 Mengge Yang
Mengge Yang Qiqi Sheng
Qiqi Sheng Shenghui Ge
Shenghui Ge Xinxin Song
Xinxin Song Jianjun Dong
Jianjun Dong Congcong Guo
Congcong Guo Lin Liao
Lin Liao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Pediatr. , 26 January 2023
Sec. Pediatric Nephrology
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1077120
This article is part of the Research Topic Insights in Pediatric Nephrology: 2022 View all 6 articles
Background and Aims: The genetic and clinical characteristics of patients with distal renal tubular acidosis (dRTA) caused by SLC4A1 mutations have not been systematically recorded before. Here, we summarized the SLC4A1 mutations and clinical characteristics associated with dRTA.
Methods: Database was searched, and the mutations and clinical manifestations of patients were summarized from the relevant articles.
Results: Fifty-three eligible articles involving 169 patients were included and 41 mutations were identified totally. Fifteen mutations involving 100 patients were autosomal dominant inheritance, 21 mutations involving 61 patients were autosomal recessive inheritance. Nephrocalcinosis or kidney stones were found in 72.27%, impairment in renal function in 14.29%, developmental disorders in 61.16%, hematological abnormalities in 33.88%, and muscle weakness in 13.45% of patients. The age of onset was younger (P < 0.01), urine pH was higher (P < 0.01), and serum potassium was lower (P < 0.001) in recessive patients than patients with dominant SLC4A1 mutations. Autosomal recessive inheritance was more often found in Asian patients (P < 0.05).
Conclusions: The children present with metabolic acidosis with high urinary pH, accompanying hypokalemia, hyperchloremia, nephrocalcinosis, growth retardation and hematological abnormalities should be suspected as dRTA and suggested a genetic testing. The patients with recessive dRTA are generally more severely affected than that with dominant SLC4A1 mutations. Autosomal recessive inheritance was more often found in Asian patients, and more attentions should be paid to the Asian patients.
The SLC4A1 gene is one of the bicarbonate anion transporters of the solute carrier family 4 (SLC4) gene family (1). This gene encodes anion exchanger member 1 (AE1), which is expressed in both erythrocytes and the acid-secreting α- intercalated cells of the kidney (2). The erythroid isoform of AE1(eAE1) is expressed in the red blood cells. The kidney anion exchanger 1 (kAE1) is highly expressed in the kidney and located in the basolateral membrane of the α-intercalated cell (3). The kAE1 protein plays an essential role in bicarbonate ion (HCO3−) in exchange with chloride ion (Cl−) during the apical secretion of hydrogen ion (H+) into the tubular lumen (4–6).
Mutations in SCL4A1 may cause distal renal tubular acidosis (dRTA). Distal renal tubular acidosis is a disease of defective urinary acidification characterized by impaired H+ secretion into the urine leading to metabolic acidosis, hypokalemia, nephrocalcinosis, and failure to thrive (7, 8). The mutations in SLC4A1 have been reported in both autosomal dominant (AD) and recessive (AR) types of dRTA. Bruce et al. first reported that dominant dRTA was attributed to SLC4A1 gene defect in 1997 (9). Mutations associated with AR types of dRTA have usually been found in homozygous and compound heterozygous conditions (10–14).
Our understanding of dRTA caused by SLC4A1 mutations is limited by low incidence and phenotypic variability, and mutations in SLC4A1 have not been systematicly documented. However, diagnosis and treatment are often delayed due to clinical variability of the disease, and hereditary kidney disease compromises the quality of life of patients. Herein we analyzed the genetic defects in SLC4A1 and clinical phenotypes of the patients to facilitate the diagnosis and treatment of dRTA with SLC4A1 mutations.
PubMed, Embase, Web of Science, the China National Knowledge Infrastructure, and Wanfang were searched from the date of inception to May 30, 2022 with the following search terms: “SLC4A1” OR “AE1” OR “Band 3”. We also scanned the references of included studies to avoid omissions in the search process. The flow diagram of the search process is provided in Supplementary Figure S1. The protocol for the systmetic review is registered with International Platform of Registered Systematic Review and Meta-analysis Protocols (INPLASY2022120031).
Eligible articles meeting the following criteria were included: (1) patients were diagnosed as dRTA. (2) mutations in SLC4A1 were confirmed using molecular genetic techniques. (3) clinical data of patients were described. The articles involving a series of patients but without detailed descriptions were excluded.
From relevant articles meeting the inclusion criteria, the following data of patients were extracted: (1) country, (2) gender, (3) age of onset, (4) mutation information, (5) clinical manifestations, (6) laboratory test results at diagnosis.
The epidemiological and clinical characteristics, and laboratory indexes of patients were described utilizing simple summary statistics. Mann-Whitney U test and t-test were used to analyze the data. The significance level was set as p < 0.05. Statistical analysis was performed using the Statistical Package for the Social Sciences version 26 for Windows (SPSS). Since certain data in some patients were missing, the total number of patients was mentioned in each analysis.
The detailed information of geographical country distribution and gender distribution is described in Figure 1. A total of 724 citations were identified through database searches and other resources, and 392 remained after duplicate removal. Fifty-three eligible articles were included ultimately, which contained 169 patients. Among them, Asian cases accounted for the largest part (118/169, 69.82%), followed by European (31/169, 18.34%), South American (10/169, 5.92%), African (8/169, 4.73%), and North American (2/169, 1.18%). Among the Asian, cases from China and Thailand accounted for 37.87% (64/169) and 14.20% (24/169) respectively. Gender distribution was female 36.96% (51/138) vs. male 63.04% (87/138).
