genotype-phenotype relationship seems to be found. For example, heterozygous genotypes including c.1803C > G were found in patient 1 and 2, but their birth weight, AST, Fib, PA and prothrombin time have a marked difference. And patient 1 and 3 carried the heterozygous variants including IVS16ins3kb, but they didn't present the same clinical manifestations. So we do not hold that there is a significant genotype-phenotype relationship between patients with NICCD. The number of cases in our study was small, so further studies with more samples may be required. c.1141delG is a novel code-shifted mutation that has not been reported in relevant clinical cases and our reference population gene database. Moreover, it is predicted that it may lead to the early occurrence of amino acid termination ciphers in protein synthesis. According to the ACMG guidelines, the pathogenicity analysis is PVS1 + PM2 + PP4, so it is likely pathogenic. Exon deletion is a rare variant type among SLC25A13 variants, which are mainly caused by large fragment deletion, including exon deletion, such as c.329–154_468 + 2352del2646bp (deletion of exon 5) (12), c.70–862_c.212 + 3527 del4532bp (deletion of exon 3) (13), Ex16 + 74_IVS17-32del516 (deletion of exon 17) (14), c.329–1687 -c.468 + 3865del5692bp (deletion of exon 5) (15), c.1019_1177 + 893del (deletion of exon 11) (16), c.1312–2860_1452 + 988del (deletion of exon 14) (17), c.1312–4144_1452 + 3373del (deletion of exon 14) (17) and c.3251 _c.15 + 18443del21709bp (deletion of exon 1) (18). Partial abnormal splicing of partially intron regions leads to exome deletions in mRNA, such as IVS4ins6 kb (skipping of exon 5) (19), IVS11+1 G > A (skipping of exon 11) (20), IVS15 + 1G > T (skipping of exon 15) (21) and IVS13+1G > A (skipping of exon 13) (22). In this study, after high-throughput sequencing analysis, patient 4 was found to carry a common variant c.852_855del and be accompanied by copy number variation. In order to clarify the definite genetic etiology of the patient, qPCR verification was performed. Eventually, another variant (exon 11 del) was found in the patient. So, this patient was confirmed as NICCD caused by compound heterozygous variants of SLC25A13. The other three variants identified in this study: c.1663_1664insGAGATTACAGGTGGCTGCCCGGG (12, 23), IVS16ins3kb (15, 24–27), and c.852_855del (23, 24, 28–31) have been reported in multiple literatures and are three of the most common variants of SLC25A13 in China.
Conventional genetic screening, such as whole exon sequencing, can only detect point mutations or small deletions. However, it was found in a study by Tokuhara (32) that about 15% of the complex heterozygotes or homozygotes of the SLC25A13 gene could not be identified by the routine methods. Fortunately, the emergence of multiplex ligation-dependent probe amplification (MLPA) (20), Southern blots restriction fragment analysis (33), custom fluorescence in situ hybridization (FISH) (34), and qPCR (21) can help us better detect this part of the variants. Therefore, additional testing is recommended for the patients who are clinically considered for NICCD but have only one heterozygous mutation detected by conventional genetic screening.
After the dietary adjustment, the liver function such as ALT, GGT, TB, DB, TBA, ALB, AFP returned to normal at the last review (Table 3). The patients were all underweight at the time of their first visit to our hospital, although the follow-up time of our study was short (the maximum is approximately seven years), the height and weight of them gradually returned to normal. Recently there were cases of liver cancer (35) in NICCD patients have been reported, it suggested that NICCD is not always harmless. Fortunately, none of the long-term complications including liver cirrhosis, hepatoadenoma or hepatocellular carcinoma, pancreatitis and so on occurred in the patients in this study. A recent study by Kido (11) found that the height and the weight of patients with NICCD were high than those with CTLN2, in the meantime long-term complications were seen in patients with CTLN2 but not in older NICCD patients. It is speculated that medical management including dietary interventions can help patients. Therefore, we emphasize here the necessity for dietary intervention in patients with NICCD to alleviate clinical manifestations and prevent long-term complications.
