 Kaoru Okazaki
Kaoru Okazaki Shinji Nakamura2
Shinji Nakamura2 Masatoshi Kondo
Masatoshi Kondo Takashi Kusaka
Takashi Kusaka- 1Department of Neonatology, Tokyo Metropolitan Children's Medical Center, Tokyo, Japan
- 2Department of Pediatrics, Faculty of Medicine, Kagawa University, Kagawa, Japan
- 3Maternal Perinatal Center, Faculty of Medicine, Kagawa University, Kagawa, Japan
Neonatologists resuscitate asphyxiated neonates by every available means, including positive ventilation, oxygen therapy, and drugs. Asphyxiated neonates sometimes present symptoms that mimic those of inflammation, such as fever and edema. The main pathophysiology of the asphyxia is inflammation caused by hypoxic-ischemic reperfusion. At birth or in the perinatal period, neonates may suffer several, hypoxic insults, which can activate inflammatory cells and inflammatory mediator production leading to the release of larger quantities of reactive oxygen species (ROS). This in turn triggers the production of oxygen stress-induced high mobility group box-1 (HMGB-1), an endogenous damage-associated molecular patterns (DAMPs) protein bound to toll-like receptor (TLR) -4, which activates nuclear factor-kappa B (NF-κB), resulting in the production of excess inflammatory mediators. ROS and inflammatory mediators are produced not only in activated inflammatory cells but also in non-immune cells, such as endothelial cells. Hypothermia inhibits pro-inflammatory mediators. A combination therapy of hypothermia and medications, such as erythropoietin and melatonin, is attracting attention now. These medications have both anti-oxidant and anti-inflammatory effects. As the inflammatory response and oxidative stress play a critical role in the pathophysiology of neonatal asphyxia, these drugs may contribute to improving patient outcomes.
1. Introduction
Neonatal asphyxia (NA) is caused by respiratory and/or circulatory failure in utero or at birth, as may be seen in uterine rupture, placental abruptio, and umbilical cord prolapse (1). Thus, the pathophysiology of NA is associated with ischemic reperfusion (IR) injuries, which cause oxidative stress and excessive systemic inflammatory response syndrome (SIRS). First, hypoxia-ischemia decreases the oxygen supply and depletes adenosine triphosphate to cause primary injuries, such as cell swelling or death, in addition to oxidative stress. Second, reperfusion causes latent injuries, such as pro-apoptotic signaling, N-methyl-D-aspartate receptor hyperexcitability, inflammation, and the production of oxygen-related species.
Neonatal asphyxia is caused by respiratory and/or circulatory failure in utero or at birth, as may be seen in uterine rupture, placental abruptio, and umbilical cord prolapse (1). Thus, the pathophysiology of neonatal asphyxia is associated with ischemic reperfusion injuries, which cause oxidative stress. First, hypoxia-ischemia decreases the oxygen supply and depletes adenosine triphosphate to cause primary injuries, such as cell swelling or death, in addition to oxidative stress. Second, reperfusion causes latent injuries, such as pro-apoptotic signaling, N-methyl-D-aspartate receptor hyperexcitability, inflammation, and the production of oxygen-related species.
Hypoxia-ischemia in neonatal asphyxia is resolved using oxygen therapy, positive ventilation, chest compression or drugs. However, a sudden increase in the supply of oxygen and blood can exacerbate oxidative stress. The secondary energy failure then causes more severe neuronal dysfunction via mitochondrial collapse, cell death, seizure, inflammation, and oxidative stress. The latter two are closely interlinked during neonatal asphyxia. The main role of inflammation is to protect tissues and organs from injuries and to prevent unfavorable factors from flowing into tissue locally or systemically via thromboembolisms caused by hypercoagulation. Inflammatory immune cells, such as leukocytes, immediately infiltrate the damaged region, strengthen inflammation, remove the stimulus, and repair the tissue (2). Inflammation is characterized by five signs, including pain, heat, redness, swelling, and loss of function. Asphyxiated neonates sometimes present symptoms that mimic those of inflammation, such as fever and edema. Hypoxia is the main cause of inflammation. A general, inflammatory, defensive reaction to hypoxia or ischemia-reperfusion damage incurred during delivery is associated with neonatal asphyxia. In the present review, we describe the relationship between inflammatory mediators and oxidative stress in neonatal asphyxia.
2. Oxidative stress due to reactive oxygen species (ROS) in neonatal asphyxia
The environment of the neonatal brain conduces to Reactive oxygen species (ROS) production. The neonatal brain includes plenty of lipids and iron (Fe2+) (3). These lipids, which are necessary for myelination, may generate free radicals via peroxidation. Iron is associated not only with growth and development, but also with the generation of ROS, such as hydroxyl radicals, through the Fenton reaction. Moreover, both lipids and iron are related to ferroptosis, an iron-dependent, non-apoptotic form of cell death (4, 5). The neonatal brain is more sensitive to ROS than the brain of older individuals.
ROS, including the superoxide anion (•O2−), hydrogen peroxide (H2O2), and hydroxyl radical (•OH), play a central role in oxidative stress. In particular, mitochondrial ROS (mit-ROS) is associated with neonatal brain injury caused by hypoxia-ischemia and hypoglycemia (6–8). The mitochondria are a major source of ischemic reperfusion-induced ROS but stores a large amount of superoxide dismutase (SOD), an important ROS scavenger. Reactive nitrogen species (RNS), such as nitric oxide (NO) and peroxynitrite (ONOO−), are also types of oxygen-related species implicated in neonatal asphyxia. Previous studies reported an association between serum or cerebrospinal fluid (CSF) NO and neonatal asphyxia although the findings are controversial (9–11). NO is generated by activated immune cells, such as neutrophils and macrophages, during an inflammatory reaction (12). ONOO−, which is generated by a reaction between NO and •O2−, leads to protein oxidation, lipid peroxidation, and DNA damage (3). Indeed, plasma nitroalbumin, a potential biological marker of nitrating species generation or nitrative stress, was found to increase significantly in neonates with moderate to severe neonatal encephalopathy a day after birth (13). Thus, nitrative stress is associated with the inflammatory reaction seen in neonatal asphyxia.
Unfortunately, the SOD function in fetuses and neonates is weak. For example, copper and zinc (Cu, Zn)-SOD activity in the cerebral cortex and white matter was remarkably lower in fetuses and neonates than in adults (15% and 50% of the adult value, respectively) because SOD activity increases with increasing gestational age (14–16). On the other hand, birth suddenly imposes considerable oxidative stress on neonates as they transition from a hypoxic uterine environment to normal ambient air. The imbalance between the increased ROS production and weak detoxification ability may cause further oxidative injury to the tissues and organs of neonates at birth. Bilirubin stimulates SOD gene expression but it is insufficient to prevent oxidative stress completely (17). SOD also attenuates cytokine-related reactions in endothelial cells, which play an important role in inflammatory reactions, including neonatal asphyxia. Thus, SOD may be strongly associated with the inhibition of the inflammatory reaction in neonatal asphyxia.
3. Inflammation and inflammatory mediators in neonatal asphyxia
Table 1 shows the relationship between neonatal asphyxia and inflammatory mediators. Excessive inflammation exacerbates tissue/organ damage through gene expression and proteins. Elevated cytokines activate various inflammatory immune cells via receptors, including toll-like receptors (TLRs), further increasing the production of cytokines and other inflammatory mediators. For instance, the concentration of cytokines, such as interleukin (IL)-1β, IL-6, IL-8, and tumor necrosis factor (TNF)-α, increased to a significantly higher degree in the serum and CSF of asphyxiated neonates than healthy neonates (18–21). Thus, neonatal asphyxia can trigger more potent inflammation as part of a defensive reaction.

Table 1. Temporal alteration of inflammatory meiators in asphyxiated neonates.
3.1. IL-6
IL-6 is associated with endothelial cell dysfunction. Increased IL-6 changes endothelial permeability, exacerbating edema of brain or general in asphyxiated neonates (22). Moreover, IL-6 deficiency protects against angiotensin II-induced endothelial dysfunction (23). The IL-6/IL-6 receptor reaction may activate signal transducer and transcription 3 activator (STAT3). In endothelial cells, the IL-6/STAT3 pathway promotes xanthine oxidase, resulting in increased ROS production. In addition, inhibitors of xanthine oxidase or SOD decrease neutrophil recruitment in tissues with hypoxic injury and neutrophil adhesion to endothelial cells (24, 25), with the resulting decrease in IL-6 suppressing the inflammation. IL-6 is also associated with fetal growth retardation (FGR). An in vitro study demonstrated that the IL-6/STAT3 signaling pathway induced premature cellular senescence via ROS and DNA damage in human fibroblasts (26). IL-6 may cause placental cell senescence, which is associated with detrimental effects, including FGR, preterm spontaneous labor or preterm (27). Chronic stimulation of cells by cytokines causes enough stress to arrest growth or induce senescence. Thus, prolonged hypoxia during the fetal period may contribute to developing FGR via IL-6 pathway.
IL-6 has neuroprotective qualities (28) and is associated with manganese (Mn)-SOD, which is encoded by the Mn-SOD gene and localizes in the mitochondrial matrix (29). Mn-SOD removes superoxide anions and ROS (28, 30, 31) while SOD plays a role in controlling the concentration of O2- produced by xanthine oxidase, NADPH oxidase (Nox), and the mitochondria (32). IL-6 binding to IL-6R (forming the IL-6/IL-6R complex) acts as an antioxidant by regulating STAT3-mediated Mn-SOD gene expression although ischemic reperfusion blocks the formation of this compound via decreased IL-6R (33). However, a previous study demonstrated that IL-6 therapy restored IL-6R and the IL-6/IL-6R compound, increasing STAT3-mediated Mn-SOD gene expression in a murine model (28). Thus, IL-6 plays a role in protecting neonates from ROS via Mn-SOD although a porcine model with FGR had significantly decreased gene expression of Mn-SOD (34). Asphyxiated neonates sometimes show persistent increases in serum CRP within the few first weeks after birth (35–37). IL-6 in these neonates may be produced as a neuroprotective factor, resulting in the persistent increase of IL-6-induced CRP. Thus IL-6 has not only adverse, but also beneficial, effects.
Of all the cytokines, IL-6 has perhaps been the most extensively investigated. Many clinical studies have addressed the relationship between the outcomes of neonatal asphyxia and the concentration of mediators in umbilical cord blood, neonatal blood, and cerebrospinal fluid (CSF) (38–42). A high IL-6 level predicts a poor outcome, such as death or abnormal neurodevelopment. The IL-6 level in neonates with hypoxic-ischemic encephalopathy (HIE) was over 300-fold higher than in non-HIE neonates (38). Serum IL-6 was found to be elevated in neonates with severe asphyxia or meconium aspiration syndrome (20, 43). In asphyxiated neonates, IL-6 production may be triggered by hypoxia, cytokines or ox-LDL (44). ROS also acts as a signaling element to regulate the secretion of proinflammatory cytokines (22),. The IL-6 CSF/plasma ratio in neonates with HIE was higher than in neonates with sepsis or normal neonates (45), suggesting that IL-6 might be produced in the central nervous system, then extravasate into circulating blood. Moreover, the CSF IL-6 level may more accurately predict outcomes than the serum or plasma IL-6 level. However, the biomarkers require patients to be in stable condition and the sampling method to be easy to perform repeatedly. CSF sampling is unsuitable for unstable asphyxiated neonates because it is invasive. On the other hand, the serum or plasma IL-6 level correlates well with outcomes, such as death, HIE severity, neurodevelopment, and the Bayley scale (38–40, 45). The IL-6 concentration in both blood and CSF strongly correlated with outcomes in asphyxiated neonates. Moreover, serum and plasma IL-6 values may reflect the CSF IL-6 value.