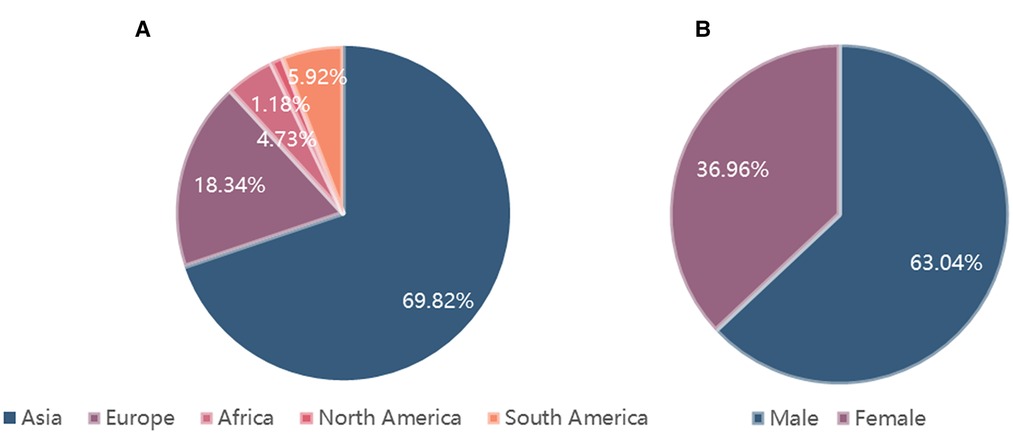
Figure 1. The detailed information of geographical country distribution and gender distribution. (A). Geographical country distribution; (B). Gender distribution.
Mutations in SLC4A1 are illustrated in Table 1 and Figure 2, and the detailed analytic data of mutation were listed in the Supplementary Table S1. Figure 3 shows the AE1 protein and the location of the variants. Totally 41 mutations were identified in 169 patients. Fifteen mutations involving 100 patients were autosomal dominant inheritance, 21 mutations involving 61 patients were autosomal recessive inheritance. Among the AR mutations, 5 mutations involving 28 patients were autosomal recessive inheritance, and 22 mutations involving 33 patients were compound heterozygosity. The hereditary mode of 7 mutations involving 8 patients were unknown. Thirty-eight patients involving 8 mutations were complicated with Southeast Asian ovalocytosis (SAO), and seven patients involving 7 mutations were complicated with hereditary spherocytosis (HS). The pattern of inheritance was unknown in 6 mutations involving 7 patients. The mutations in Arg589 were the most frequently observed in AD mutations and the most common in all mutations. G701D was the most common in AR mutations. Among 101 Asian patients, 62 patients are autosomal dominant inheritance and 49 patients are autosomal recessive inheritance. Among 51 Non-Asian patients, 39 patients are autosomal dominant inheritance and 12 patients are autosomal recessive inheritance. Autosomal recessive inheritance was more often found in Asian patients (P < 0.05).
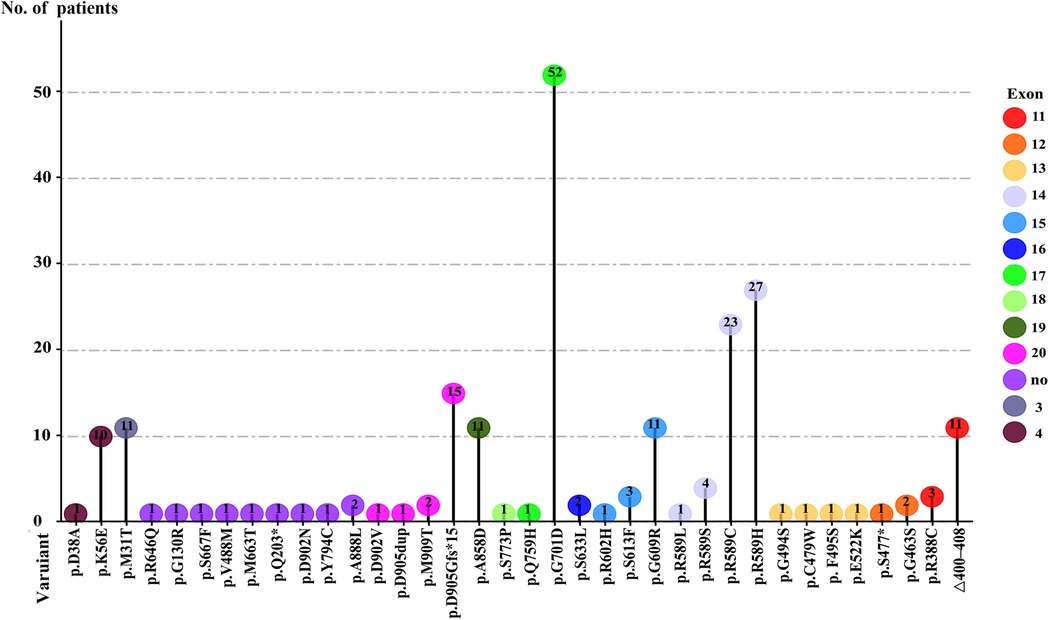
Figure 2. Frenquency of mutations at each mutation site.
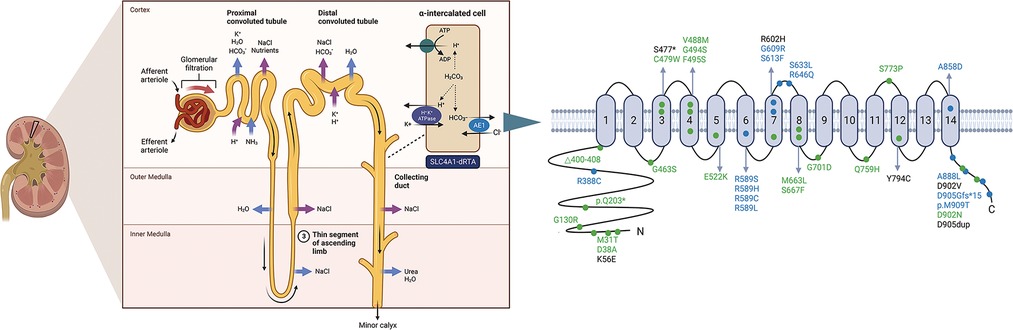
Figure 3. Ae1 protein and the location of the variants. Blue and green dots indicate dominant and recessive mutations, respectively. Black dots indicate the heredity is unknown. Created with Biorender.com.