In summary, our study reviewed the clinical manifestations, genetic test results, and prognosis of NICCD patients from 4 unrelated families. There are 6 variants of SLC25A13 were identified, and three novel variants were found: c.1803C > G, c.1141delG, and exon 11 del. This result clarified their genetic etiology and provided a strong basis for the clinical diagnosis of patients. At the same time, the detection of the novel variants enriched the variant spectrum of the SLC25A13, which further laid the foundation for the correlation study of the genotype and phenotype of NICCD.
Data availability statement
The Genetic screening data presented in the study are deposited in the Genome Sequence Archive or Human (https://ngdc.cncb.ac.cn/gsa-human/), with accession number HRA003718.
Ethics statement
The studies involving human participants were reviewed and approved by Human Ethics Committee of Huazhong University of Science and Technology. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
WKN, SSN: conceptualized and designed the study, wrote, and reviewed the manuscript. ZB, CF, ZJL, HZH: did the follow-up study, collected the data, and managed the patient. All authors contributed to the article and approved the submitted version.
Acknowledgments
We really appreciate for the help of all the physicians in the course of the medical treatment.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Song Y-Z, Deng M, Chen F-P, Wen F, Guo L, Cao SL, et al. Genotypic and phenotypic features of citrin deficiency: five-year experience in a Chinese pediatric center. Int J Mol Med. (2011) 28(1):33–40. doi: 10.3892/ijmm.2011.653
2. Liu W-W, Zhong X-MX. Advances in citrin deficiency. Chin J Med. (2022) 57(09):957–61. doi: 10.3760/cma.j.cn511374-20201212-00872
3. Lin W-X, Yaqub MR, Zhang Z-H, Mao M, Zeng H-S, Chen F-P, et al. Molecular epidemiologic study of citrin deficiency by screening for four reported pathogenic Slc25a13 variants in the shaanxi and guangdong provinces, China. Transl Pediatr. (2021) 10(6):1658–67. doi: 10.21037/tp-21-58
4. Numakura C, Tamiya G, Ueki M, Okada T, Maisawa S-I, Kojima-Ishii K, et al. Growth impairment in individuals with citrin deficiency. J Inherit Metab Dis. (2019) 42(3):501–8. doi: 10.1002/jimd.12051
5. Ryosuke M, Jun S, Koki O, Kenitiro K, Hironori K, Yoshiteru A, et al. Neonatal intrahepatic cholestasis caused by citrin deficiency with No hepatic steatosis: a case report. BMC Pediatr. (2021) 21(1):237. doi: 10.1186/s12887-021-02717-w
6. Gottesman LE, Del Vecchio MT, Aronoff SC. Etiologies of conjugated hyperbilirubinemia in infancy: a systematic review of 1692 subjects. BMC Pediatr. (2015) 15:192. doi: 10.1186/s12887-015-0506-5
7. Zhao M-X, Wang J-S, Gong J-Y. Analysis of 6 cases with hepatocerebral mitochondrial DNA depletion syndrome and literature review. Chin J Pediatr. (2022) 60(05):457–61. doi: 10.3760/cma.j.cn112140-20210827-00711
8. Bao L-S, Liu F, Wu Y-Y, Wang J-N. The neonatal mitochondrial DNA depletion syndrome: a case report. Chin J Neonatol. (2022) 37(01):77–8. doi: 10.3760/cma.j.issn.2096-2932.2022.01.018
9. Grünert SC, Schumann A, Freisinger P, Rosenbaum-Fabian S, Schmidts M, Mueller AJ, et al. Citrin deficiency mimicking mitochondrial depletion syndrome. BMC Pediatr. (2020) 20(1):518. doi: 10.1186/s12887-020-02409-x
10. Lin Y-M, Yu K, Li L-F, Zheng Z-Z, Lin W-H, Fu Q-L. Mutation analysis of ASS1, ASL, SLC25A13 genes in six Chinese patients with citrullinemia. Chin J Med Genet. (2017) 34(5):676–9. doi: 10.3760/cma.j.issn1003-9406.2017.05.012