3.2. Nuclear factor-kappa B (NF-κB)
NF-κB forms a family of transcription factors that play an essential role in the immune inflammatory response. The complex, formed by NF-κB and inhibitor κB (IκB), has an inactive form in cytosol. Oxidative stress activates IκB kinase (IKK) via TNF-α, IL-1 or ROS (46). The activated IKK then decomposes IκBα, resulting in decomposition of the complex and translocation of NF-κB into the nucleus where it combines with the DNA to activate the NF-κB pathway. ROS in oxidative stress activate inflammatory cells via TLR-4 mediated NF-κB activation (47). Hypoxia-induced-IL-1β also increases the gene expression of NF-κB. Thus, both ROS and hypoxia can activate the NF-κB pathway. The activated NF-κB pathway in turn increases the gene expression of inflammatory mediators and regulates RNS, including iNOS synthesis and NO production, in the neurons of the hippocampus and cortex (48, 49).
One major role of NF-κB is to enhance the inflammatory response by increasing the gene expression of inflammatory mediators, such as TNF-α, IL-1β, and IL-6. ROS-induced oxidative stress activated NF-κB in neurons, astrocytes, microglia, and endothelial cells (50, 51). On the other hand, its activation possibly indicates the mechanism underlying the neuroprotective effect in a murine model of ischemic brain overexpressing Cu,Zn-SOD (52). NF-κB acts not only as a protein transcription factor but also as a regulator of innate immunity via toll-like receptors (TLRs) (53, 54). NF-κB may attenuate ROS to promote cellular survival. Thus, increased expression of NF-κB may not always be detrimental to neonates.
Resuscitation with pure oxygen increased NF-κB activation in a murine model (55). In vitro, the NF-κB activation level of peripheral blood mononuclear cells in asphyxiated neonates with neurological sequelae was higher than in non-asphyxiated neonates (56). Indeed, NF-κB orchestrates inflammatory responses. Serum high- mobility group box 1 (HMGB1) also increased in neonatal asphyxia (57). HMGB1, a damage-associated molecular patterns (DAMPs) protein, binds to TLR-4, activating the nuclear factor kappa B (NF-κB) pathway, resulting in an enormous increase in inflammatory mediators (58). However, HMGB1 also play a role in brain development during fetus (59). HMGB1 may act as a mediator during brain development and brain damage. In addition, cell-penetrating, anti-NF-κB peptide treatments administered intranasally attenuated NF-κB signaling, microglia activation, and brain damage in a neonatal, infection-sensitized, HI animal model (60). Anti-NF-κB peptide treatments can regulate robust inflammatory responses.
3.3. Hypoxia-inducible factor (HIF) -1
HIF-1, a heterodimer composed of the inducible HIF-1α and constitutive HIF-1β subunits, is tightly regulated by cellular oxygen tension and is essential to surmounting the hypoxic environment in neonates. Hypoxia increases the expression of HIF-1 (61). Increased HIF-1 is associated with ROS production via NOS or Nox. HIF-1-induced NOS produces small amounts of ROS in physiological cell signaling. During inflammation, the increase in HIF-1 induces NOS and reduces arginine, leading to increased ONOO− and O2- production (NOS uncoupling). In addition, HIF-1α promotes the production and activation of Nox, an enzymatic factor in ROS production, which includes Nox-1 to 5 and dual oxidase-1 and -2 (62–64). HIF-1-induced Nox promotes ROS production by transferring electrons from NADPH to oxygen. The resulting increase in Nox-derived ROS, then further promotes HIF-1α production (65, 66). Ischemia also activates Nox in immune as well as non-immune cells, such as neurons, vascular endothelial cells, and microglia. In severe hypoxia, accumulated HIF-1α causes necrosis and apoptosis (67) with calcium and calpain and exacerbates cerebral edema by increasing the permeability of the blood-brain barrier (BBB) (67). Movement of the activated immune cells through the BBB promotes an inflammatory immune reaction in the brain involving ROS and cytokines. Thus, HIF-1 may exacerbate neonatal asphyxia by further increasing both ROS and NOS production.
On the other hand, HIF-1 may protect the fetus from hypoxic injury (61). HIF-1α expression is increased in fetuses with physiological hypoxia and is highest in the brain, decreasing slightly with increasing gestational age (68). HIF-1α also increases erythropoietin (EPO) and vascular endothelial growth factor (VEGF). These promote the development of the brain and other organs in the fetus (69). HIF-1 preconditioning before or at birth may help to attenuate hypoxia-related injuries (69, 70). Hypoxia induced mit-ROS inhibits HIF-1α hydroxylation, resulting in HIF-1α accumulation (71). In a postnatal hypoxic state, such as neonatal asphyxia, accumulated HIF-1α binds to HIF-1β subunits, forming a complex that then binds to hypoxia-response elements (HRE) and activates more than 100 genes which help the neonate adapt to the hypoxic environment (61, 72, 73).
Serum HIF-1α was found to increase under conditions of severe asphyxia in accordance with the severity (74). HIF-1α related proteins, such as microRNA (miRNA)-373 (HIF-1α dependent miRNA) and VEGF, also increased in neonatal HIE (74). Downregulation of HIF-1α increased cellular apoptosis in a neonatal HIE rat model (75). Prolonging the HIF-EPO signaling pathway in vitro was associated with attenuation of neuronal apoptosis during therapeutic hypothermia (76). Thus, HIF-1 does not always have an adverse effect. Indeed, HIF-1 may increase blood circulation in the brain via production of EPO and VEGF, which has an anti-apoptotic and neovascularizing effect, respectively (67). However, serum EPO was higher in HIE neonates than in non-HIE neonates (77) and was associated with poor outcomes, including death and abnormal MRI findings (77). Serum VEGF was lower in neonates with moderate to severe HIE receiving therapeutic hypothermia (77, 78). Whether increased HIF-1 is associated with a good or poor outcome in neonatal asphyxia is still moot although the answer may depend on the HIF-1 concentration involved.
3.4. Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1)
Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) is thought to be a biomarker of several diseases, including vascular inflammation and atherosclerosis. LOX-1 is expressed on the surface of the vessel wall cells as a scavenger receptor that mediates the cellular effects of oxidized low-density lipoprotein (OxLDL), resulting in ROS generation (79–81). LOX-1 increases with an increase in OxLDL, free radicals, proinflammatory cytokines, high glucose, and endothelial cell dysfunction and plays a prominent role in neonatal asphyxia (81). Thus, LOX-1 may be a useful biomarker in asphyxiated neonates. Recently, Akamatsu et al. demonstrated that serum LOX-1 was significantly higher in moderately and severely asphyxiated neonates than in normal neonates (82). Ox-LDL/LOX-1 markedly increased VEGF mRNA expression in vitro (83). In addition, both ROS and ischemic reperfusion induced LOX-1 in endothelial cells and microglia (79, 84, 85). LOX-1 regulates microglial activation in neonatal HIE (86) and is also associated with ferroptosis, an iron-dependent form of cell death (4, 5), which may be involved in neonatal hypoxic-ischemic brain damage (4, 87). A high LOX-1 level contributes to neuronal apoptosis, breakdown of the BBB, and increased inflammatory mediators and ROS (88–90). Indeed, anti-LOX-1 therapy was found to decrease oxidative stress and inflammation (91, 92); the drug used in the study has the potential to become a novel therapy for neonatal asphyxia.
3.5. Toll-like receptors (TLRs)
TLRs, or transmembrane proteins, are a crucial pattern recognition receptor on the surface of both immune cells (e.g., macrophages, neutrophils) and non-immune cells (e.g., cardiac myocytes, hepatocytes, vascular smooth muscle cells) (93). TLR signaling can activate the innate immune system, inducing the initial inflammatory response. Endogenous ligands to TLR include proteins which are actively or passively released by cells. DAMPs, which are potent endogenous ligands and include HMGB1 and heat shock proteins, are an SOS signal transmitted by various cells during hypoxia (94). After tissue injury, they are released extracellularly to activate the immune system and result in a pro-inflammatory cascade. DAMPs activate TLR-2 and TLR-4 signaling after oxidative stress, leading to neuronal apoptosis and ROS production (95).
TLRs are present in both glial cells and neurons and are associated with the pathophysiology of neonatal hypoxia-ischemia. Hypoxia-ischemia increased the expression of TLRs, such as TLR-1, TLR-2, TLR-4, and TLR-7, but decreased TLR-5 and TLR-8 expression in neonatal mouse brains (96). HIF-1α bound to the promoter region of TLR-4 during hypoxia, increasing TLR-4 expression on the surface of macrophages, which peaked at eight hours after hypoxic exposure in vitro (97, 98). Furthermore, HIF-1α activated the NF-κB pathway via TLR-4 on the surface of microglia during hypoxia, leading to the production of inflammatory mediators, such as TNF-α, IL-1β, and inducible NOS (99). These mediators injure oligodendrocytes and neurons in HIE. In addition, TLR-2 and TLR-4 deficient mice had a smaller infarct size and decreased reperfusion-induced ROS production and leukocyte infiltration (100, 101). Bilirubin scavenged ROS produced by TLR-4-dependent activation of Nox, inhibiting the up-regulation of iNOS (102, 103). In an ovine model of asphyxia, TLR-2 and TLR-4 expression in the cortex and subcortex increased to a significantly higher degree after resuscitation with 100% oxygen than with 21% oxygen (104). ROS then activated inflammatory cells via TLR-4-mediated NF-κB activation (47). In asphyxiated neonates, LPS-induced TLR-4 expression in neutrophils increased on day 3 after a hypoxic event (105).
4. Immune cells related to inflammation in neonatal asphyxia
4.1. Neutrophils
Neutrophils play a prominent role in oxidative tissue/organ injury. Primed neutrophils produce ROS and inflammatory mediators. ROS regulates both apoptotic and necrotic cell death depending on the severity of the oxidative stress (106). First, neutrophils are primed by inflammatory mediators, such as chemokines. The priming times in neutrophils depend on the kind of inflammatory mediator involved. For instance, platelet-activating factor (PAF) primes neutrophils transiently, but granulocyte-macrophage colony-stimulating factor (GM-CSF) prolongs the priming process (107). The PAF level in the CSF of asphyxiated neonates was found to be abnormally high but decreased after head-cooling (107). Serum GM-CSF was significantly higher in neonates with moderate to severe brain injury than in those with mild or no injury 96 h after birth (108). GM-CSF not only extends the preservation of the primed state of neutrophils but also delays neutrophil apoptosis by increasing the stability of Mcl-1 (an anti-apoptosis protein) (109, 110). On consequence, GM-CSF may prolong the production of ROS generated by neutrophils, thereby enhancing tissue injury. Indeed, a high serum GM-CSF level is related to abnormal cranial MRI findings and developmental delay at age 2 years (19). In addition, primed, circulating neutrophils move into sites of hypoxic injury where they accumulate.
Inflammatory mediators, such as IL-6, IL-8, and TNF-α, also have an important role in immune cell extravasation. Neutrophils and other inflammatory cells produce ROS at hypoxic injury sites in addition to secreting inflammatory mediators (111, 112). Moreover, inflammatory mediators may promote transmigration of neutrophils over endothelial cells and the BBB, prolong cell survival, and increase degranulation (113, 114). Interestingly, the accumulated time course of inflammatory cells was consistent with increased ROS generation and tissue/organ injury (115). In particular, IL-8 is a potent chemokine which enhances neutrophil functions in inflammatory reactions, including their migration from circulating blood, chemotaxis to inflammation sites, activation in areas of inflammation, and priming of immune cells (116). Increased IL-8 in the serum and cerebrospinal fluid of asphyxiated neonates was associated with poor outcomes (19, 20). IL-8 does not directly activate Nox but rather phosphorylates Nox components and moves to the lipid rafts of neutrophils, leading to extracellular ROS generation (109, 117).
