
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 09 June 2023
Sec. Pediatric Hematology and Hematological Malignancies
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1087002
This article is part of the Research TopicCase Reports in Pediatric Hematology and Hematological Malignancies 2022View all 14 articles
 Lucia Leonardi1Alessia Testa1,†Mariavittoria Feleppa1,†
Lucia Leonardi1Alessia Testa1,†Mariavittoria Feleppa1,† Roberto Paparella1*†
Roberto Paparella1*† Francesca Conti2Antonio Marzollo3
Francesca Conti2Antonio Marzollo3 Alberto Spalice1
Alberto Spalice1 Fiorina Giona4
Fiorina Giona4 Maria Gnazzo5Gian Marco Andreoli1Francesco Costantino1
Maria Gnazzo5Gian Marco Andreoli1Francesco Costantino1 Luigi Tarani1
Luigi Tarani1
Kabuki syndrome (KS) is a rare multisystemic disease due to mutations in the KMT2D or KDM6A genes, which act as epigenetic modulators of different processes, including immune response. The syndrome is characterized by anomalies in multiple organ systems, and it is associated with autoimmune and inflammatory disorders, and an underlying immunological phenotype characterized by immunodeficiency and immune dysregulation. Up to 17% of KS patients present with immune thrombocytopenia characterized by a severe, chronic or relapsing course, and often associated to other hematological autoimmune diseases including autoimmune hemolytic anemia, eventually resulting in Evans syndrome (ES). A 23-year-old woman, clinically diagnosed with KS and presenting from the age of 3 years with ES was referred to the Rare Diseases Centre of our Pediatric Department for corticosteroid-induced hyperglycemia. Several ES relapses and recurrent respiratory infections in the previous years were reported. Severe hypogammaglobulinemia, splenomegaly and signs of chronic lung inflammation were diagnosed only at the time of our observation. Supportive treatment with amoxicillin-clavulanate prophylaxis and recombinant human hyaluronidase-facilitated subcutaneous immunoglobulin replacement were immediately started. In KS patients, the failure of B-cell development and the lack of autoreactive immune cells suppression can lead to immunodeficiency and autoimmunity that may be undiagnosed for a long time. Our patient's case is paradigmatic since she presented with preventable morbidity and severe lung disease years after disease onset. This case emphasizes the importance of suspecting immune dysregulation in KS. Pathogenesis and immunological complications of KS are discussed. Moreover, the need to perform immunologic evaluations is highlighted both at the time of KS diagnosis and during disease follow-up, in order to allow proper treatment while intercepting avoidable morbidity in these patients.
Kabuki syndrome (KS) is a rare, multiple congenital anomaly/intellectual disability syndrome with an estimated prevalence of 1:32,000 in Japan (1), where it was first described in 1981. The prevalence outside Japan is presumably similar to that seen in the Japanese population, but is not known (2). In 2010, Ng et al. identified heterozygous mutations in KMT2D as the main genetic cause of KS (3). More recently, mutations in KDM6A have been reported in almost 5% of KS cases. However, genetic basis of the syndrome is still unknown in up to 20%–25% of the patients (4). Both KMT2D, inherited as autosomal dominant, and KDM6A, inherited as X-linked, are involved in embryogenesis, development and immune response, functioning as epigenetic modulators explaining the characteristic phenotype of KS patients (5, 6).
Immune abnormalities may occur later in childhood, being characterized by immune dysregulation with increased risk of autoimmune diseases and immunodeficiency and, therefore, of infections (7). Among autoimmune diseases, immune thrombocytopenic purpura (ITP), hemolytic anemia [often combined in Evans syndrome (ES)], thyroiditis and vitiligo are reported (8–10). Autoimmune cytopenia associated with KS usually present with a chronic and relapsing course and a poor response to conventional therapy (11–14). Non-malignant lymphoproliferation has also been described as a feature of KS, similarly to other inborn errors of immunity (15, 16).
We report the case of a 23-year-old woman with KS and a history of several relapses of ES, referred to the Rare Diseases Centre of our Pediatric Department for corticosteroid-induced hyperglycemia. Severe hypogammaglobulinemia was also diagnosed at the time of our first observation.