11. Kido J, Häberle J, Sugawara K, Tanaka T, Nagao M, Sawada T, et al. Clinical manifestation and long-term outcome of citrin deficiency: report from a nationwide study in Japan. J Inherit Metab Dis. (2022) 45(3):431–44. doi: 10.1002/jimd.12483
12. Lin W-X, Zeng H-S, Zhang Z-H, Mao M, Zheng Q-Q, Zhao S-T, et al. Molecular diagnosis of pediatric patients with citrin deficiency in China: slc25a13 mutation Spectrum and the geographic distribution. Sci Rep. (2016) 6:29732. doi: 10.1038/srep29732
13. Wong L-JC, Dimmock D, Geraghty MT, Quan R, Lichter-Konecki U, Wang J, et al. Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions. Clin Chem. (2008) 54(7):1141–8. doi: 10.1373/clinchem.2008.103721
14. Takaya J, Kobayashi K, Ohashi A, Ushikai M, Tabata A, Fujimoto S, et al. Variant clinical courses of 2 patients with neonatal intrahepatic cholestasis who have a novel mutation of Slc25a13. Metab Clin Exp. (2005) 54(12):1615–9. doi: 10.1016/j.metabol.2005.06.009
15. Zheng Q-Q, Zhang Z-H, Zeng H-S, Lin W-X, Yang H-W, Yin Z-N, et al. Identification of a large Slc25a13 deletion via sophisticated molecular analyses using peripheral blood lymphocytes in an infant with neonatal intrahepatic cholestasis caused by citrin deficiency (niccd): a clinical and molecular study. Biomed Res Int. (2016) 2016:4124263. doi: 10.1155/2016/4124263
16. Zeng H-S, Lin W-X, Zhao S-T, Zhang Z-H, Yang H-W, Chen F-P, et al. Slc25a13 cdna cloning analysis using peripheral blood lymphocytes facilitates the identification of a large deletion mutation: molecular diagnosis of an infant with neonatal intrahepatic cholestasis caused by citrin deficiency. Mol Med Rep. (2016) 14(6):5189–94. doi: 10.3892/mmr.2016.5873
17. Lau NKC, Lee HHC, Chen SPL, Ng CWY, Mak CM, Chong YK, et al. In-house multiplex ligation-dependent probe amplification assay for citrin deficiency: analytical validation and novel exonic deletions in Slc25a13. Pathology. (2021) 53(7):867–74. doi: 10.1016/j.pathol.2021.02.010
18. Zhang Z-H, Lin W-X, Zheng Q-Q, Guo L, Song Y-Z. Molecular diagnosis of citrin deficiency in an infant with intrahepatic cholestasis: identification of a 21.7 kb gross deletion that completely silences the transcriptional and translational expression of the affected allele. Oncotarget. (2017) 8(50):87182–93. doi: 10.18632/oncotarget.19901
19. Song Y-Z, Zhang Z-H, Lin W-X, Zhao X-J, Deng M, Ma Y-L, et al. Slc25a13 gene analysis in citrin deficiency: sixteen novel mutations in east Asian patients, and the mutation distribution in a large pediatric cohort in China. PLoS One. (2013) 8(9):e74544. doi: 10.1371/journal.pone.0074544
20. Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by Multiplex ligation-dependent probe amplification. Nucleic Acids Res. (2002) 30(12):e57. doi: 10.1093/nar/gnf056
21. Sieber OM, Lamlum H, Crabtree MD, Rowan AJ, Barclay E, Lipton L, et al. Whole-Gene apc deletions cause classical familial adenomatous polyposis, but not attenuated polyposis or “multiple” colorectal adenomas. Proc Natl Acad Sci U S A. (2002) 99(5):2954–8. doi: 10.1073/pnas.042699199
22. Yasuda T, Yamaguchi N, Kobayashi K, Nishi I, Horinouchi H, Jalil MA, et al. Identification of two novel mutations in the Slc25a13 gene and detection of seven mutations in 102 patients with adult-onset type ii citrullinemia. Hum Genet. (2000) 107(6):537–45. doi: 10.1007/s004390000430
23. Kobayashi K, Bang Lu Y, Xian Li M, Nishi I, Hsiao K-J, Choeh K, et al. Screening of nine Slc25a13 mutations: their frequency in patients with citrin deficiency and high carrier rates in Asian populations. Mol Genet Metab. (2003) 80(3):356–9. doi: 10.1016/S1096-7192(03)00140-9