4.2. Endothelial cells
The collapse of endothelial cell function directly induces an accumulation of inflammatory cells in extravascular, hypoxic injury sites. Immediately after ischemia, small amounts of ROS, such as xanthine oxidase-derived superoxide anion, cause endothelial cell barrier dysfunction (increased permeability) and increase adhesion molecule expression on the surface of endothelial cells and neutrophils. This promotes the extravasation of circulating immune cells. ROS further promotes the gene expression of proinflammatory proteins (e.g., cytokines and adhesion molecules) in endothelial cells and the BBB (118) and triggers an increase in matrix metalloproteinases downstream, causing a breakdown in the BBB (119). SOD mimetics reduced production of pro-inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6) as well as of cell adhesion molecule expression on endothelial cells, decreasing neutrophil extravasation (120–122). In endothelial cells, SOD is associated with decreased ROS production and cell adhesion molecule expression via cytokine-related processes.
Adhesion molecules on the endothelial cell surface are important for enabling the migration of inflammatory cells to affected areas. IL-6 induces the expression of adhesion molecules, including intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1, and e-selectin. Further, oxidative stress and RAS-related C3 botulinus toxin substrate-1 (Rac1) activation increased ICAM-1 expression via the IL-6/STAT3 pathway in endothelial cells, increasing inflammatory cell infiltration (123). On the other hand, IL-6 can directly reduce eNOS expression (124). IL-6 dose-dependently reduced eNOS expression in cultured endothelial cells in vitro (125) and suppressed the IL-6/STAT3 pathway, resulting in decreased ICAM-1 expression (126). In addition, ROS promotes coagulation in endothelial cells although NO inhibits platelet aggregation (127). VCAM-1 is expressed on the endothelial cell surface and promotes neutrophil transmigration; this priming produces ROS. On the other hand, cross-linking of VCAM-1 activates calcium flux and Rac-1 in endothelial cells (128), which in turn activates Nox and results in ROS generation. The VCAM-1 signaling via ROS mediates changes in actin in endothelial cells and leads to the creation of open intercellular passageways via changes in endothelial cell structure (129), enabling many neutrophils promptly to transmigrate to sites of injury. Antioxidants, such as bilirubin and vitamin E, were found to inhibit VCAM-1-dependent neutrophil recruitment both in vivo and in vitro (130). VCAM-1 blockade inhibited leukocyte recruitment and IR injuries in the liver (129, 131).
5. Treatment
5.1. Therapeutic hypothermia
Therapeutic hypothermia, including cooling of the body or head, influences inflammatory mediator levels in neonatal asphyxia (Table 2). Asphyxiated neonates receiving therapeutic hypothermia demonstrated a temporary increase in serum cytokines [e.g., IL-6, IL-8, IL-10, granulocyte-colony stimulating factor, monocyte chemoattractant protein (MCP)-1] and a temporary decrease in serum cytokines [e.g., VEGF, macrophage inflammatory proteins (MIP-1α)] (78, 132). Thus, hypothermia can mitigate the inflammatory response in neonatal asphyxia.
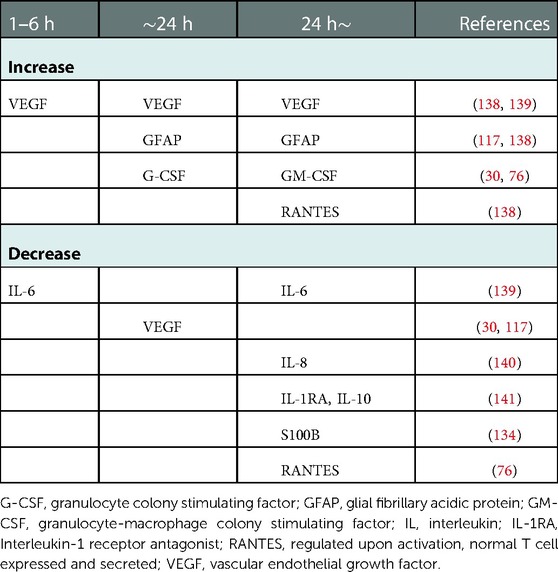
Table 2. Temporary changes in inflammatory mediators in asphyxiated neonates treated with therapeutic hypothermia.
Serum granulocyte colony stimulating factor (G-CSF) markedly increased and remained high in severe neonatal asphyxia treated with head cooling (78). G-CSF may mediate anti-apoptosis in neurons (133, 134), and high enough quantities may prevent excessive neuronal death due to hypoxia in severe neonatal asphyxia (135). Brain edema, inflammation, and disruption of the BBB were also mitigated by G-CSF administration (133). On the other hand, VEGF, an HIF-1 related mediator, decreased and remained low (78, 136). VEGF acts as an enhancer of BBB permeability and may cause cerebral edema (137); a high VEGF level may thus exacerbate pre-existing edema (137). Lowering VEGF in severely asphyxiated neonates using head cooling might prevent cerebral edema even in the presence of severe hypoxia. However, decreased VEGF is reportedly associated with poor outcomes, including death and abnormal MRI findings (77). VEGF may be a key mediator, and further research is needed to clarify its association with neonatal asphyxia.
5.2. Erythropoietin
EPO, an HIF-1 related mediator, has anti-apoptotic, anti-inflammatory, anti-oxidative, and anti-excitotoxic effects. Its therapeutic efficacy is currently being debated; previous studies have investigated the anti-apoptotic effect of EPO during hypothermia, and other, similar studies are currently being conducted (11, 138–141). However, a recent, multicentric, double-blind, randomized, placebo-controlled trial found that EPO had no significant effect on mortality or neurodevelopmental impairment in a large cohort of asphyxiated neonates and was in fact associated with a high rate of serious adverse events (142). Further studies on the safety, effects, and viability of EPO therapy are clearly needed. At present, many studies aiming to develop medications for use with hypothermia to treat neonatal asphyxia are being conducted (Table 3).
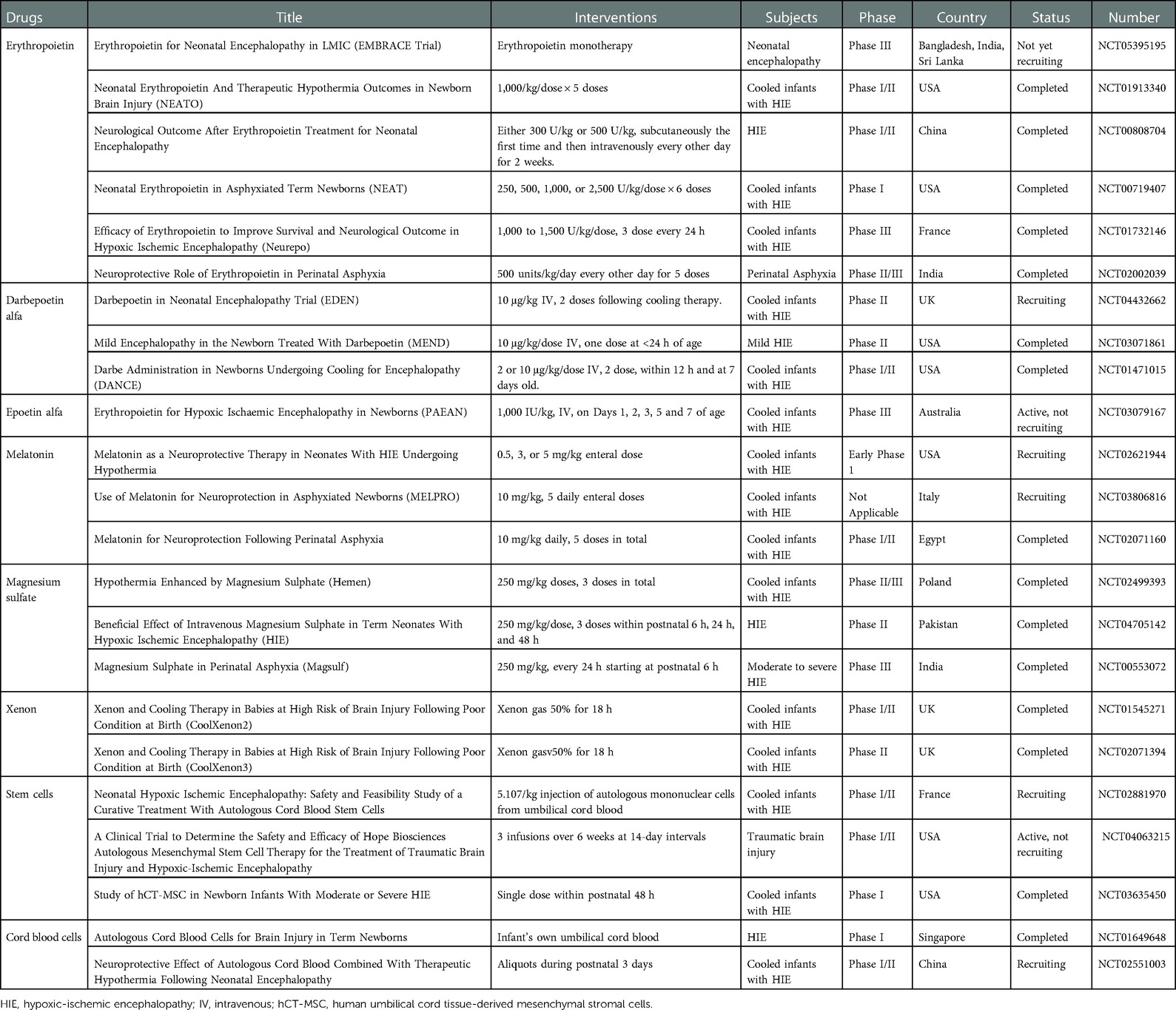
Table 3. Unreported or ongoing clinical trial of pharmacotherapy for neonatal asphyxia.
5.3. Melatonin
Melatonin has potential as a treatment for NA in combination with therapeutic hypothermia. Melatonin acts as an antioxidant, anti-inflammatory mediator, and antiapoptotic agent (143); it not only directly neutralizes ROS and RNS, but also reduces NF-κB translocation and proinflammatory cytokine production (144). Melatonin can also freely cross the placenta and BBB because of its low molecular weight (232.28 g/mol). A pharmacokinetic study of enterally administered melatonin in neonates treated with hypothermia (33.5°C) showed that its half-life (mean 50.9 h) and clearance (mean 0.046 L/h/kg) were greater while its distribution volume (mean 1.80 L/kg) was smaller than in adults (145). Moreover, melatonin is safe. A randomized trial of a combination of melatonin and therapeutic hypothermia reduced oxidative stress and improved neurodevelopmental outcomes in neonates with moderate to severe HIE at age 6 months (145). Thus, melatonin has the potential to be a viable treatment for NA.