A 23-year-old woman, clinically diagnosed with KS at the age of 5 years, was referred to our Rare Diseases Centre for hyperglycemia following high-dose prednisone treatment due to an ES relapse. The patient was born at term from cesarean section due to fetal distress. She was the third daughter of healthy Italian non-consanguineous parents, with no family history of immunological, autoimmune or rare diseases. Neonatal examination highlighted bifid uvula. At the age of three, she was diagnosed with autoimmune hemolytic anemia (AIHA) with a positive direct antiglobulin test, needing long-term corticosteroid therapy. During infancy she also presented with several episodes of sinusitis, otitis media and three episodes of pneumonia that required hospitalization. Moreover, she was diagnosed with ostium secundum atrial septal defect, surgically corrected at the age of 5 years. During hospitalization, genetic counseling highlighted the presence of major criteria for clinical diagnosis of KS including moderate intellectual disability, postnatal short stature, skeletal abnormalities, dermatoglyphic anomalies and facial dysmorphism (Figure 1).
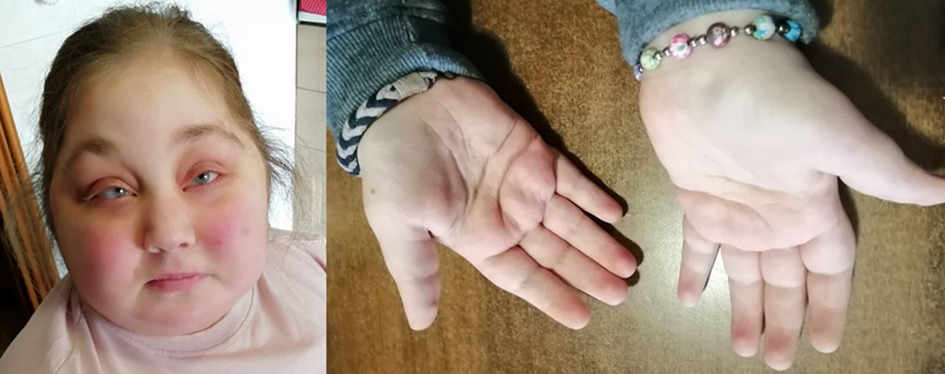
Figure 1. Patient's facial features (arched and broad eyebrows, long palpebral fissures, eversion of the lower eyelid, depressed nasal tip and short columella) and persistence of fetal fingertip pads, typical of Kabuki syndrome.
Shortly afterward, the patient presented with ITP which, associated with AIHA, led to ES diagnosis. Over the years, the patient underwent several courses of corticosteroids due to ES relapses; however, since the age of 12, she had been poorly followed in any aspect of her syndrome except for neuromuscular rehabilitation.
At the time of our observation, at the age of 23 years, the patient presented with a peculiar KS facial appearance, short stature, obesity (BMI 43 kg/m2), mild developmental delay, multiple skeletal defects (dorsal kyphosis, lumbar hyperlordosis, bilateral pronated feet) and dermatoglyphic anomalies. Moreover, she presented with clinical side effects of systemic glucocorticoids therapy including Striae rubrae, central obesity, and arterial hypertension. Severe hyperglycemia (glucose level >400 mg/dl) with elevated HbA1C level (>9%) led to glucocorticoid-induced diabetes mellitus diagnosis. GAD65, ICA and IA-2A autoantibodies were not detected. Serum glucose concentration had always been lower than 160 mg/dl before prednisone treatment.
Short-term glycemic control with a target range of 100–180 mg/dl throughout the day was chosen. Routine blood glucose monitoring revealed a typical pattern of steroid-induced diabetes characterized by near-normal fasting glucose levels followed by hyperglycemia during the day. Standard dietary counseling and short-acting insulin were therefore initiated.