24. Tabata A, Sheng J-S, Ushikai M, Song Y-Z, Gao H-Z, Lu Y-B, et al. Identification of 13 novel mutations including a retrotransposal insertion in Slc25a13 gene and frequency of 30 mutations found in patients with citrin deficiency. J Hum Genet. (2008) 53(6):534–45. doi: 10.1007/s10038-008-0282-2
25. Radha Rama Devi A, Naushad SM. Slc25a13 C.1610_1612delinsat mutation in an Indian patient and literature review of 79 cases of citrin deficiency for genotype-phenotype associations. Gene. (2018) 668:190–5. doi: 10.1016/j.gene.2018.05.076
26. Chen R, Wang X-H, Fu H-Y, Zhang S-R, Abudouxikuer K, Saheki T, et al. Different regional distribution of Slc25a13 mutations in Chinese patients with neonatal intrahepatic cholestasis. World J Gastroenterol. (2013) 19(28):4545–51. doi: 10.3748/wjg.v19.i28.4545
27. Zhang M-H, Gong J-Y, Wang J-S. Citrin deficiency presenting as acute liver failure in an eight-month-old infant. World J Gastroenterol. (2015) 21(23):7331–4. doi: 10.3748/wjg.v21.i23.7331
28. Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, Lee JR, et al. The gene mutated in adult-onset type ii citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet. (1999) 22(2):159–63. doi: 10.1038/9667
29. Song Y-Z, Deng M, Chen F-P, Wen F, Guo L, Cao S-L, et al. Genotypic and phenotypic features of citrin deficiency: five-year experience in a Chinese pediatric center. Int J Mol Med. (2011) 28(1):33–40. doi: 10.3892/ijmm.2011.653
30. Zhang Z-H, Lin W-X, Deng M, Zhao X-J, Song Y-Z. Molecular analysis of Slc25a13 gene in human peripheral blood lymphocytes: marked transcript diversity, and the feasibility of cdna cloning as a diagnostic tool for citrin deficiency. Gene. (2012) 511(2):227–34. doi: 10.1016/j.gene.2012.09.049
31. Fu H-Y, Zhang S-R, Wang X-H, Saheki T, Kobayashi K, Wang J-S. The mutation spectrum of the Slc25a13 gene in Chinese infants with intrahepatic cholestasis and aminoacidemia. J Gastroenterol. (2011) 46(4):510–8. doi: 10.1007/s00535-010-0329-y
32. Tokuhara D, Iijima M, Tamamori A, Ohura T, Takaya J, Maisawa S, et al. Novel diagnostic approach to citrin deficiency: analysis of citrin protein in lymphocytes. Mol Genet Metab. (2007) 90(1):30–6. doi: 10.1016/j.ymgme.2006.09.009
33. Monaco AP, Bertelson CJ, Middlesworth W, Colletti CA, Aldridge J, Fischbeck KH, et al. Detection of deletions spanning the duchenne muscular dystrophy locus using a tightly linked DNA segment. Nature. (1985) 316(6031):842–5. doi: 10.1038/316842a0
34. Bendavid C, Kleta R, Long R, Ouspenskaia M, Muenke M, Haddad BR, et al. Fish diagnosis of the common 57-Kb deletion in ctns causing cystinosis. Hum Genet. (2004) 115(6):510–4. doi: 10.1007/s00439-004-1170-2
Keywords: citrin deficiency, NICCD, SLC25A13, novel variant, prognosis
Citation: Wang K, Zou B, Chen F, Zhang J, Huang Z and Shu S (2023) Case report: Three novel variants on SLC25A13 in four infants with neonatal intrahepatic cholestasis caused by citrin deficiency. Front. Pediatr. 11:1103877. doi: 10.3389/fped.2023.1103877
Received: 21 November 2022; Accepted: 9 January 2023;
Published: 29 March 2023.
Edited by:
Kenneth K.Y. Wong, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Jun Kido, Kumamoto University Hospital, JapanFuad Al Mutairi, Ministry of National Guard Health Affairs (MNGHA), Saudi Arabia
© 2023 Wang, Zou, Chen, Zhang, Huang and Shu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sainan Shu c2h1c2FpbmFuQDE2My5jb20=
Specialty Section: This article was submitted to Pediatric Gastroenterology, Hepatology and Nutrition, a section of the journal Frontiers in Pediatrics