5.4. Magnesium sulfate
Magnesium has immunomodulatory activity (146). Therefore, magnesium sulfate was examined for its potential to improve outcomes in neonatal asphyxia by inhibiting cellular calcium influx and excitatory amino acid release in neurons via blockade of the NMDA-receptor channel, which has a magnesium-dependent calcium gate (147, 148). Magnesium deficiency opens the NMDA-receptor channel and leads to an influx of calcium. The increased, intracellular calcium ions stimulate production of substance P, which promotes pro-inflammatory cytokines (149), which prime phagocytes, such as macrophages, and increase ROS production (150). Thus, magnesium deficiency is linked to both increased inflammation and oxidative stress (150). On the other hand, magnesium administration rapidly increased the intracellular magnesium level (146), leading to an increased intracellular IκBα concentration, which inhibited NF-κB activation (146). Consequently, magnesium may decrease inflammation and oxidative stress via reduced NF-κB. Interestingly, it was reported that suppressing the NMDA-receptor via magnesium blockade was essential to forming long-term memory in Drosophila (151). Magnesium therapy may inhibit or delay ischemic cell death and decrease neurodevelopmental deficits. A previous RCT showed that magnesium improved short-term outcomes, such as cranial imaging findings, oral feeding establishment, and neurological outcomes at discharge (152, 153). A few RCT also reported that postnatal magnesium sulfate improved developmental outcomes, but the studies did not enroll a large cohort (154). A recent review and meta-analysis failed to find any long-term benefits but found instead an increased trend in mortality in patients receiving magnesium (155, 156). Research into the effect of the combination therapy consisting of hypothermia and magnesium sulfate in neonates with HIE is still ongoing, but results indicating favorable, long-term outcomes are expected like other treatments (Table 3).
5.5. Xenon
Xenon, a noble gas, was first used as a general anesthetic with few adverse effects (157). Xenon's neuro-protective qualities attracted a great deal of attention (158). The molecular mechanism of Xenon's activity involves inhibiting the NMDA receptor, stabilizing the membrane potential, inhibiting Bax-induced apoptosis, increasing the anti-apoptosis genes, Bcl-x-L and Bcl 2, and inhibiting activated inflammatory cells (e.g., macrophage) (159). Many preclinical studies using animal models reported that a combination of hypothermia and inhaled xenon had neuroprotective effects in neonatal HIE (160–162). However, in the sole human study to date, inhaled Xenon failed to improve clinical signs and MRI findings (TOBY-Xe) (163). One problem with this study was that xenon inhalation was begun after postnatal hour 6, which is outside the therapeutic window. This delay may have affected the results. In the future, more human studies are needed to assess the effects of inhaled xenon within the first six hours of life.
5.6. Hydrogen (H2)
Hydrogen gas (H2), an expected novel therapeutic agent, selectively reduces •OH as a radical scavenger (164). In addition, H2 decreases inflammatory cytokines, including TNF-α and NF-κB, as an anti-inflammatory agent (165, 166) and induces anti-apoptotic molecules as an anti-apoptotic agent (167). Thus, H2 may be associated with preventing lung or brain from ROS or inflammations. Indeed, H2 prevented brain injuries in an adult rat model of ischemic reperfusion (168). The combination between H2 inhalation and hypothermia (33.5 ± 0.5 °C) in a neonatal pig model of hypoxia-ischemia improved short-term neurological outcomes, such as walking (169). Moreover, it is suitable that the cost of H2 is lower though that of Xenon is higher for using in clinical practices. Thus, the clinical use of H2 is expected as a novel therapy for neonatal HIE. However, there is still no human studies in neonates though some human studies are conducted in adults, such as coronavirus disease 2019 (COVID-19) (NCT04378712).
5.7. Stem cell therapy
Stem cells for neonatal therapy can be derived from autologous umbilical cord blood (umbilical cord blood and Wharton's jelly) or amniotic fluid. There are many kinds of stem cells, such as mesenchymal stem cells (MSCs) and hematopoietic stem cells. These stem cells are capable not only of multipotent differentiation, but also of inhibiting inflammation and oxidative stress, reducing apoptosis, and decreasing mitochondrial dysfunction (170). MSCs, which are commonly used in neonatal animal HIE studies, are characterized by stronger resistance to oxidative stress-induced death and higher activity of both SOD and catalase, which result in reducing oxidative stress (171). MSCs also decrease proinflammatory factors, secrete anti-inflammatory factors, such as IL-10, and inhibit the proliferation and activation of inflammatory cells in the injured cortex (172, 173). Interestingly, MSCs rescued neurons and endothelial cells from dysfunction and apoptosis through tunneling nanotube-mediated mitochondrial transfer (174, 175). In addition, MSCs increased the number of neurons in the hippocampus and improved neurogenic and developmental outcomes, such as cognitive, behavioral, and sensorimotor functions (176–178). In neonatal animal models, other stem cells also demonstrated benefits. For instance, hematopoietic stem cells increased brain blood flow and the vessel diameter, and endothelial progenitor cells decreased neuronal inflammation and cell apoptosis (179, 180).
Indeed, many preclinical studies have demonstrated the effects of stem cell therapy. Phase I studies proved the feasibility and safety of stem cell therapy (181–183). Stem cell therapy for neonatal HIE is expected to confer antioxidant, anti-inflammatory, and anti-apoptosis effects. However, at present, no human study has demonstrated the efficacy of stem cell therapy for neonatal HIE (184). Engraftment of stem cells to sites of injury can induce multipotent differentiation and paracrine growth or anti-inflammatory factors. However, MSCs engraftment has not yet been successful (185). The results of many, currently ongoing clinical trials using MSCs, muse cells, and other cell types await publication (184, 186).
6. Summary
In NA, inflammatory mediators may promote ROS production and enhance ROS function (Figure 1). ROS can also promote inflammatory mediator production and activate inflammatory cells. Therefore, a novel therapy capable of regulating ROS production and controlling the inflammatory response is much to be desired. However, measuring temporary changes in inflammatory mediator levels soon after birth is challenging. First, the precise timing of NA onset in utero cannot be determined although inflammatory mediators may demonstrate drastic, albeit temporary, changes in quantity depending on the time from NA onset. Second, cytokines have pleiotropy and redundancy. However, we can know the timing of hypothermia therapy start. In the future, investigating temporary fluctuations in the level of inflammatory mediators from the start time may lead to the development of a novel, effective therapy.
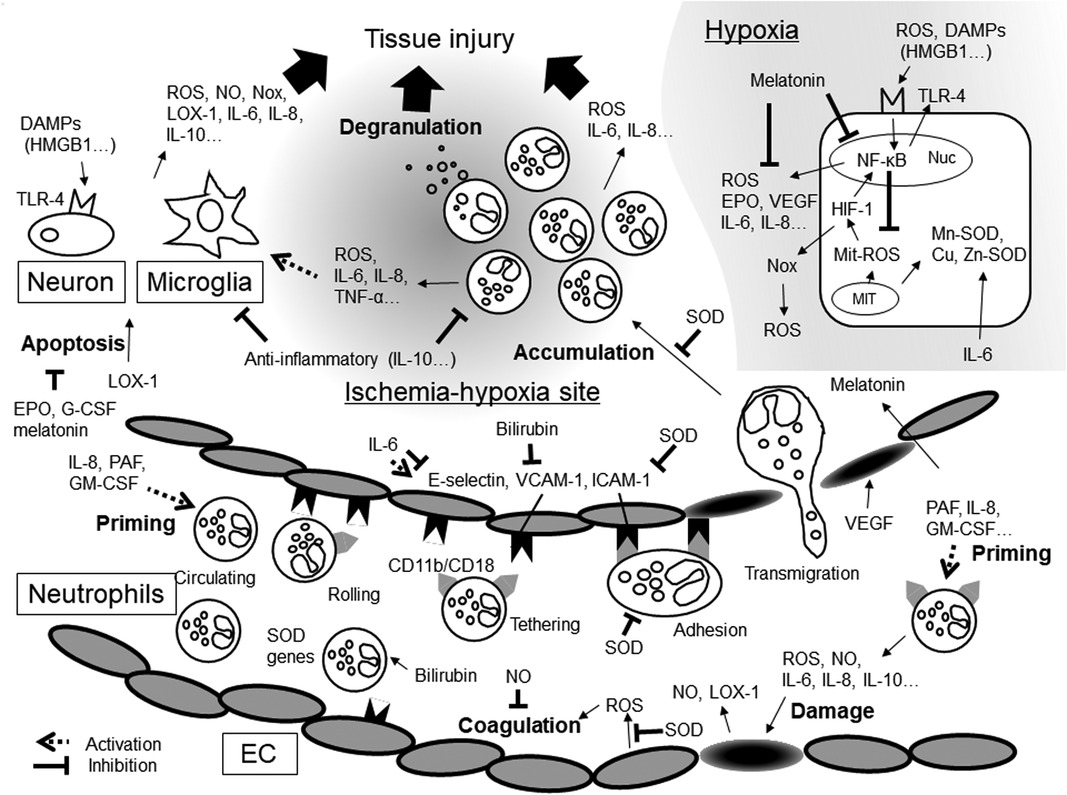
Figure 1. Inflammatory mediators and oxidative stresses in hypoxic-ischemia. DAMPs, damage-associated molecular patterns; EC, endothelial cells; EPO, erythropoietin; G-CSF, granulocyte-macrophage colony stimulating factor; GM-CSF, granulocyte-macrophage colony stimulating factor; HIF-1, hypoxia-inducible factors-1; HMGB1, high- mobility group box 1; ICAM-1, intercellular adhesion molecule; IL, interleukin; LOX-1, lectin-like oxidized low-density lipoprotein receptor-1; MIT, mitochondria; NF-κB, Nuclear factor-kappa B; NO, nitric oxide; Nox, NADPH oxidase; Nuc, nuclear cell; PAF, platelet-activating factor; ROS, reactive oxygen species; SOD, superoxide dismutase; TLR-4, Toll-like receptors-4; TNF-α, tumor necrosis factor-α; VCAM-1, vascular cell adhesion molecule; VEGF, vascular endothelial growth factor.
Author contributions
KO and TK contributed equally to this work. KO and TK substantially contributed to its conceptualization. SN, KK, YK, and MK contributed to writing the draft. All the authors critically reviewed and revised the manuscript and approved the final version for submission.
Funding
This study was financially supported by JSPS KAKENHI Grants-in-Aid Nos. 19K08253, 19K08349, 21K02709, 22K07822, and 20H00102. The sponsors had no role in the study design, in the collection, analysis, or interpretation of data, in the writing of the manuscript, or in the decision to submit the manuscript for publication.