Further investigations indicated mild bilateral sensorineural hearing loss, renal anomalies and alternating strabismus. Echocardiography documented bilateral ventricular wall thickening, first-degree diastolic dysfunction of the left ventricle, and right ventricular systolic function below normal range. Abdominal ultrasound showed hepatic steatosis, mild splenomegaly (spleen longitudinal diameter = 12.6 cm), and bicornuate uterus. High-resolution computed tomography (HRCT) of the lungs revealed diffuse ground-glass opacities, interlobular septal and bronchial wall thickening, subsolid nodules with blurred edges, peribronchial calcifications and low-attenuation areas due to air trapping (Figure 2A). These findings were interpreted as interstitial lung disease and chronic inflammation, likely due to both recurrent respiratory infections and immune dysregulation, while monoclonal lymphoproliferative disorders were excluded. Given the diagnosis of ES, the chronic lung disease and the splenomegaly, an immunological evaluation was performed highlighting severe hypogammaglobulinemia: IgG 200 mg/dl (reference value 700–1,600); IgA 2 mg/dl (reference value 68–400) and IgM 149 mg/dl (reference value 40–259). T-cell phenotype showed: CD3+ = 574 cells/µl (CD4+ = 298 cells/µl, 29%; CD8+ = 219 cells/µl, 21%). B- cell phenotype was characterized by a normal circulating B-cell number (CD19+ B-cells = 364 cells/µl), with a reduction in IgD+CD27+ memory (3%) and IgD−CD27+ switched memory (2.5%) B-cells percentage, as well as an expanded population of CD21low B-cells (12%). Lymphocyte level ranged between 700 and 1100 cells/µl in sequential analyses. Antibiotic prophylaxis with amoxicillin-clavulanate and replacement treatment with recombinant human hyaluronidase-facilitated subcutaneous immunoglobulin (fSCIg) were initiated, therefore limiting further immunological studies. With regard to the lung imaging, although solid data regarding diagnostic and prognostic markers of granulomatous -lymphocytic interstitial lung disease (GLILD) are currently lacking, and considerably variable histopathological findings in GLILD patients have been described, the diagnostic hypothesis of GLILD was considered. Lung biopsy and bronchoscopy, however, were not performed because of the high risk of the procedure due to disease severity and comorbidities, while lung diffusion testing was limited by the patient's poor compliance.
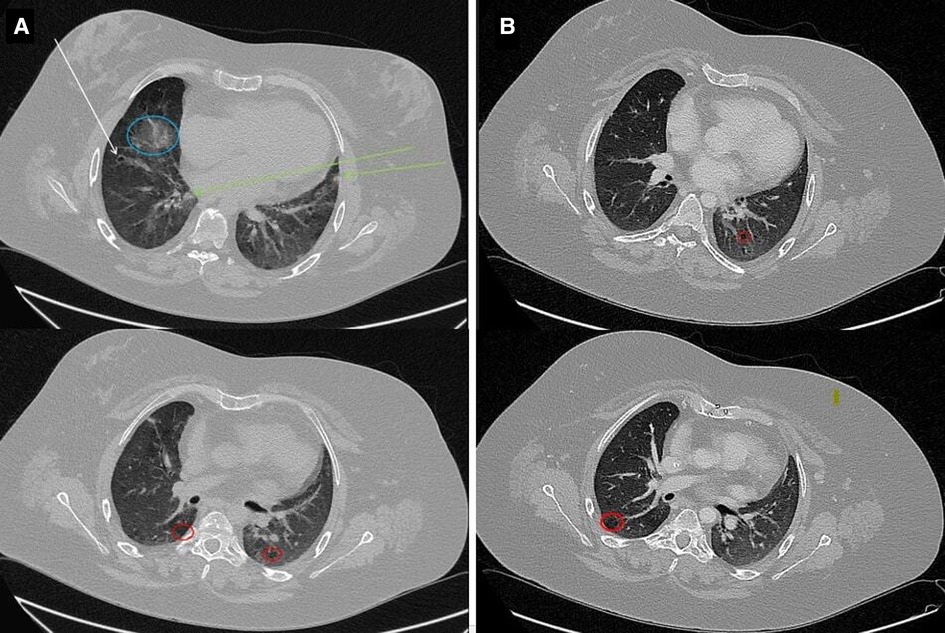
Figure 2. Chest HRCT. (A) Images show interstitial lung disease and chronic inflammation. Note the numerous findings, including ground-glass opacity (blue circle), bronchiectasis (white arrow), nodules with blurred edges (green arrows), and regions of air trapping (red circles). (B) Radiological disease improvement with persistence of air-trapping.
Genetic testing was conducted in accordance with the Helsinki Declaration, after obtaining informed consent from the patient's parents. Sequencing analysis for Kabuki syndrome panel detected a nonsense variant in the KMT2D (NM_003482.3) gene: c.8974G>T, p.Glu2992Ter. This variant, confirmed by Sanger analysis, was not reported in the database of human variations GnomAD (https://gnomad.broadinstitute.org/) and was classified as likely pathogenic according to the American College of Medical Genetics and Genomics (ACMG) criteria (17).