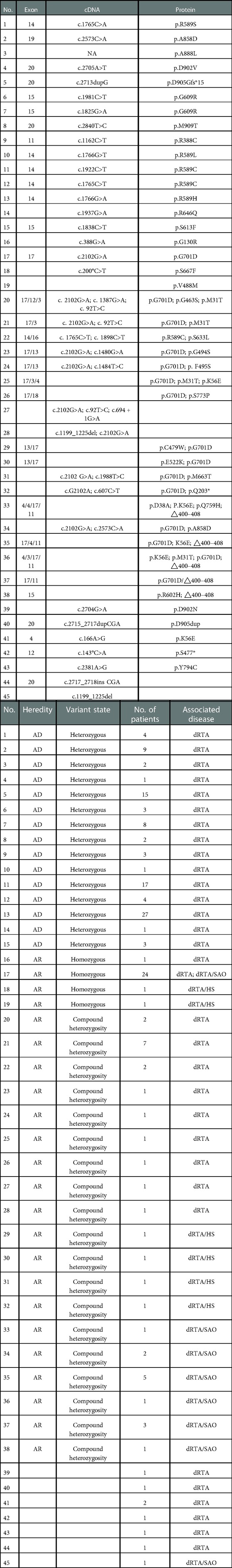
Table 1. Mutations in SLC4A1.
The age of onset ranged from 0 to 72 years among 129 patients (median 5 years, IQR 2–12). The mean age of onset in patients with autosomal dominant mutations (82 patients), autosomal recessive mutations (22 patients) and compound heterozygosity (21 patients) was 16.33, 6.36 and 4.29 years, respectively. The clinical manifestations of patients diagnosed as dRTA with mutations in SLC4A1 were described in 121 patients (Table 2). Nephrocalcinosis and kidney stones were presented in 65 (53.72%) and 23 (19.01%) patients respectively, and impairment in renal function was found in 17 (14.29%) patients. Developmental disorders were found in 74 (61.16%) patients, including growth retardation (31.40%) and rickets (29.75%). Hematological abnormalities were observed in 41 (33.88%) patients, including spherocytosis, ovalocytosis and anemia. Muscle weakness was observed in 16 (13.45%) patients. Alkaline urine (PH > 6.5) was presented in 64 (64/79, 81.01%) patients, and the median urine PH in 79 patients was 7.00 (IQR 6.7–7.5). The median blood PH was 7.27 (IQR 7.25–7.30). Hypokalemia was seen in 72 (72/109, 66.06%) patients, and the mean of serum potassium in 109 patients was 3.16 mmol/L. Hyperchloremia was seen in 50 (50/53, 94.34%) patients, and the mean of serum potassium in 109 patients was 113.02 mmol/L.
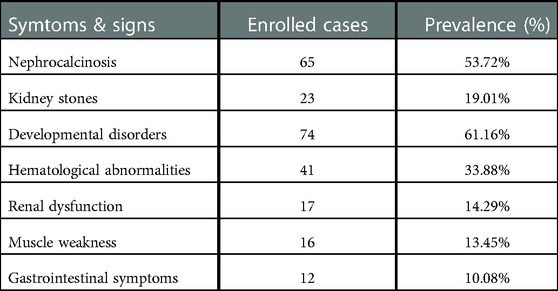
Table 2. The clinical manifestations of patients diagnosed as dRTA with mutations in SLC4A1.
The comparisons of biochemical indexes and age of onset between AD and AR SLC4A1-dRTA are shown in Figure 4. The age of onset was younger (P < 0.01), alkaline urine was more severe (P < 0.01), and serum potassium was lower (P < 0.001) in recessive patients than patients with dominant SLC4A1 mutations. Among 61 patients with AR dRTA, hematological abnormalities were observed in 30 (30/61,49.18%) patients, and most of them were from Southeast Asia. However, only 11(10.89%) patients presented with hematological abnormalities in 101 patients with AD dRTA. The comparisons of biochemical indexes and age of onset between Asian and Non-Asian patients are shown in Figure 5. The age of onset was younger (P < 0.01), alkaline urine was more severe (P < 0.001), and serum potassium was lower (P < 0.01) in Asian patients than Non-Asian patients. There are no differences in biochemical indexes and age of onset between male and female (P > 0.05). As shown in Figure 6, the recessive forms of dRTA often involves other organs than the kidney, incluing developmental disorders, hematological abnormalities (P < 0.001) and gastrointestinal symptoms (P < 0.05).
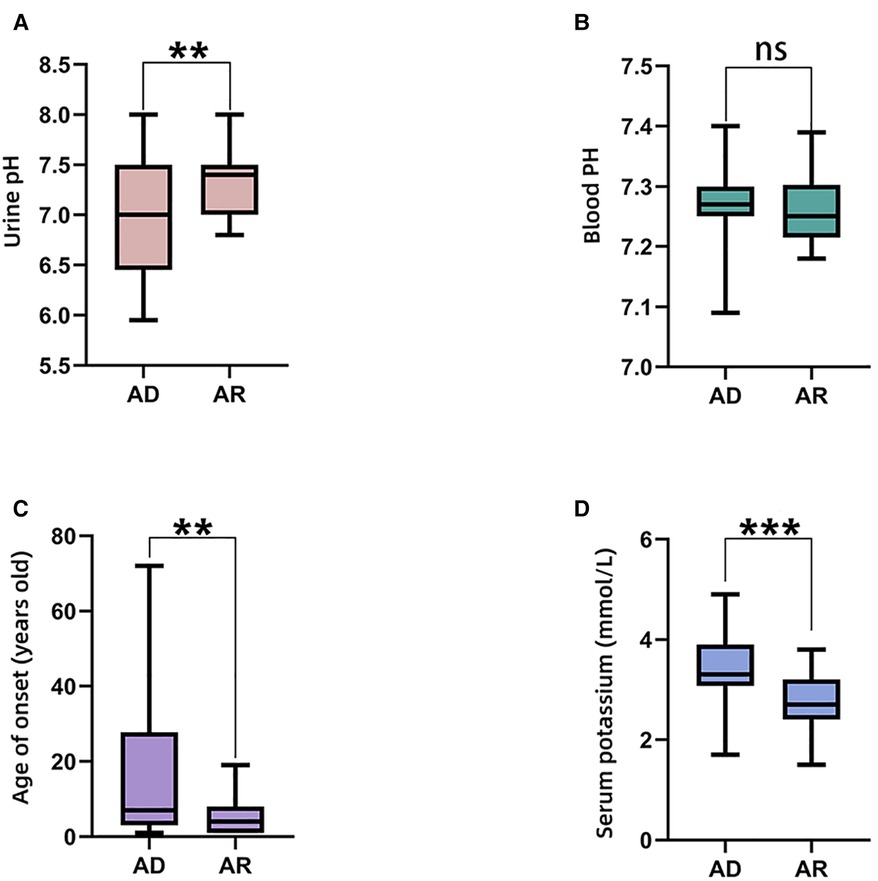
Figure 4. The comparisons of biochemical indexes and age of onset between AD and AR. (A). Urine pH; (B). Blood PH; (C). Age of onset; (D). Serum potassium. *P < 0.05; **P < 0.01; ***P < 0.001; ns: P > 0.05.