Acknowledgments
We thank JR Valera for his assistance with editing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Moshiro R, Mdoe P, Perlman JM. A global view of neonatal asphyxia and resuscitation. Front Pediatr. (2019) 7:489. doi: 10.3389/fped.2019.00489
3. Qin X, Cheng J, Zhong Y, Mahgoub OK, Akter F, Fan Y, et al. Mechanism and treatment related to oxidative stress in neonatal hypoxic-ischemic encephalopathy. Front Mol Neurosci. (2019) 12:88. doi: 10.3389/fnmol.2019.00088
4. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
5. Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 Wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. (2017) 171(3):628–41.e26. doi: 10.1016/j.cell.2017.09.044
6. Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Huttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen Species generation. Mol Neurobiol. (2013) 47(1):9–23. doi: 10.1007/s12035-012-8344-z
7. Thornton C, Jones A, Nair S, Aabdien A, Mallard C, Hagberg H. Mitochondrial dynamics, mitophagy and biogenesis in neonatal hypoxic-ischaemic brain injury. FEBS Lett. (2018) 592(5):812–30. doi: 10.1002/1873-3468.12943
8. McGowan JE, Chen L, Gao D, Trush M, Wei C. Increased mitochondrial reactive oxygen Species production in newborn brain during hypoglycemia. Neurosci Lett. (2006) 399(1–2):111–4. doi: 10.1016/j.neulet.2006.01.034
9. Ergenekon E, Gucuyener K, Erbas D, Suheyl Ezgu F, Atalay Y. Cerebrospinal fluid and Serum nitric oxide levels in asphyxiated newborns. Biol Neonate. (1999) 76(4):200–6. doi: 10.1159/000014159
10. Ergenekon E, Gucuyener K, Erbas D, Aral S, Koc E, Atalay Y. Cerebrospinal fluid and Serum vascular endothelial growth factor and nitric oxide levels in newborns with hypoxic ischemic encephalopathy. Brain Dev. (2004) 26(5):283–6. doi: 10.1016/S0387-7604(03)00166-9
11. Elmahdy H, El-Mashad AR, El-Bahrawy H, El-Gohary T, El-Barbary A, Aly H. Human recombinant erythropoietin in asphyxia neonatorum: pilot trial. Pediatrics. (2010) 125(5):e1135–42. doi: 10.1542/peds.2009-2268
12. Dedon PC, Tannenbaum SR. Reactive nitrogen Species in the chemical biology of inflammation. Arch Biochem Biophys. (2004) 423(1):12–22. doi: 10.1016/j.abb.2003.12.017
13. Wayenberg JL, Ransy V, Vermeylen D, Damis E, Bottari SP. Nitrated plasma albumin as a marker of nitrative stress and neonatal encephalopathy in perinatal asphyxia. Free Radic Biol Med. (2009) 47(7):975–82. doi: 10.1016/j.freeradbiomed.2009.07.003
14. Qanungo S, Mukherjea M. Ontogenic profile of some antioxidants and lipid peroxidation in human placental and fetal tissues. Mol Cell Biochem. (2000) 215(1–2):11–9. doi: 10.1023/a:1026511420505
15. Davis JM, Auten RL. Maturation of the antioxidant system and the effects on preterm birth. Semin Fetal Neonatal Med. (2010) 15(4):191–5. doi: 10.1016/j.siny.2010.04.001
16. Nishida A, Misaki Y, Kuruta H, Takashima S. Developmental expression of copper, zinc-superoxide dismutase in human brain by chemiluminescence. Brain Dev. (1994) 16(1):40–3. doi: 10.1016/0387-7604(94)90111-2
17. Weinberger B, Archer FE, Kathiravan S, Hirsch DS, Kleinfeld AM, Vetrano AM, et al. Effects of bilirubin on neutrophil responses in newborn infants. Neonatology. (2013) 103(2):105–11. doi: 10.1159/000343097
18. Savman K, Blennow M, Gustafson K, Tarkowski E, Hagberg H. Cytokine response in cerebrospinal fluid after birth asphyxia. Pediatr Res. (1998) 43(6):746–51. doi: 10.1203/00006450-199806000-00006
19. Sweetman DU, Strickland T, Melo AM, Kelly LA, Onwuneme C, Watson WR, et al. Neonatal encephalopathy is associated with altered IL-8 and GM-CSF which correlates with outcomes. Front Pediatr. (2020) 8:556216. doi: 10.3389/fped.2020.556216
20. Okazaki K, Nishida A, Kato M, Kozawa K, Uga N, Kimura H. Elevation of cytokine concentrations in asphyxiated neonates. Biol Neonate. (2006) 89(3):183–9. doi: 10.1159/000089180
21. Aly H, Khashaba MT, El-Ayouty M, El-Sayed O, Hasanein BM. IL-1beta, IL-6 and TNF-alpha and outcomes of neonatal hypoxic ischemic encephalopathy. Brain Dev. (2006) 28(3):178–82. doi: 10.1016/j.braindev.2005.06.006
22. Ali MH, Schlidt SA, Chandel NS, Hynes KL, Schumacker PT, Gewertz BL. Endothelial permeability and IL-6 production during hypoxia: role of ros in signal transduction. Am J Physiol. (1999) 277(5):L1057–65. doi: 10.1152/ajplung.1999.277.5.L1057
23. Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 Deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. (2007) 27(12):2576–81. doi: 10.1161/ATVBAHA.107.153080
24. Zimmerman BJ, Grisham MB, Granger DN. Role of oxidants in ischemia/reperfusion-induced granulocyte infiltration. Am J Physiol. (1990) 258(2 Pt 1):G185–90. doi: 10.1152/ajpgi.1990.258.2.G185
25. Ichikawa H, Flores S, Kvietys PR, Wolf RE, Yoshikawa T, Granger DN, et al. Molecular mechanisms of anoxia/reoxygenation-induced neutrophil adherence to cultured endothelial cells. Circ Res. (1997) 81(6):922–31. doi: 10.1161/01.res.81.6.922
26. Kojima H, Kunimoto H, Inoue T, Nakajima K. The STAT3-IGFBP5 axis is critical for IL-6/Gp130-induced premature senescence in human fibroblasts. Cell Cycle. (2012) 11(4):730–9. doi: 10.4161/cc.11.4.19172
27. Sultana Z, Maiti K, Dedman L, Smith R. Is there a role for placental senescence in the genesis of obstetric complications and fetal growth restriction? Am J Obstet Gynecol. (2018) 218(2S):S762–73. doi: 10.1016/j.ajog.2017.11.567
28. Jung JE, Kim GS, Chan PH. Neuroprotection by interleukin-6 is mediated by signal transducer and activator of transcription 3 and antioxidative signaling in ischemic stroke. Stroke. (2011) 42(12):3574–9. doi: 10.1161/STROKEAHA.111.626648
29. Tsan MF, White JE, Del Vecchio PJ, Shaffer JB. IL-6 enhances TNF-alpha- and IL-1-induced increase of mn superoxide dismutase mRNA and O2 tolerance. Am J Physiol. (1992) 263(1 Pt 1):L22–6. doi: 10.1152/ajplung.1992.263.1.L22
30. Chan PH. Role of oxidants in ischemic brain damage. Stroke. (1996) 27(6):1124–9. doi: 10.1161/01.str.27.6.1124
31. Palma FR, He C, Danes JM, Paviani V, Coelho DR, Gantner BN, et al. Mitochondrial superoxide dismutase: what the established, the intriguing, and the novel reveal about a key cellular redox switch. Antioxid Redox Signal. (2020) 32(10):701–14. doi: 10.1089/ars.2019.7962
32. Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. (2011) 15(6):1583–606. doi: 10.1089/ars.2011.3999
33. Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH. Regulation of mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci. (2009) 29(21):7003–14. doi: 10.1523/JNEUROSCI.1110-09.2009
34. Che L, Xuan Y, Hu L, Liu Y, Xu Q, Fang Z, et al. Effect of postnatal nutrition restriction on the oxidative Status of neonates with intrauterine growth restriction in a pig model. Neonatology. (2015) 107(2):93–9. doi: 10.1159/000368179
35. Munteanu AI, Manea AM, Jinca CM, Boia M. Basic biochemical and hematological parameters in perinatal asphyxia and their correlation with hypoxic ischemic encephalopathy. Exp Ther Med. (2021) 21(3):259. doi: 10.3892/etm.2021.9690
36. Cilla A, Arnaez J, Benavente-Fernandez I, Ochoa C, Vega C, Lubian-Lopez S, et al. Effect of hypothermia and severity of hypoxic-ischemic encephalopathy in the levels of C-reactive protein during the first 120 hours of life. Am J Perinatol. (2020) 37(7):722–30. doi: 10.1055/s-0039-1688818
37. Saito J, Shibasaki J, Shimokaze T, Kishigami M, Ohyama M, Hoshino R, et al. Temporal relationship between Serum levels of interleukin-6 and C-reactive protein in therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy. Am J Perinatol. (2016) 33(14):1401–6. doi: 10.1055/s-0036-1583192
38. Chiesa C, Pellegrini G, Panero A, De Luca T, Assumma M, Signore F, et al. Umbilical cord interleukin-6 levels are elevated in term neonates with perinatal asphyxia. Eur J Clin Invest. (2003) 33(4):352–8. doi: 10.1046/j.1365-2362.2003.01136.x
39. Chalak LF, Sanchez PJ, Adams-Huet B, Laptook AR, Heyne RJ, Rosenfeld CR. Biomarkers for severity of neonatal hypoxic-ischemic encephalopathy and outcomes in newborns receiving hypothermia therapy. J Pediatr. (2014) 164(3):468–74.e1. doi: 10.1016/j.jpeds.2013.10.067
40. Orrock JE, Panchapakesan K, Vezina G, Chang T, Harris K, Wang Y, et al. Association of brain injury and neonatal cytokine response during therapeutic hypothermia in newborns with hypoxic-ischemic encephalopathy. Pediatr Res. (2016) 79(5):742–7. doi: 10.1038/pr.2015.280
41. Leifsdottir K, Mehmet H, Eksborg S, Herlenius E. Fas-ligand and interleukin-6 in the cerebrospinal fluid are early predictors of hypoxic-ischemic encephalopathy and long-term outcomes after birth asphyxia in term infants. J Neuroinflammation. (2018) 15(1):223. doi: 10.1186/s12974-018-1253-y
42. Tekgul H, Yalaz M, Kutukculer N, Ozbek S, Kose T, Akisu M, et al. Value of biochemical markers for outcome in term infants with asphyxia. Pediatr Neurol. (2004) 31(5):326–32. doi: 10.1016/j.pediatrneurol.2004.05.004
43. Okazaki K, Kondo M, Kato M, Kakinuma R, Nishida A, Noda M, et al. Serum cytokine and chemokine profiles in neonates with meconium aspiration syndrome. Pediatrics. (2008) 121(4):e748–53. doi: 10.1542/peds.2007-1697
44. Didion SP. Cellular and oxidative mechanisms associated with interleukin-6 signaling in the vasculature. Int J Mol Sci. (2017) 18(12):2563. doi: 10.3390/ijms18122563
45. Silveira RC, Procianoy RS. Interleukin-6 and tumor necrosis factor-alpha levels in plasma and cerebrospinal fluid of term newborn infants with hypoxic-ischemic encephalopathy. J Pediatr. (2003) 143(5):625–9. doi: 10.1067/S0022-3476(03)00531-6
46. Lingappan K. NF-κB in oxidative stress. Curr Opin Toxicol. (2018) 7:81–6. doi: 10.1016/j.cotox.2017.11.002
47. Asehnoune K, Strassheim D, Mitra S, Kim JY, Abraham E. Involvement of reactive oxygen Species in toll-like receptor 4-dependent activation of NF-κB. J Immunol. (2004) 172(4):2522–9. doi: 10.4049/jimmunol.172.4.2522