At 6-month follow-up evaluation after fSCIg and antibiotic initiation, no infections were reported. Bimonthly fSCIg have been administered at the dosage of 0.4 g/kg. The frequency of infusions was adjusted according to IgG levels and clinical history over time. Pre-infusion IgG levels of 700–800 mg/dl, associated to an optimal clinical outcome, were finally reached with a monthly fSCIg replacement dosage of 0.4 g/kg. Insulin was no longer needed since blood glucose target concentration was reached only by diet. HRCT of the lungs performed during follow-up demonstrated an important decrease of the previously described opacities and nodules, with the sole persistence of air-trapping (Figure 2B). Considering the radiological disease improvement obtained with immunoglobulin replacement, the diagnosis of GLILD was eventually ruled out. Hemoglobin level remained at about 12 emsp14;g/dl in the absence of any transfusion and without features of hemolysis. ITP, which initially was partially responsive to high-dose prednisone treatment, improved after immunoglobulin replacement, with a platelet count durably greater than 70,000/µL; relapses, previously documented at any attempt of corticosteroid withdrawal, became fewer and better controlled with low-dose corticosteroids (Figure 3). Alternative therapies, including rituximab have been therefore postponed.
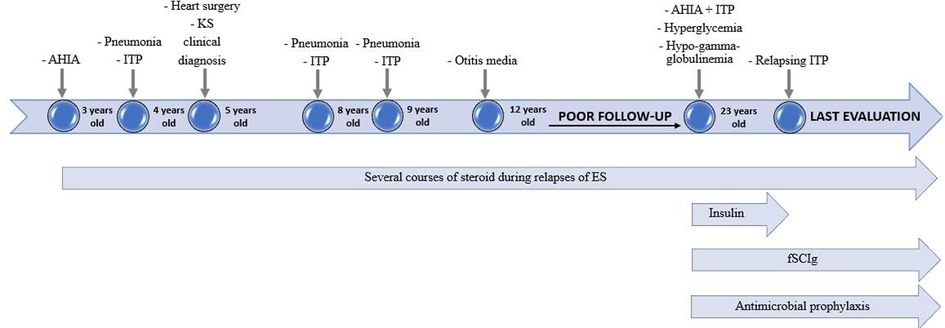
Figure 3. Timeline of principal clinical events and therapeutic strategies.
We hereby report the case of a 23-year-old woman affected by KS presenting with hypogammaglobulinemia, chronic lung disease due to recurrent respiratory infections and immune dysregulation, and several relapses of ES progressively increasing in frequency despite corticosteroid courses.
An international consensus in 2019 defined KS diagnostic criteria (18, 19). The syndrome is characterized by dysmorphic facies (defined by an eversion of the lower lateral eyelids, arched eyebrows with sparse lateral part, depressed nasal tip and prominent ears), postnatal hypotonia, growth deficiency, several skeletal and visceral malformation (including cleft palate, congenital heart defects), dermatoglyphic anomalies and mild to moderate grade of intellectual impairment. Beyond multiorgan anomalies and intellectual disability, autoimmune disorders, including ITP, AIHA, vitiligo and thyroiditis may complicate KS presentation. In KS patients, ITP seems to be the most frequent among immune-mediated cytopenias, but cases of AIHA and/or neutropenia, either combined with ITP or presenting as single-line cytopenia, are also described (7, 12, 20).
Primary autoimmune cytopenias in childhood, especially ITP, are often self-limiting disorders or responsive to first-line therapy (21). Conversely, early-onset, long-lasting, multilineage, refractory cytopenias should strongly suggest an underlying monogenic immune defect (20, 22–24). Consistently with the abovementioned findings, when associated with KS, autoimmune cytopenias, and more specifically ITP, are characterized by severe presentation with chronic or relapsing symptoms, poorly responsive to conventional treatments (13). Moreover, in several patients with KS, non-malignant lymphoproliferation and recurrent respiratory infections, due to hypogammaglobulinemia, have been reported (25, 26).