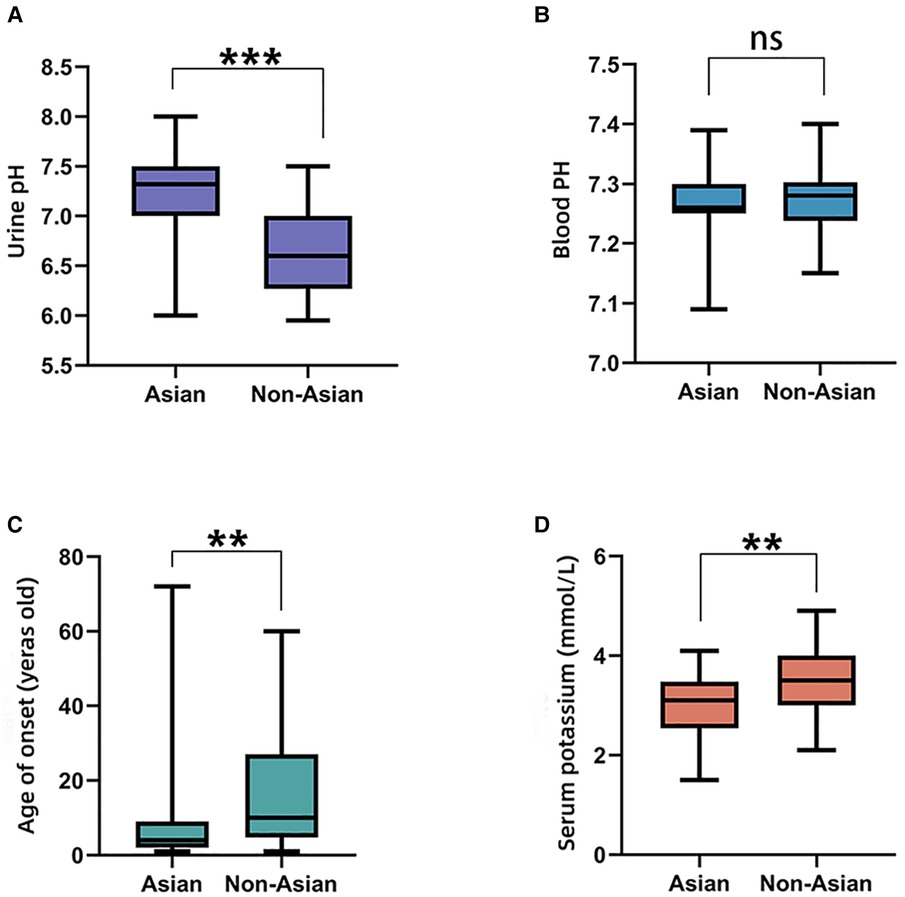
Figure 5. The comparisons of biochemical indexes and age of onset between Asian and Non-Asian. (A). Urine pH; (B). Blood PH; (C). Age of onset; (D). Serum potassium. *P < 0.05; **P < 0.01; ***P < 0.001; ns: P > 0.05.
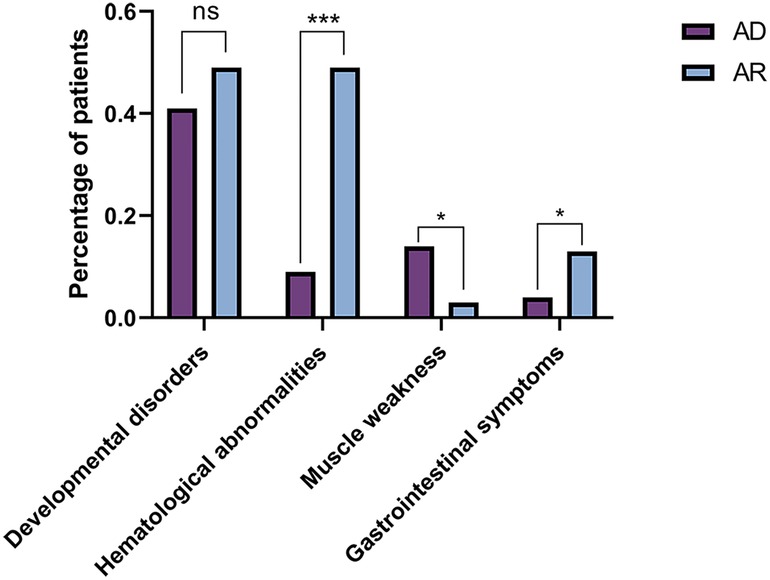
Figure 6. The comparisons of patients involving other organs between AD and AR. *P < 0.05; ***P < 0.001; ns: P > 0.05.