48. Serou MJ, DeCoster MA, Bazan NG. Interleukin-1 beta activates expression of cyclooxygenase-2 and inducible nitric oxide synthase in primary hippocampal neuronal culture: platelet-activating factor as a preferential mediator of cyclooxygenase-2 expression. J Neurosci Res. (1999) 58(4):593–8. doi: 10.1002/(SICI)1097-4547(19991115)58:4%3C593::AID-JNR12%3E3.0.CO;2-4
49. Hu X, Nesic-Taylor O, Qiu J, Rea HC, Fabian R, Rassin DK, et al. Activation of nuclear factor-KappaB signaling pathway by interleukin-1 after hypoxia/ischemia in neonatal rat hippocampus and Cortex. J Neurochem. (2005) 93(1):26–37. doi: 10.1111/j.1471-4159.2004.02968.x
50. Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. (1991) 10(8):2247–58. doi: 10.1002/j.1460-2075.1991.tb07761.x
51. Ridder DA, Schwaninger M. Nf-Kappab signaling in cerebral ischemia. Neuroscience. (2009) 158(3):995–1006. doi: 10.1016/j.neuroscience.2008.07.007
52. Huang CY, Fujimura M, Noshita N, Chang YY, Chan PH. SOD1 Down-Regulates NF-kappaB and c-myc expression in mice after transient focal cerebral ischemia. J Cereb Blood Flow Metab. (2001) 21(2):163–73. doi: 10.1097/00004647-200102000-00008
53. Salminen A, Huuskonen J, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Activation of innate immunity system during aging: NF-κB signaling is the molecular culprit of inflamm-aging. Ageing Res Rev. (2008) 7(2):83–105. doi: 10.1016/j.arr.2007.09.002
54. Baltimore D. Discovering NF-κB. Cold Spring Harb Perspect Biol. (2009) 1(1):a000026. doi: 10.1101/cshperspect.a000026
55. Døhlen G, Antal EA, Castellheim A, Thaulow E, Kielland A, Saugstad OD. Hyperoxic resuscitation after hypoxia-ischemia induces cerebral inflammation that is attenuated by tempol in a reporter mouse model with very young mice. J Perinat Med. (2013) 41(3):251–7. doi: 10.1515/jpm-2012-0135
56. Hasegawa K, Ichiyama T, Isumi H, Nakata M, Sase M, Furukawa S. NF-kappaB activation in peripheral blood mononuclear cells in neonatal asphyxia. Clin Exp Immunol. (2003) 132(2):261–4. doi: 10.1046/j.1365-2249.2003.02127.x
57. Okazaki K, Kondo M, Kato M, Kakinuma R, Nishida A, Noda M, et al. Elevation of high-mobility group box 1 concentration in asphyxiated neonates. Neonatology. (2008) 94(2):105–9. doi: 10.1159/000116635
58. Palumbo R, Galvez BG, Pusterla T, De Marchis F, Cossu G, Marcu KB, et al. Cells migrating to sites of tissue damage in response to the danger signal HMGB1 require NF-κB activation. J Cell Biol. (2007) 179(1):33–40. doi: 10.1083/jcb.200704015
59. Fang P, Schachner M, Shen YQ. HMGB1 In development and diseases of the central nervous system. Mol Neurobiol. (2012) 45(3):499–506. doi: 10.1007/s12035-012-8264-y
60. Yang D, Sun YY, Lin X, Baumann JM, Dunn RS, Lindquist DM, et al. Intranasal delivery of cell-penetrating anti-NF-κB peptides (Tat-NBD) alleviates infection-sensitized hypoxic-ischemic brain injury. Exp Neurol. (2013) 247:447–55. doi: 10.1016/j.expneurol.2013.01.015
61. Ke Q, Costa M. Hypoxia-Inducible factor-1 (HIF-1). Mol Pharmacol. (2006) 70(5):1469–80. doi: 10.1124/mol.106.027029
62. Rupin A, Paysant J, Sansilvestri-Morel P, Lembrez N, Lacoste JM, Cordi A, et al. Role of NADPH oxidase-mediated superoxide production in the regulation of E-selectin expression by endothelial cells subjected to anoxia/reoxygenation. Cardiovasc Res. (2004) 63(2):323–30. doi: 10.1016/j.cardiores.2004.03.018
63. Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, et al. Hypoxia-Dependent regulation of nonphagocytic NADPH oxidase subunit Nox4 in the pulmonary vasculature. Circ Res. (2007) 101(3):258–67. doi: 10.1161/CIRCRESAHA.107.148015
64. Kahles T, Brandes RP. Which NADPH oxidase isoform is relevant for ischemic stroke? The case for nox 2. Antioxid Redox Signal. (2013) 18(12):1400–17. doi: 10.1089/ars.2012.4721
65. Kleikers PW, Wingler K, Hermans JJ, Diebold I, Altenhofer S, Radermacher KA, et al. NADPH Oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J Mol Med (Berl). (2012) 90(12):1391–406. doi: 10.1007/s00109-012-0963-3
66. Kietzmann T, Gorlach A. Reactive oxygen Species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol. (2005) 16(4–5):474–86. doi: 10.1016/j.semcdb.2005.03.010
67. Fan X, Heijnen CJ, van der Kooij MA, Groenendaal F, van Bel F. The role and regulation of hypoxia-inducible factor-1alpha expression in brain development and neonatal hypoxic-ischemic brain injury. Brain Res Rev. (2009) 62(1):99–108. doi: 10.1016/j.brainresrev.2009.09.006
68. Madan A, Varma S, Cohen HJ. Developmental stage-specific expression of the alpha and beta subunits of the HIF-1 protein in the mouse and human Fetus. Mol Genet Metab. (2002) 75(3):244–9. doi: 10.1006/mgme.2001.3293
69. Trollmann R, Gassmann M. The role of hypoxia-inducible transcription factors in the hypoxic neonatal brain. Brain Dev. (2009) 31(7):503–9. doi: 10.1016/j.braindev.2009.03.007
70. Jones NM, Bergeron M. Hypoxic preconditioning induces changes in HIF-1 target genes in neonatal rat brain. J Cereb Blood Flow Metab. (2001) 21(9):1105–14. doi: 10.1097/00004647-200109000-00008
71. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. (2016) 167(2):457–70.e13. doi: 10.1016/j.cell.2016.08.064
72. Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, et al. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. (2003) 93(11):1074–81. doi: 10.1161/01.RES.0000102937.50486.1B
73. Semenza GL, Prabhakar NR. The role of hypoxia-inducible factors in carotid body (patho) physiology. J Physiol. (2018) 596(15):2977–83. doi: 10.1113/JP275696
74. Liu CY, Al-Ward H, Ngaffo Mekontso F, Liu N, Zeng HQ, Liu M, et al. Experimental study on the correlation between miRNA-373 and HIF-1alpha, MMP-9, and VEGF in the development of hie. Biomed Res Int. (2021) 2021:5553486. doi: 10.1155/2021/5553486
75. Li L, Qu Y, Li J, Xiong Y, Mao M, Mu D. Relationship between HIF-1alpha expression and neuronal apoptosis in neonatal rats with hypoxia-ischemia brain injury. Brain Res. (2007) 1180:133–9. doi: 10.1016/j.brainres.2007.08.059
76. Toriuchi K, Kakita H, Tamura T, Takeshita S, Yamada Y, Aoyama M. Prolonged astrocyte-derived erythropoietin expression attenuates neuronal damage under hypothermic conditions. J Neuroinflammation. (2020) 17(1):141. doi: 10.1186/s12974-020-01831-3
77. Sweetman DU, Onwuneme C, Watson WR, Murphy JF, Molloy EJ. Perinatal asphyxia and erythropoietin and VEGF: serial Serum and cerebrospinal fluid responses. Neonatology. (2017) 111(3):253–9. doi: 10.1159/000448702
78. Okazaki K, Kusaka T, Kondo M, Kozawa K, Yoshizumi M, Kimura H. Temporal alteration of Serum G-CSF and VEGF levels in perinatal asphyxia treated with head cooling. Cytokine. (2012) 60(3):812–4. doi: 10.1016/j.cyto.2012.08.001
79. Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, et al. An endothelial receptor for oxidized low-density lipoprotein. Nature. (1997) 386(6620):73–7. doi: 10.1038/386073a0
80. Kume N, Kita T. Apoptosis of vascular cells by oxidized LDL: involvement of caspases and LOX-1 and its implication in atherosclerotic plaque rupture. Circ Res. (2004) 94(3):269–70. doi: 10.1161/01.RES.0000119804.92239.97
81. Pirillo A, Norata GD, Catapano AL. LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm. (2013) 2013:152786. doi: 10.1155/2013/152786
82. Akamatsu T, Sugiyama T, Aoki Y, Kawabata K, Shimizu M, Okazaki K, et al. A pilot study of soluble form of LOX-1 as a novel biomarker for neonatal hypoxic-ischemic encephalopathy. J Pediatr. (2019) 206:49–55.e3. doi: 10.1016/j.jpeds.2018.10.036
83. Kanata S, Akagi M, Nishimura S, Hayakawa S, Yoshida K, Sawamura T, et al. Oxidized LDL binding to LOX-1 upregulates VEGF expression in cultured bovine chondrocytes through activation of ppar-gamma. Biochem Biophys Res Commun. (2006) 348(3):1003–10. doi: 10.1016/j.bbrc.2006.07.133
84. Zhang WF, Xu YY, Xu KP, Wu WH, Tan GS, Li YJ, et al. Inhibitory effect of selaginellin on high glucose-induced apoptosis in differentiated PC12 cells: role of NADPH oxidase and LOX-1. Eur J Pharmacol. (2012) 694(1–3):60–8. doi: 10.1016/j.ejphar.2012.08.011
85. Minami M, Kume N, Kataoka H, Morimoto M, Hayashida K, Sawamura T, et al. Transforming growth factor-beta(1) increases the expression of lectin-like oxidized low-density lipoprotein receptor-1. Biochem Biophys Res Commun. (2000) 272(2):357–61. doi: 10.1006/bbrc.2000.2778
86. Akamatsu T, Sugiyama T, Oshima T, Aoki Y, Mizukami A, Goishi K, et al. Lectin-Like oxidized low-density lipoprotein receptor-1-related microglial activation in neonatal hypoxic-ischemic encephalopathy: morphologic consideration. Am J Pathol. (2021) 191(7):1303–13. doi: 10.1016/j.ajpath.2021.04.009
87. Lin W, Zhang T, Zheng J, Zhou Y, Lin Z, Fu X. Ferroptosis is involved in hypoxic-ischemic brain damage in neonatal rats. Neuroscience. (2022) 487:131–42. doi: 10.1016/j.neuroscience.2022.02.013
88. Li Y, Duan Z, Gao D, Huang S, Yuan H, Niu X. The new role of LOX-1 in hypertension induced neuronal apoptosis. Biochem Biophys Res Commun. (2012) 425(4):735–40. doi: 10.1016/j.bbrc.2012.07.143
89. Schreurs MP, Hubel CA, Bernstein IM, Jeyabalan A, Cipolla MJ. Increased oxidized low-density lipoprotein causes blood-brain barrier disruption in early-onset preeclampsia through LOX-1. FASEB J. (2013) 27(3):1254–63. doi: 10.1096/fj.12-222216
90. Kazmierski R, Michalak S, Wencel-Warot A, Nowinski WL. Serum tight-junction proteins predict hemorrhagic transformation in ischemic stroke patients. Neurology. (2012) 79(16):1677–85. doi: 10.1212/WNL.0b013e31826e9a83
91. Kattoor AJ, Goel A, Mehta JL. LOX-1: regulation, signaling and its role in atherosclerosis. Antioxidants (Basel). (2019) 8(7):218. doi: 10.3390/antiox8070218
92. Wendt TS, Gonzales RJ. Beneficial effects of LOX-1 inhibition on brain endothelial proinflammatory mediators and barrier proteins following OxLDL plus acute ischemic injury. FASEB J. (2022) 36:S1. doi: 10.1096/fasebj.2022.36.S1.R5618