KS is indeed a paradigmatic example of immunological dysregulation due to mutation in the KMT2D or KDM6A genes, involved in epigenetic modulation of immune system function. The KMT2D gene, also known as MLL2, encodes for a histone H3 lysine 4 (H3K4) mono-methyltransferase, essential for cell differentiation and embryonic development (27). Methylation on H3K4 is found in actively transcribed genes. The KDM6A gene, also known as UTX, encodes for the lysine-specific demethylase 6A linked with demethylation of lysine residues on histone, in particular H3K27, resulting in a gene de-repression. Methylation on H3K27 is associated with transcriptional repression, therefore the action of the KDM6A product allows chromatin opening and active gene transcription (28). Mutations of these enzymes cause an impairment of epigenetic activation of certain genes, leading to the distinctive developmental abnormalities of KS. The immune characteristics of KS patients with KMT2D mutations may derive from a loss of H3K4 methylation at crucial transcription factors, that finally dysregulates B and T differentiation. A disrupted terminal B-cell differentiation and an impaired somatic immunoglobulin hypermutation have been observed in patients with KMT2D mutations, likely as result of impaired epigenetic regulation of the IGH locus. KS patients can in fact present with hypogammaglobulinemia and reduced number of memory B-cells associated to expansion of CD21 B-cell population (29).
Moreover, a clustering of missense mutations in the terminal region of the KMT2D gene might increase the risk for autoimmune diseases, depending either on defective regulatory T-cells (Tregs) generation or intrinsic B tolerance breakage (10, 12, 29, 30). Immunodeficiency may worsen with age and more than 80% of KS patients, mostly with KMT2D mutations, display hypogammaglobulinemia and diminished memory B-cell populations (7). However, autoimmune diseases or hypogammaglobulinemia may not occur until later in childhood, being often underestimated or undiagnosed for a long time, leading to a substantial diagnostic delay. Our patient's case is paradigmatic, since she was referred to our center several years after the onset of refractory autoimmune disease, while also presenting with severe lung disease, suggesting a long-term, undiagnosed, hypogammaglobulinemia. Immunological study was indeed performed for the first time in this patient in occasion of our first observation.
Susceptibility to infections, especially of middle ear and upper respiratory tract, is common among patients with KS (31). Hearing loss, mainly due to recurrent middle ear infections, occurs approximately in up to 80% of patients, and can be conductive, sensorineural or mixed (1, 32, 33). An early immunoglobulin replacement therapy could therefore have a positive effect on hearing function by preventing recurrent otitis media. It is thus advisable to perform a routine and periodic immunologic evaluation in KS patients, in order to prevent chronic diseases and to reduce morbidity and mortality rate. Moreover, because most of KS cases are reported in the pediatric age, the actual frequency of immune alterations in adults with KS may be underestimated.
In addition, as aforementioned, autoimmune cytopenias in KS are severe and often refractory to conventional treatment with corticosteroids. Thus alternative therapies, including rituximab, sirolimus and mycophenolate mofetil (13, 15, 34) should be considered. Moreover, although glucocorticoid-induced diabetes mellitus generally resolves when corticosteroids are discontinued, periodic blood glucose assessment should always be performed in case of long-term steroid treatment (35) to prevent complications, such as diabetic ketoacidosis, due to the impairment of pancreatic endocrine function. In this scenario, multidisciplinary evaluation coordinated by a case manager and immunological evaluations during follow-up are crucial. In our case, patient's overall clinical condition improved after fSCIg replacement initiation. No infections, as well as a lower recurrence of AIHA and ITP episodes were observed. In conclusion, this case highlights the need of improving awareness in suspecting immune dysregulation in KS patients. Moreover, it is advisable that KS patients undergo immunologic evaluations at diagnosis and during follow-up, aiming to provide adequate treatment and avoid preventable and severe morbidity.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://databases.lovd.nl/shared/variants/0000881406#00023885, 0000881406.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was also obtained from the patient’s legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