To our knowledge, this is the first study to systematically and comprehensively analyze reported dRTA patients with SLC4A1 mutations. In this work, we summarized the mutation spectrum and clinical characteristics in patients with SLC4A1 mutations. SLC4A1 is located on chromosome 17 and encodes anion exchanger 1 protein in erythrocyte cell membranes and α-intercalated cells of the kidney (15).
There are several possible explanations for the pathogenesis of the acid secreting defect secondary to SLC4A1. A mouse model lacking AE1 exhibits spontaneous hyperchloremic metabolic acidosis with reduced acid excretion, and inappropriately alkaline urine (16). Basolateral Cl-/HCO3- exchange activity was reduced, while alternate bicarbonate transport pathways were upregulated. Dysregulated expression and localization of the aquaporin-2 water channel were observed in mice lacking AE1, accompanied by severe urinary concentration defect (16). KAE1 forms functional homodimers or etero-oligomers, and oligomerization of a dominant negative mutant AE1 with a wild-type polypeptide in the heterozygote A-IC likely explains some dominant disease. The mutant/wild-type heterodimer fails to traffic to the cell surface or traffics normally but may not function normally at the cell surface (16–18). Dominant dRTA might be caused by inappropriate targeting to A-IC apical membrane, with resultant apical bicarbonate secretion likely short-circuiting luminal acid secretion (19, 20). Whereas, the study by Mumtaz R et al. generated a mouse corresponding to the dominant dRTA mutation in human AE1 R589H, suggesting normal targeting of the pathologic R607H variant, but the mutant mice exhibited reduced expression of V-type ATPase and compromised targeting of this proton pump to the plasma membrane upon acid challenge (21). Homozygous patients may not have any kAE1 isoform at the plasma membrane of a-intercalated cells in the distal nephron since these cells lack glycophorin A and develop dRTA consequently (17). Previous study has also reported that the integrity of the cytosolic COOH terminus played a crucial role in normal kAE1 targeting to the cell surface. Truncation of the last 11 amino acids increased the kAE1 endocytosis rate and reduced recycle to the plasma membrane. The decreased abundance at the plasma membrane and altered recycling provide a possible physiological mechanism of distal renal tubular acidosis in patients carrying SLC4A1 mutations (22).
The defect of kAE1 is associated with inherited distal renal tubular acidosis (dRTA) (9, 23–26). In addition, eAE1 is an important structural component of the red cell membrane, which ensures the normal skeleton structure and ion transport of erythrocytes (27). Consequently, SLC4A1 mutations that alter AE1 composition are often associated with disorders of red cell membrane integrity including SAO and HS, which are usually autosomal recessive inheritance (28, 29). SAO is a morphological erythrocyte abnormality caused by a mutational deletion of 27 bp in exon 11 of SLC4A1, resulting in a frame 9 amino acid deletion at the junction between the N-terminal domain of eAE1 and the first transmembrane segment, involving codons 400–408 (30). These patients are usually homozygous for a single SLC4A1 mutation or compound heterozygotes of two different SLC4A1 mutations found throughout Southeast Asia most commonly, one of which is usually the SAO mutation.
SLC4A1-dRTA exhibits different behaviors with regard to different ethnicities. The G701D mutation was first identified in a Thai family (11), which was recessive and frequently observed in Southeast Asia in both homozygote and compound heterozygote with SAO mutation (31). These patients usually accompanied with anemia, jaundice, and splenomegaly; spherocytosis and ovalocytosis could be found in in blood smear (14, 32). Our study showed that almost half of the patients with AR dRTA had hematological abnormalities, while it was uncommon in patients with AD dRTA. Dominant mutant proteins tend to maintain normal Cl-/HCO3- exchange function in erythrocytes, hemolytic anemia was rarely observed consequently (9). The Arg589 is a hotspot mutation site, which is located in the intracellular domain between the sixth and the seventh transmembrane regions of the AE1 protein, more commonly seen in Caucasian (33–35). Even though SAO and G701D are more common in Southeast Asians (10, 14, 31). Since Southeast Asia historically have had a high incidence of Plasmodium falciparum malaria, a presumable supposition is that SAO is considered to have evolved, which might provide a protection against the clinical effects of Plasmodium falciparum malaria (36, 37).
Our study showed that the age of onset varies from birth to adulthood, mostly in childhood. Common clinical manifestations include vomit, failure to thrive, nephrocalcinosis, nephrolithiasis, rickets and muscle weakness. The spectrum of phenotypic severity in dRTA is wide, ranging from growth impairment to renal function impairment. In dRTA, ɑ-intercalated cells in the collecting duct are unable to secrete H+ ions and acidify urine, and result in metabolic acidosis and hypokalemia. Severe hypokalemia could result in vomit and muscle weakness (12, 26, 38). The metabolic acidosis is mainly buffered by the bone, and bone demineralization leads to failure to thrive and rickets. The excess of calcium in the blood and decreased expression of kidney calcium transporter proteins cause hypercalciuria, which results in nephrocalcinosis and/or nephrolithiasis, eventually leads to renal insufficiency (25, 36). The patients often visit different departments due to various clinical manifestations, therefore clinical workers should raise general awareness of the disease.
Our review of the clinical characteristics of these patients revealed the differences between the dominant and recessive forms of patients with SLC4A1 mutations. The clinical phenotypes of dRTA caused by SLC4A1 mutations are of great heterogenity. The percentage of homozygous subjects is greater in the Asian population and for this reason they present a more severe clinical picture. Consistent with already published data, the clinical manifestations and biochemical phenotypes of AD are usually milder compared with that of patients with AR mutations (39). Compared with the patients with autosomal dominant mutations, the patients with autosomal recessive mutations had more severe alkaline urine and hypokalemia, and the age of onset was much younger. Probably because the dominant mutants retain wild-type kAE1 protein intracellularly while the recessive kAE1 mutants do not. The patients with the recessive mutants show more severe trafficking defects (37). Based on the above genotype-phenotype correlations, a molecular diagnosis is necessary due to the implications for treatment, prognosis and family risk. For the patients with AR mutations, more attention needs to be paid.