93. Arumugam TV, Okun E, Tang SC, Thundyil J, Taylor SM, Woodruff TM. Toll-Like receptors in ischemia-reperfusion injury. Shock. (2009) 32(1):4–16. doi: 10.1097/SHK.0b013e318193e333
94. Castellheim A, Brekke OL, Espevik T, Harboe M, Mollnes TE. Innate immune responses to danger signals in systemic inflammatory response syndrome and sepsis. Scand J Immunol. (2009) 69(6):479–91. doi: 10.1111/j.1365-3083.2009.02255.x
95. Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: toll-like receptors. Free Radic Biol Med. (2010) 48(9):1121–32. doi: 10.1016/j.freeradbiomed.2010.01.006
96. Stridh L, Smith PL, Naylor AS, Wang X, Mallard C. Regulation of toll-like receptor 1 and -2 in neonatal mice brains after hypoxia-ischemia. J Neuroinflammation. (2011) 8:45. doi: 10.1186/1742-2094-8-45
97. Ock J, Jeong J, Choi WS, Lee WH, Kim SH, Kim IK, et al. Regulation of toll-like receptor 4 expression and its signaling by hypoxia in cultured microglia. J Neurosci Res. (2007) 85(9):1989–95. doi: 10.1002/jnr.21322
98. Kim SY, Choi YJ, Joung SM, Lee BH, Jung YS, Lee JY. Hypoxic stress up-regulates the expression of toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology. (2010) 129(4):516–24. doi: 10.1111/j.1365-2567.2009.03203.x
99. Yao L, Kan EM, Lu J, Hao A, Dheen ST, Kaur C, et al. Toll-Like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: role of TLR4 in hypoxic microglia. J Neuroinflammation. (2013) 10:23. doi: 10.1186/1742-2094-10-23
100. Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, et al. TLR2 Has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. (2007) 359(3):574–9. doi: 10.1016/j.bbrc.2007.05.157
101. Favre J, Musette P, Douin-Echinard V, Laude K, Henry JP, Arnal JF, et al. Toll-Like receptors 2-deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol. (2007) 27(5):1064–71. doi: 10.1161/ATVBAHA.107.140723
102. Wang WW, Smith DL, Zucker SD. Bilirubin inhibits iNOS expression and No production in response to endotoxin in rats. Hepatology. (2004) 40(2):424–33. doi: 10.1002/hep.20334
103. Idelman G, Smith DLH, Zucker SD. Bilirubin inhibits the up-regulation of inducible nitric oxide synthase by scavenging reactive oxygen Species generated by the toll-like receptor 4-dependent activation of NADPH oxidase. Redox Biol. (2015) 5:398–408. doi: 10.1016/j.redox.2015.06.008
104. Markus T, Hansson S, Amer-Wahlin I, Hellstrom-Westas L, Saugstad OD, Ley D. Cerebral inflammatory response after fetal asphyxia and hyperoxic resuscitation in newborn sheep. Pediatr Res. (2007) 62(1):71–7. doi: 10.1203/PDR.0b013e31811ead6e
105. Eliwan HO, Watson RW, Aslam S, Regan I, Philbin B, O'Hare FM, et al. Neonatal brain injury and systemic inflammation: modulation by activated protein C ex vivo. Clin Exp Immunol. (2015) 179(3):477–84. doi: 10.1111/cei.12453
106. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen Species and NF-kappaB signaling. Cell Res. (2011) 21(1):103–15. doi: 10.1038/cr.2010.178
107. Akisu M, Huseyinov A, Yalaz M, Cetin H, Kultursay N. Selective head cooling with hypothermia suppresses the generation of platelet-activating factor in cerebrospinal fluid of newborn infants with perinatal asphyxia. Prostaglandins Leukot Essent Fatty Acids. (2003) 69(1):45–50. doi: 10.1016/s0952-3278(03)00055-3
108. Yasova Barbeau D, Krueger C, Huene M, Copenhaver N, Bennett J, Weaver M, et al. Heart rate variability and inflammatory markers in neonates with hypoxic-ischemic encephalopathy. Physiol Rep. (2019) 7(15):e14110. doi: 10.14814/phy2.14110
109. Vogt KL, Summers C, Chilvers ER, Condliffe AM. Priming and De-priming of neutrophil responses in vitro and in vivo. Eur J Clin Invest. (2018) 48(Suppl 2):e12967. doi: 10.1111/eci.12967
110. Hornstein T, Lehmann S, Philipp D, Detmer S, Hoffmann M, Peter C, et al. Staurosporine resistance in inflammatory neutrophils is associated with the inhibition of caspase- and proteasome-mediated mcl-1 degradation. J Leukoc Biol. (2016) 99(1):163–74. doi: 10.1189/jlb.3A1114-537RR
111. Hammerman C, Ischemia KM, Injury R. The ultimate pathophysiologic paradox. Clin Perinatol. (1998) 25(3):757–77. doi: 10.1016/S0095-5108(18)30110-6
112. Hallenbeck JM, Dutka AJ, Tanishima T, Kochanek PM, Kumaroo KK, Thompson CB, et al. Polymorphonuclear leukocyte accumulation in brain regions with low blood flow during the early postischemic period. Stroke. (1986) 17(2):246–53. doi: 10.1161/01.str.17.2.246
113. Linden JR, Kunkel D, Laforce-Nesbitt SS, Bliss JM. The role of galectin-3 in phagocytosis of Candida Albicans and Candida parapsilosis by human neutrophils. Cell Microbiol. (2013) 15(7):1127–42. doi: 10.1111/cmi.12103
114. Lawrence SM, Corriden R, Nizet V. Age-Appropriate functions and dysfunctions of the neonatal neutrophil. Front Pediatr. (2017) 5:23. doi: 10.3389/fped.2017.00023
115. Smith EF 3rd, Egan JW, Bugelski PJ, Hillegass LM, Hill DE, Griswold DE. Temporal relation between neutrophil accumulation and myocardial reperfusion injury. Am J Physiol. (1988) 255(5 Pt 2):H1060–8. doi: 10.1152/ajpheart.1988.255.5.H1060
116. Mackay CR. Chemokines: immunology's high impact factors. Nat Immunol. (2001) 2(2):95–101. doi: 10.1038/84298
117. Guichard C, Pedruzzi E, Dewas C, Fay M, Pouzet C, Bens M, et al. Interleukin-8-Induced priming of neutrophil oxidative burst requires sequential recruitment of NADPH oxidase components into lipid rafts. J Biol Chem. (2005) 280(44):37021–32. doi: 10.1074/jbc.M506594200
118. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. (2010) 87(5):779–89. doi: 10.1189/jlb.1109766
119. Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. (2009) 43(4):348–64. doi: 10.1080/10715760902751902
120. Cuzzocrea S, Mazzon E, Dugo L, Caputi AP, Aston K, Riley DP, et al. Protective effects of a new stable, highly active SOD mimetic, M40401 in splanchnic artery occlusion and reperfusion. Br J Pharmacol. (2001) 132(1):19–29. doi: 10.1038/sj.bjp.0703775
121. Salvemini D, Mazzon E, Dugo L, Riley DP, Serraino I, Caputi AP, et al. Pharmacological manipulation of the inflammatory cascade by the superoxide dismutase mimetic, M40403. Br J Pharmacol. (2001) 132(4):815–27. doi: 10.1038/sj.bjp.0703841
122. Masini E, Cuzzocrea S, Mazzon E, Marzocca C, Mannaioni PF, Salvemini D. Protective effects of M40403, a selective superoxide dismutase mimetic, in myocardial ischaemia and reperfusion injury in vivo. Br J Pharmacol. (2002) 136(6):905–17. doi: 10.1038/sj.bjp.0704774
123. Wung BS, Ni CW, Wang DL. ICAM-1 Induction by TNFalpha and IL-6 is mediated by distinct pathways via rac in endothelial cells. J Biomed Sci. (2005) 12(1):91–101. doi: 10.1007/s11373-004-8170-z
124. Ali MI, Chen X, Didion SP. Heterozygous enos deficiency is associated with oxidative stress and endothelial dysfunction in diet-induced obesity. Physiol Rep. (2015) 3(12):e12630. doi: 10.14814/phy2.12630
125. Saura M, Zaragoza C, Bao C, Herranz B, Rodriguez-Puyol M, Lowenstein CJ. Stat3 mediates interleukin-6 [correction of interelukin-6] inhibition of human endothelial nitric-oxide synthase expression. J Biol Chem. (2006) 281(40):30057–62. doi: 10.1074/jbc.M606279200
126. Wung BS, Hsu MC, Wu CC, Hsieh CW. Resveratrol suppresses IL-6-induced ICAM-1 gene expression in endothelial cells: effects on the inhibition of STAT3 phosphorylation. Life Sci. (2005) 78(4):389–97. doi: 10.1016/j.lfs.2005.04.052
127. Kvietys PR, Granger DN. Role of reactive oxygen and nitrogen Species in the vascular responses to inflammation. Free Radic Biol Med. (2012) 52(3):556–92. doi: 10.1016/j.freeradbiomed.2011.11.002
128. van Wetering S, van den Berk N, van Buul JD, Mul FP, Lommerse I, Mous R, et al. VCAM-1-Mediated rac signaling controls endothelial cell-cell contacts and leukocyte transmigration. Am J Physiol Cell Physiol. (2003) 285(2):C343–52. doi: 10.1152/ajpcell.00048.2003
129. Cook-Mills JM, Marchese ME, Abdala-Valencia H. Vascular cell adhesion molecule-1 expression and signaling during disease: regulation by reactive oxygen Species and antioxidants. Antioxid Redox Signal. (2011) 15(6):1607–38. doi: 10.1089/ars.2010.3522
130. Keshavan P, Deem TL, Schwemberger SJ, Babcock GF, Cook-Mills JM, Zucker SD. Unconjugated bilirubin inhibits VCAM-1-mediated transendothelial leukocyte migration. J Immunol. (2005) 174(6):3709–18. doi: 10.4049/jimmunol.174.6.3709
131. Kavanagh DP, Durant LE, Crosby HA, Lalor PF, Frampton J, Adams DH, et al. Haematopoietic stem cell recruitment to injured murine liver sinusoids Depends on (alpha)4(Beta)1 integrin/VCAM-1 interactions. Gut. (2010) 59(1):79–87. doi: 10.1136/gut.2008.168054
132. Okazaki K, Nishida A, Kimura H. Inflammatory mediators in neonatal asphyxia and infection. In: Buonocore G, Bracci R, Weindling M, editors. Neonatology: A practical approach to neonatal diseases. Switzerland: Springer, Cham (2016). p. 1619–39. doi: 10.1007/978-3-319-18159-2_248-1
133. Solaroglu I, Tsubokawa T, Cahill J, Zhang JH. Anti-Apoptotic effect of granulocyte-colony stimulating factor after focal cerebral ischemia in the rat. Neuroscience. (2006) 143(4):965–74. doi: 10.1016/j.neuroscience.2006.09.014
134. Solaroglu I, Cahill J, Jadhav V, Zhang JH. A novel neuroprotectant granulocyte-colony stimulating factor. Stroke. (2006) 37(4):1123–8. doi: 10.1161/01.STR.0000208205.26253.96
135. Yata K, Matchett GA, Tsubokawa T, Tang J, Kanamaru K, Zhang JH. Granulocyte-Colony stimulating factor inhibits apoptotic neuron loss after neonatal hypoxia-ischemia in rats. Brain Res. (2007) 1145:227–38. doi: 10.1016/j.brainres.2007.01.144
136. Chavez-Valdez R, Miller S, Spahic H, Vaidya D, Parkinson C, Dietrick B, et al. Therapeutic hypothermia modulates the relationships between indicators of severity of neonatal hypoxic ischemic encephalopathy and Serum biomarkers. Front Neurol. (2021) 12:748150. doi: 10.3389/fneur.2021.748150