LL contributed to conception and design of the manuscript. AT, MF, and RP wrote the first draft of the manuscript and collected data. FG, MG, and GA provided and analyzed data. FCon, AM, AS, FCos, and LT supervised, reviewed and wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
The authors thank the patient's family for their cooperation and for providing photos.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Niikawa N, Kuroki Y, Kajii T, Matsuura N, Ishikiriyama S, Tonoki H, et al. Kabuki make-up (Niikawa–Kuroki) syndrome: a study of 62 patients. Am J Med Genet. (1988) 31:565–89. doi: 10.1002/ajmg.1320310312
2. Cheon C-K, Ko JM. Kabuki syndrome: clinical and molecular characteristics. Korean J Pediatr. (2015) 58:317. doi: 10.3345/kjp.2015.58.9.317
3. Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. (2010) 42:790–3. doi: 10.1038/ng.646
4. Bögershausen N, Wollnik B. Unmasking Kabuki syndrome: unmasking Kabuki syndrome. Clin Genet. (2013) 83:201–11. doi: 10.1111/cge.12051
5. Banka S, Veeramachaneni R, Reardon W, Howard E, Bunstone S, Ragge N, et al. How genetically heterogeneous is Kabuki syndrome? MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. (2012) 20:381–8. doi: 10.1038/ejhg.2011.220
6. Camacho-Ordonez N, Ballestar E, Timmers H, Grimbacher B. What can clinical immunology learn from inborn errors of epigenetic regulators? J Allergy Clin Immunol. (2021) 147:1602–18. doi: 10.1016/j.jaci.2021.01.035
7. Hoffman JD, Ciprero KL, Sullivan KE, Kaplan PB, McDonald-McGinn DM, Zackai EH, et al. Immune abnormalities are a frequent manifestation of Kabuki syndrome. Am J Med Genet A. (2005) 135A:278–81. doi: 10.1002/ajmg.a.30722
9. McGaughran J, Aftimos S, Jefferies C, Winship I. Clinical phenotypes of nine cases of Kabuki syndrome from New Zealand. Clin Dysmorphol. (2001) 10:257–62. doi: 10.1097/00019605-200110000-00004
10. Margot H, Boursier G, Duflos C, Sanchez E, Amiel J, Andrau J-C, et al. Immunopathological manifestations in Kabuki syndrome: a registry study of 177 individuals. Genet Med. (2020) 22:181–8. doi: 10.1038/s41436-019-0623-x
11. Giordano P, Lassandro G, Sangerardi M, Faienza MF, Valente F, Martire B. Autoimmune haematological disorders in two Italian children with Kabuki syndrome. Ital J Pediatr. (2014) 40:10. doi: 10.1186/1824-7288-40-10
12. Ming JE, Russell KL, McDonald-McGinn DM, Zackai EH. Autoimmune disorders in Kabuki syndrome: autoimmune disorders in Kabuki syndrome. Am J Med Genet A. (2005) 132A:260–2. doi: 10.1002/ajmg.a.30332
13. Torii Y, Yagasaki H, Tanaka H, Mizuno S, Nishio N, Muramatsu H, et al. Successful treatment with rituximab of refractory idiopathic thrombocytopenic purpura in a patient with Kabuki syndrome. Int J Hematols. (2009) 90:174–6. doi: 10.1007/s12185-009-0387-1
14. Fischer A, Provot J, Jais J-P, Alcais A, Mahlaoui N, Adoue D, et al. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol. (2017) 140:1388–93.e8. doi: 10.1016/j.jaci.2016.12.978
15. Marzollo A, Colavito D, Sartori S, Fanelli GN, Putti MC. Cerebral lymphoproliferation in a patient with Kabuki syndrome. J Clin Immunol. (2018) 38:475–7. doi: 10.1007/s10875-018-0516-9
16. Tesch VK, Abolhassani H, Shadur B, Zobel J, Mareika Y, Sharapova S, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol. (2020) 145:1452–63. doi: 10.1016/j.jaci.2019.12.896
17. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
18. Boniel S, Szymańska K, Śmigiel R, Szczałuba K. Kabuki syndrome—clinical review with molecular aspects. Genes. (2021) 12:468. doi: 10.3390/genes12040468