For patients with dRTA, the clinical management is a critical issue that needs to be taken seriously. Since acid-base homeostasis is essential for normal growth and development, the treatments of dRTA should not be limited to correcting the biochemical abnormalities, but also to preventing disease-related abnormalities such as failure to thrive, growth retardation, rickets, osteoporosis, nephrolithiasis, and nephrocalcinosis (40, 41). Since the progression of nephrocalcinosis may lead to chronic kidney disease and end-stage kidney disease in dRTA patients, prevention of nephrocalcinosis is particularly important (40). As citrate salts can prevent nephrolithiasis, potassium citrate is usually recommended (23, 40).
Our results also suggested over half children presented with growth retardation, which has been a major problem in children with renal tubular acidosis, deserving our concerns. At an early stage, alkali and potassium supplementation could correct metabolic acidosis, regulating acid-base balance and significantly improve growth and skeletal deformities (13, 42). And lifelong treatment is recommended (23). Although calcium and active vitamin D are beneficial in the treatment of osteoporosis and osteomalacia, they promote the formation of kidney stones and should be avoided (43). It was reported that combined conventional alkali supplementation with recombinant human growth hormone (rhGH) therapies might have a beneficial effect on growth in dRTA patients, however, further high-quality clinical studies involving more patients are needed to confirm this observation (44). For patients with erythrocyte membrane disorders, be of benefit in patients with severe and moderate hemolytic anemia, but is not necessary in mild cases generally (45).
Our study has several limitations. First, the number of reported cases is not large enough to permit definite genotype-phenotype correlations. Second, this study might have selective bias, since typical or more severe patients tend to be diagnosed and reported. Additionally, the biochemical indexes were from different laboratory, which might cause a slight deviation. And further high-quality studies are needed for the purpose of explaining the more precise molecular mechanism.
In summary, our study firstly summarized mutations and clinical characteristics of dRTA caused by SLC4A1 mutations. The patients with the presence of metabolic acidosis, hypokalemia, hyperchloremia, nephrocalcinosis and growth retardation should be prompt a genetic test as soon as possible. The patients with recessive dRTA are generally more severely affected and the age of onset is earlier than that with dominant SLC4A1 mutations, and autosomal recessive inheritance was more often found in Asian patients. Early identification and early treatment are very essential for the prognosis of patients, especially the Asian patients.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
MGY contributed to the conception and design of the study; SHG and XXS performed the literature search and study selection; MGY performed the data extraction and statistical analyses; MGY, LL and JJD drafted the manuscript and CCG revised it critically; QQS and CCG polished the language. All authors gave final approval for submitted manuscript content and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Schematics were created with BioRender.com. All authors contributed to the article and approved the submitted versio.
This work was funded by National Natural Science Foundation of China Grants (NO. 82170847, 82105044); Key project of Shandong Provincial Natural Science Foundation of China Grants (NO. ZR2020KH004); Shandong Provincial Natural Science Foundation of China Grants (No. ZR2020QH310).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1077120/full#supplementary-material.
1. Yenchitsomanus PT. Human anion exchanger1 mutations and distal renal tubular acidosis. Southeast Asian J Trop Med Public Health. (2003) 34(3):651–8.15115146
2. Tanner MJ. Band 3 anion exchanger and its involvement in erythrocyte and kidney disorders. Curr Opin Hematol. (2002) 9(2):133–9. doi: 10.1097/00062752-200203000-00009
3. Duangtum N, Junking M, Phadngam S, Sawasdee N, Castiglioni A, Charngkaew K, et al. gamma-COPI mediates the retention of kAE1 G701D protein in Golgi apparatus—a mechanistic explanation of distal renal tubular acidosis associated with the G701D mutation. Biochem J. (2017) 474(15):2573–84. doi: 10.1042/bcj20170088
4. Alper SL, Natale J, Gluck S, Lodish HF, Brown D. Subtypes of intercalated cells in rat kidney collecting duct defined by antibodies against erythroid band 3 and renal vacuolar H+-ATPase. Proc Natl Acad Sci USA. (1989) 86(14):5429–33. doi: 10.1073/pnas.86.14.5429
5. Kollert-Jöns A, Wagner S, Hübner S, Appelhans H, Drenckhahn D. Anion exchanger 1 in human kidney and oncocytoma differs from erythroid AE1 in its NH2 terminus. Am J Physiol. (1993) 265(6 Pt 2):F813–21. doi: 10.1152/ajprenal.1993.265.6.F813
6. Tanner MJ. The structure and function of band 3 (AE1): recent developments (review). Mol Membr Biol. (1997) 14(4):155–65. doi: 10.3109/09687689709048178
7. Alper SL. Genetic diseases of acid-base transporters. Annu Rev Physiol. (2002) 64:899–923. doi: 10.1146/annurev.physiol.64.092801.141759