19. Adam MP, Banka S, Bjornsson HT, Bodamer O, Chudley AE, Harris J, et al. Kabuki syndrome: international consensus diagnostic criteria. J Med Genet. (2019) 56:89–95. doi: 10.1136/jmedgenet-2018-105625
20. Kawame H, Hannibal MC, Hudgins L, Pagon RA. Phenotypic spectrum and management issues in Kabuki syndrome. J Pediatr. (1999) 134:480–5. doi: 10.1016/S0022-3476(99)70207-6
21. Provan D, Arnold DM, Bussel JB, Chong BH, Cooper N, Gernsheimer T, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. (2019) 3:3780–817. doi: 10.1182/bloodadvances.2019000812
22. Hadjadj J, Aladjidi N, Fernandes H, Leverger G, Magérus-Chatinet A, Mazerolles F, et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood. (2019) 134:9–21. doi: 10.1182/blood-2018-11-887141
23. Zama D, Conti F, Moratti M, Cantarini ME, Facchini E, Rivalta B, et al. Immune cytopenias as a continuum in inborn errors of immunity: an in-depth clinical and immunological exploration. Immun Inflamm Dis. (2021) 9:583–94. doi: 10.1002/iid3.420
24. Rivalta B, Zama D, Pancaldi G, Facchini E, Cantarini ME, Miniaci A, et al. Evans syndrome in childhood: long term follow-up and the evolution in primary immunodeficiency or rheumatological disease. Front Pediatr. (2019) 7:304. doi: 10.3389/fped.2019.00304
25. Blanchette V. Childhood chronic immune thrombocytopenic purpura (ITP). Blood Rev. (2002) 16:23–6. doi: 10.1054/blre.2001.0176
26. Warnatz K, Voll RE. Pathogenesis of autoimmunity in common variable immunodeficiency. Front Immunol. (2012) 3:210. doi: 10.3389/fimmu.2012.00210
27. Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. (2005) 6:227. doi: 10.1186/gb-2005-6-8-227
28. Hong S, Cho Y-W, Yu L-R, Yu H, Veenstra TD, Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci. (2007) 104:18439–44. doi: 10.1073/pnas.0707292104
29. Stagi S, Gulino AV, Lapi E, Rigante D. Epigenetic control of the immune system: a lesson from Kabuki syndrome. Immunol Res. (2016) 64:345–59. doi: 10.1007/s12026-015-8707-4
30. Lindsley AW, Saal HM, Burrow TA, Hopkin RJ, Shchelochkov O, Khandelwal P, et al. Defects of B-cell terminal differentiation in patients with type-1 Kabuki syndrome. J Allergy Clin Immunol. (2016) 137:179–87.e10. doi: 10.1016/j.jaci.2015.06.002
31. Di Candia F, Fontana P, Paglia P, Falco M, Rosano C, Piscopo C, et al. Clinical heterogeneity of Kabuki syndrome in a cohort of Italian patients and review of the literature. Eur J Pediatr. (2022) 181:171–87. doi: 10.1007/s00431-021-04108-w
32. Peterson-Falzone SJ, Golabi M, Lalwani AK. Otolaryngologic manifestations of Kabuki syndrome. Int J Pediatr Otorhinolaryngol. (1997) 38:227–36. doi: 10.1016/S0165-5876(96)01443-7
33. Barozzi S, Di Berardino F, Atzeri F, Filipponi E, Cerutti M, Selicorni A, et al. Audiological and vestibular findings in the Kabuki syndrome. Am J Med Genet A. (2009) 149A:171–6. doi: 10.1002/ajmg.a.32610
34. Deyà-Martínez A, Esteve-Solé A, Vélez-Tirado N, Celis V, Costa J, Cols M, et al. Sirolimus as an alternative treatment in patients with granulomatous-lymphocytic lung disease and humoral immunodeficiency with impaired regulatory T cells. Pediatr Allergy Immunol. (2018) 29:425–32. doi: 10.1111/pai.12890
Keywords: Kabuki syndrome, Evans syndrome, autoimmunity, immunodeficiency, hypogammaglobulinemia, immune dysregulation
Citation: Leonardi L, Testa A, Feleppa M, Paparella R, Conti F, Marzollo A, Spalice A, Giona F, Gnazzo M, Andreoli GM, Costantino F and Tarani L (2023) Immune dysregulation in Kabuki syndrome: a case report of Evans syndrome and hypogammaglobulinemia. Front. Pediatr. 11:1087002. doi: 10.3389/fped.2023.1087002
Received: 1 November 2022; Accepted: 25 May 2023;
Published: 9 June 2023.
Edited by:
Daniele Zama, Sant'Orsola-Malpighi Polyclinic, ItalyReviewed by:
Emma Westermann-Clark, University of South Florida, United States© 2023 Leonardi, Testa, Feleppa, Paparella, Conti, Marzollo, Spalice, Giona, Gnazzo, Andreoli, Costantino and Tarani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberto Paparella cm9iZXJ0by5wYXBhcmVsbGFAdW5pcm9tYTEuaXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.