8. Weber S, Soergel M, Jeck N, Konrad M. Atypical distal renal tubular acidosis confirmed by mutation analysis. Pediatr Nephrol. (2000) 15(3-4):201–4. doi: 10.1007/s004670000454
9. Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, et al. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest. (1997) 100(7):1693–707. doi: 10.1172/JCI119694
10. Yenchitsomanus PT, Vasuvattakul S, Kirdpon S, Wasanawatana S, Susaengrat W, Sreethiphayawan S, et al. Autosomal recessive distal renal tubular acidosis caused by G701D mutation of anion exchanger 1 gene. Am J Kidney Dis. (2002) 40(1):21–9. doi: 10.1053/ajkd.2002.33909
11. Tanphaichitr VS, Sumboonnanonda A, Ideguchi H, Shayakul C, Brugnara C, Takao M, et al. Novel AE1 mutations in recessive distal renal tubular acidosis. Loss-of-function is rescued by glycophorin A. J Clin Invest. (1998) 102(12):2173–9. doi: 10.1172/JCI4836
12. Sritippayawan S, Sumboonnanonda A, Vasuvattakul S, Keskanokwong T, Sawasdee N, Paemanee A, et al. Novel compound heterozygous SLC4A1 mutations in Thai patients with autosomal recessive distal renal tubular acidosis. Am J Kidney Dis. (2004) 44(1):64–70. doi: 10.1053/j.ajkd.2004.03.033
13. Park E, Phaymany V, Yi ES, Phangmanixay S, Cheong HI, Choi Y. Primary autosomal recessive distal renal tubular acidosis caused by a common homozygous SLC4A1 mutation in two Lao families. J Korean Med Sci. (2018) 33(13):e95. doi: 10.3346/jkms.2018.33.e95
14. Choo KE, Nicoli TK, Bruce LJ, Tanner MJ, Ruiz-Linares A, Wrong OM. Recessive distal renal tubular acidosis in sarawak caused by AE1 mutations. Pediatr Nephrol. (2006) 21(2):212–7. doi: 10.1007/s00467-005-2061-z
15. Anacleto FE, Bruce LJ, Clayton P, Hegde S, Resontoc LP, Wrong O. Distal renal tubular acidosis in filipino children, caused by mutations of the anion-exchanger SLC4A1 (AE1, band 3). Gene. Nephron. (2010) 114(2):19–24. doi: 10.1159/000274484
16. Deejai N, Sawasdee N, Nettuwakul C, Wanachiwanawin W, Sritippayawan S, Yenchitsomanus PT, et al. Impaired trafficking and instability of mutant kidney anion exchanger 1 proteins associated with autosomal recessive distal renal tubular acidosis. BMC Med Genomics. (2022) 15(1):228. doi: 10.1186/s12920-022-01381-y
17. Cordat E, Kittanakom S, Yenchitsomanus PT, Li J, Du K, Lukacs GL, et al. Dominant and recessive distal renal tubular acidosis mutations of kidney anion exchanger 1 induce distinct trafficking defects in MDCK cells. Traffic. (2006) 7(2):117–28. doi: 10.1111/j.1600-0854.2005.00366.x
18. Dahl NK, Jiang L, Chernova MN, Stuart-Tilley AK, Shmukler BE, Alper SL. Deficient HCO3- transport in an AE1 mutant with normal Cl- transport can be rescued by carbonic anhydrase II presented on an adjacent AE1 protomer. J Biol Chem. (2003) 278(45):44949–58. doi: 10.1074/jbc.M308660200
19. Rungroj N, Devonald MA, Cuthbert AW, Reimann F, Akkarapatumwong V, Yenchitsomanus PT, et al. A novel missense mutation in AE1 causing autosomal dominant distal renal tubular acidosis retains normal transport function but is mistargeted in polarized epithelial cells. J Biol Chem. (2004) 279(14):13833–8. doi: 10.1074/jbc.M400188200
20. Toye AM, Banting G, Tanner MJ. Regions of human kidney anion exchanger 1 (kAE1) required for basolateral targeting of kAE1 in polarised kidney cells: mis-targeting explains dominant renal tubular acidosis (dRTA). J Cell Sci. (2004) 117(Pt 8):1399–410. doi: 10.1242/jcs.00974
21. Mumtaz R, Trepiccione F, Hennings JC, Huebner AK, Serbin B, Picard N, et al. Intercalated cell depletion and vacuolar H(+)-ATPase mistargeting in an Ae1 R607H knockin model. J Am Soc Nephrol. (2017) 28(5):1507–20. doi: 10.1681/ASN.2016020169
22. Almomani E, Lashhab R, Alexander RT, Cordat E. The carboxyl-terminally truncated kidney anion exchanger 1 R901X dRTA mutant is unstable at the plasma membrane. Am J Physiol Cell Physiol. (2016) 310(9):C764–72. doi: 10.1152/ajpcell.00305.2015
23. Batlle D, Haque SK. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant. (2012) 27(10):3691–704. doi: 10.1093/ndt/gfs442
24. Guo W, Song Q, Zhang R, Xin Q, Liu Z, Lang Y, et al. Genotypic and phenotypic analysis in 51 Chinese patients with primary distal renal tubular acidosis. Clin Genet. (2021) 100(4):440–6. doi: 10.1111/cge.14011
25. Atmis B, Cevizli D, Melek E, Bisgin A, Unal I, Anarat A, et al. Evaluation of phenotypic and genotypic features of children with distal kidney tubular acidosis. Pediatr Nephrol. (2020) 35(12):2297–306. doi: 10.1007/s00467-020-04685-2
26. Zhou F, Mao J, Ye Q, Zhu X, Zhang Y, Ye Y, et al. Clinical features and genetic findings in Chinese children with distal renal tubular acidosis. Int J Clin Exp Pathol. (2018) 11(7):3523–32.31949730
27. Liu SC, Zhai S, Palek J, Golan DE, Amato D, Hassan K, et al. Molecular defect of the band 3 protein in Southeast Asian ovalocytosis. N Engl J Med. (1990) 323(22):1530–8. doi: 10.1056/NEJM199011293232205
28. Ribeiro ML, Alloisio N, Almeida H, Gomes C, Texier P, Lemos C, et al. Severe hereditary spherocytosis and distal renal tubular acidosis associated with the total absence of band 3. Blood. (2000) 96(4):1602–4. doi: 10.1182/blood.V96.4.1602
29. Sawasdee N, Udomchaiprasertkul W, Noisakran S, Rungroj N, Akkarapatumwong V, Yenchitsomanus PT. Trafficking defect of mutant kidney anion exchanger 1 (kAE1) proteins associated with distal renal tubular acidosis and Southeast Asian ovalocytosis. Biochem Biophys Res Commun. (2006) 350(3):723–30. doi: 10.1016/j.bbrc.2006.09.113
30. Jarolim P, Palek J, Amato D, Hassan K, Sapak P, Nurse GT, et al. Deletion in erythrocyte band 3 gene in malaria-resistant Southeast Asian ovalocytosis. Proc Natl Acad Sci USA. (1991) 88(24):11022–6. doi: 10.1073/pnas.88.24.11022
31. Ungsupravate D, Sawasdee N, Khositseth S, Udomchaiprasertkul W, Khoprasert S, Li J, et al. Impaired trafficking and intracellular retention of mutant kidney anion exchanger 1 proteins (G701D and A858D) associated with distal renal tubular acidosis. Mol Membr Biol. (2010) 27(2/3):92–103. doi: 10.3109/09687681003588020
32. Vasuvattakul S, Yenchitsomanus PT, Vachuanichsanong P, Thuwajit P, Kaitwatcharachai C, Laosombat V, et al. Autosomal recessive distal renal tubular acidosis associated with Southeast Asian ovalocytosis. Kidney Int. (1999) 56(5):1674–82. doi: 10.1046/j.1523-1755.1999.00756.x
33. Karet FE, Gainza FJ, Györy AZ, Unwin RJ, Wrong O, Tanner MJ, et al. Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci USA. (1998) 95(11):6337–42. doi: 10.1073/pnas.95.11.6337
34. Jarolim P, Shayakul C, Prabakaran D, Jiang L, Stuart-Tilley A, Rubin HL, et al. Autosomal dominant distal renal tubular acidosis is associated in three families with heterozygosity for the R589H mutation in the AE1 (band 3) cl-/HCO3- exchanger. J Biol Chem. (1998) 273(11):6380–8. doi: 10.1074/jbc.273.11.6380
35. Palazzo V, Provenzano A, Becherucci F, Sansavini G, Mazzinghi B, Orlandini V, et al. The genetic and clinical spectrum of a large cohort of patients with distal renal tubular acidosis. Kidney Int. (2017) 91(5):1243–55. doi: 10.1016/j.kint.2016.12.017
36. Bruce LJ, Wrong O, Toye AM, Young MT, Ogle G, Ismail Z, et al. Band 3 mutations, renal tubular acidosis and South-East Asian ovalocytosis in Malaysia and Papua New Guinea: loss of up to 95% band 3 transport in red cells. Biochem J. (2000) 350(Pt 1(Pt 1)):41–51. doi: 10.1042/bj3500041
37. Yenchitsomanus PT, Kittanakom S, Rungroj N, Cordat E, Reithmeier RA. Molecular mechanisms of autosomal dominant and recessive distal renal tubular acidosis caused by SLC4A1 (AE1) mutations. J Mol Genet Med. (2005) 1(2):49–62. doi: 10.4172/1747-0862.1000013
38. Fry AC, Su Y, Yiu V, Cuthbert AW, Trachtman H, Karet Frankl FE. Mutation conferring apical-targeting motif on AE1 exchanger causes autosomal dominant distal RTA. J Am Soc Nephrol. (2012) 23(7):1238–49. doi: 10.1681/ASN.2012020112
39. Zhang Z, Liu KX, He JW, Fu WZ, Yue H, Zhang H, et al. Identification of two novel mutations in the SLC4A1 gene in two unrelated Chinese families with distal renal tubular acidosis. Arch Med Res. (2012) 43(4):298–304. doi: 10.1016/j.arcmed.2012.05.001
40. Vallés PG, Batlle D. Hypokalemic distal renal tubular acidosis. Adv Chronic Kidney Dis. (2018) 25(4):303–20. doi: 10.1053/j.ackd.2018.05.003
41. Watanabe T. Improving outcomes for patients with distal renal tubular acidosis: recent advances and challenges ahead. Pediatric Health Med Ther. (2018) 9:181–90. doi: 10.2147/PHMT.S174459
42. Sakuraya K, Nozu K, Oka I, Fujinaga S, Nagano C, Ohtomo Y, et al. A different clinical manifestation in a Japanese family with autosomal dominant distal renal tubular acidosis caused by SLC4A1 mutation. CEN Case Rep. (2020) 9(4):442–5. doi: 10.1007/s13730-020-00500-x
43. Cheidde L, Vieira TC, Lima PR, Saad ST, Heilberg IP. A novel mutation in the anion exchanger 1 gene is associated with familial distal renal tubular acidosis and nephrocalcinosis. Pediatrics. (2003) 112(6 Pt 1):1361–7. doi: 10.1542/peds.112.6.1361
44. Liu J, Shen Q, Li G, Zhai Y, Fang X, Xu H. Clinical and genetic analysis of distal renal tubular acidosis in three Chinese children. Renal Fail. (2018) 40(1):520–6. doi: 10.1080/0886022x.2018.1487858
Keywords: distal renal tubular acidosis, SLC4A1, metabolic acidosis, mutation, clinical characteristics
Citation: Yang M, Sheng Q, Ge S, Song X, Dong J, Guo C and Liao L (2023) Mutations and clinical characteristics of dRTA caused by SLC4A1 mutations: Analysis based on published patients. Front. Pediatr. 11:1077120. doi: 10.3389/fped.2023.1077120
Received: 22 October 2022; Accepted: 6 January 2023;
Published: 26 January 2023.
Edited by:
Michael L. Moritz, University of Pittsburgh, United StatesReviewed by:
Francesco Trepiccione, University of Campania Luigi Vanvitelli, Italy© 2023 Yang, Sheng, Ge, Song, Dong, Guo and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Liao bGlhb2xpbkBzZHUuZWR1LmNu Congcong Guo Z3VvY29uZ2NvbmdAc2RmbXUuZWR1LmNu
Specialty Section: This article was submitted to Pediatric Nephrology, a section of the journal Frontiers in Pediatrics
Abbreviations SLC4A1, solute carrier family 4, member 1; SLC4, solute carrier family 4; dRTA, distal renal tubular acidosis; rhGH, recombinant human growth hormone; AE1, anion exchanger member 1; eAE1, erythroid isoform of AE1; kAE1, kidney anion exchanger 1; AD, autosomal dominant; AR, autosomal recessive; SAO, Southeast Asian ovalocytosis; HS, hereditary spherocytosis.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.