137. Gialanella B. Aphasia assessment and functional outcome prediction in patients with aphasia after stroke. J Neurol. (2011) 258(2):343–9. doi: 10.1007/s00415-010-5868-x
138. Malla RR, Asimi R, Teli MA, Shaheen F, Bhat MA. Erythropoietin monotherapy in perinatal asphyxia with moderate to severe encephalopathy: a randomized placebo-controlled trial. J Perinatol. (2017) 37(5):596–601. doi: 10.1038/jp.2017.17
139. Maiwald CA, Annink KV, Rudiger M, Benders M, van Bel F, Allegaert K, et al. Effect of allopurinol in addition to hypothermia treatment in neonates for hypoxic-ischemic brain injury on neurocognitive outcome (albino): study protocol of a blinded randomized placebo-controlled parallel group multicenter trial for superiority (phase III). BMC Pediatr. (2019) 19(1):210. doi: 10.1186/s12887-019-1566-8
140. Garg B, Sharma D, Bansal A. Systematic review seeking erythropoietin role for neuroprotection in neonates with hypoxic ischemic encephalopathy: presently where do we stand. J Matern Fetal Neonatal Med. (2018) 31(23):3214–24. doi: 10.1080/14767058.2017.1366982
141. Wu YW, Mathur AM, Chang T, McKinstry RC, Mulkey SB, Mayock DE, et al. High-Dose erythropoietin and hypothermia for hypoxic-ischemic encephalopathy: a phase ii trial. Pediatrics. (2016) 137(6):e20160191. doi: 10.1542/peds.2016-0191
142. Wu YW, Comstock BA, Gonzalez FF, Mayock DE, Goodman AM, Maitre NL, et al. Trial of erythropoietin for hypoxic-ischemic encephalopathy in newborns. N Engl J Med. (2022) 387(2):148–59. doi: 10.1056/NEJMoa2119660
143. Alonso-Alconada D, Alvarez A, Arteaga O, Martinez-Ibarguen A, Hilario E. Neuroprotective effect of melatonin: a novel therapy against perinatal hypoxia-ischemia. Int J Mol Sci. (2013) 14(5):9379–95. doi: 10.3390/ijms14059379
144. Mohan N, Sadeghi K, Reiter RJ, Meltz ML. The neurohormone melatonin inhibits cytokine, mitogen and ionizing radiation induced NF-kappa B. Biochem Mol Biol Int. (1995) 37(6):1063–70. Retrieved from: https://europepmc.org/article/med/8747536.8747536
145. Aly H, Elmahdy H, El-Dib M, Rowisha M, Awny M, El-Gohary T, et al. Melatonin use for neuroprotection in perinatal asphyxia: a randomized controlled pilot study. J Perinatol. (2015) 35(3):186–91. doi: 10.1038/jp.2014.186
146. Sugimoto J, Romani AM, Valentin-Torres AM, Luciano AA, Ramirez Kitchen CM, Funderburg N, et al. Magnesium decreases inflammatory cytokine production: a novel innate immunomodulatory mechanism. J Immunol. (2012) 188(12):6338–46. doi: 10.4049/jimmunol.1101765
147. Peruche B, Krieglstein J. Mechanisms of drug actions against neuronal damage caused by ischemia–an overview. Prog Neuropsychopharmacol Biol Psychiatry. (1993) 17(1):21–70. doi: 10.1016/0278-5846(93)90032-n
148. Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. (1984) 307(5950):462–5. doi: 10.1038/307462a0
149. Weglicki WB. Hypomagnesemia and inflammation: clinical and basic aspects. Annu Rev Nutr. (2012) 32:55–71. doi: 10.1146/annurev-nutr-071811-150656
150. Nielsen FH. Magnesium deficiency and increased inflammation: current perspectives. J Inflamm Res. (2018) 11:25–34. doi: 10.2147/JIR.S136742
151. Miyashita T, Oda Y, Horiuchi J, Yin JC, Morimoto T, Saitoe M. Mg(2+) block of drosophila NMDA receptors is required for long-term memory formation and CREB-dependent gene expression. Neuron. (2012) 74(5):887–98. doi: 10.1016/j.neuron.2012.03.039
152. Ichiba H, Tamai H, Negishi H, Ueda T, Kim TJ, Sumida Y, et al.; kansai magnesium study group. Randomized controlled trial of magnesium sulfate infusion for severe birth asphyxia. Pediatr Int. (2002) 44(5):505–9. doi: 10.1046/j.1442-200x.2002.01610.x
153. Gathwala G, Khera A, Singh J, Balhara B. Magnesium for neuroprotection in birth asphyxia. J Pediatr Neurosci. (2010) 5(2):102–4. doi: 10.4103/1817-1745.76094
154. Bhat MA, Charoo BA, Bhat JI, Ahmad SM, Ali SW, Mufti MU. Magnesium sulfate in severe perinatal asphyxia: a randomized, placebo-controlled trial. Pediatrics. (2009) 123(5):e764–9. doi: 10.1542/peds.2007-3642
155. Tagin M, Shah PS, Lee KS. Magnesium for newborns with hypoxic-ischemic encephalopathy: a systematic review and meta-analysis. J Perinatol. (2013) 33(9):663–9. doi: 10.1038/jp.2013.65
156. Westermaier T, Stetter C, Kunze E, Willner N, Raslan F, Vince GH, et al. Magnesium treatment for neuroprotection in ischemic diseases of the brain. Exp Transl Stroke Med. (2013) 5(1):6. doi: 10.1186/2040-7378-5-6
157. Cullen SC, Gross EG. The anesthetic properties of xenon in animals and human beings, with additional observations on krypton. Science. (1951) 113(2942):580–2. doi: 10.1126/science.113.2942.580
158. Franks NP, Dickinson R, de Sousa SL, Hall AC, Lieb WR. How does xenon produce anaesthesia? Nature. (1998) 396(6709):324. doi: 10.1038/24525
159. Maze M. Preclinical neuroprotective actions of xenon and possible implications for human therapeutics: a narrative review. Can J Anaesth. (2016) 63(2):212–26. doi: 10.1007/s12630-015-0507-8
160. Chakkarapani E, Dingley J, Liu X, Hoque N, Aquilina K, Porter H, et al. Xenon enhances hypothermic neuroprotection in asphyxiated newborn pigs. Ann Neurol. (2010) 68(3):330–41. doi: 10.1002/ana.22016
161. Faulkner S, Bainbridge A, Kato T, Chandrasekaran M, Kapetanakis AB, Hristova M, et al. Xenon augmented hypothermia reduces early lactate/N-acetylaspartate and cell death in perinatal asphyxia. Ann Neurol. (2011) 70(1):133–50. doi: 10.1002/ana.22387
162. Dingley J, Tooley J, Porter H, Thoresen M. Xenon provides short-term neuroprotection in neonatal rats when administered after hypoxia-ischemia. Stroke. (2006) 37(2):501–6. doi: 10.1161/01.STR.0000198867.31134.ac
163. Azzopardi D, Robertson NJ, Bainbridge A, Cady E, Charles-Edwards G, Deierl A, et al. Moderate hypothermia within 6 h of birth plus inhaled xenon versus moderate hypothermia alone after birth asphyxia (TOBY-Xe): a proof-of-concept, open-label, randomised controlled trial. Lancet Neurol. (2016) 15(2):145–53. doi: 10.1016/S1474-4422(15)00347-6
164. Htun Y, Nakamura S, Kusaka T. Hydrogen and therapeutic gases for neonatal hypoxic-ischemic encephalopathy: potential neuroprotective adjuncts in translational research. Pediatr Res. (2021) 89(4):753–9. doi: 10.1038/s41390-020-0998-z
165. Gharib B, Hanna S, Abdallahi OM, Lepidi H, Gardette B, De Reggi M. Anti-inflammatory properties of molecular hydrogen: investigation on parasite-induced liver inflammation. C R Acad Sci III. (2001) 324(8):719–24. doi: 10.1016/s0764-4469(01)01350-6
166. Tian Y, Guo S, Zhang Y, Xu Y, Zhao P, Zhao X. Effects of hydrogen-rich saline on hepatectomy-induced postoperative cognitive dysfunction in old mice. Mol Neurobiol. (2017) 54(4):2579–84. doi: 10.1007/s12035-016-9825-2
167. Kawamura T, Huang CS, Tochigi N, Lee S, Shigemura N, Billiar TR, et al. Inhaled hydrogen gas therapy for prevention of lung transplant-induced ischemia/reperfusion injury in rats. Transplantation. (2010) 90(12):1344–51. doi: 10.1097/TP.0b013e3181fe1357
168. Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. (2007) 13(6):688–94. doi: 10.1038/nm1577
169. Htun Y, Nakamura S, Nakao Y, Mitsuie T, Nakamura M, Yamato S, et al. Hydrogen ventilation combined with mild hypothermia improves short-term neurological outcomes in a 5-day neonatal hypoxia-ischaemia piglet model. Sci Rep. (2019) 9(1):4088. doi: 10.1038/s41598-019-40674-8
170. Damianos A, Xu K, Kalin GT, Kalinichenko VV. Placental tissue stem cells and their role in neonatal diseases. Semin Fetal Neonatal Med. (2022) 27(1):101322. doi: 10.1016/j.siny.2021.101322
171. Valle-Prieto A, Conget PA. Human mesenchymal stem cells efficiently manage oxidative stress. Stem Cells Dev. (2010) 19(12):1885–93. doi: 10.1089/scd.2010.0093
172. Zhang R, Liu Y, Yan K, Chen L, Chen XR, Li P, et al. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J Neuroinflammation. (2013) 10:106. doi: 10.1186/1742-2094-10-106
173. van Velthoven CT, Kavelaars A, van Bel F, Heijnen CJ. Mesenchymal stem cell transplantation changes the gene expression profile of the neonatal ischemic brain. Brain Behav Immun. (2011) 25(7):1342–8. doi: 10.1016/j.bbi.2011.03.021
174. Yang F, Zhang Y, Liu S, Xiao J, He Y, Shao Z, et al. Tunneling nanotube-mediated mitochondrial transfer rescues nucleus Pulposus cells from mitochondrial dysfunction and apoptosis. Oxid Med Cell Longev. (2022) 2022:3613319. doi: 10.1155/2022/3613319
175. Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A. (2006) 103(5):1283–8. doi: 10.1073/pnas.0510511103
176. Boruczkowski D, Pujal JM, Zdolinska-Malinowska I. Autologous cord blood in children with cerebral palsy: a review. Int J Mol Sci. (2019) 20(10):2433. doi: 10.3390/ijms20102433
177. van Velthoven CT, Kavelaars A, van Bel F, Heijnen CJ. Mesenchymal stem cell treatment after neonatal hypoxic-ischemic brain injury improves behavioral outcome and induces neuronal and oligodendrocyte regeneration. Brain Behav Immun. (2010) 24(3):387–93. doi: 10.1016/j.bbi.2009.10.017
178. Donega V, van Velthoven CT, Nijboer CH, van Bel F, Kas MJ, Kavelaars A, et al. Intranasal mesenchymal stem cell treatment for neonatal brain damage: long-term cognitive and sensorimotor improvement. PLoS One. (2013) 8(1):e51253. doi: 10.1371/journal.pone.0051253
179. Tsuji M, Taguchi A, Ohshima M, Kasahara Y, Sato Y, Tsuda H, et al. Effects of intravenous administration of umbilical cord blood CD34(+) cells in a mouse model of neonatal stroke. Neuroscience. (2014) 263:148–58. doi: 10.1016/j.neuroscience.2014.01.018
180. Grandvuillemin I, Garrigue P, Ramdani A, Boubred F, Simeoni U, Dignat-George F, et al. Long-Term recovery after endothelial colony-forming cells or human umbilical cord blood cells administration in a rat model of neonatal hypoxic-ischemic encephalopathy. Stem Cells Transl Med. (2017) 6(11):1987–96. doi: 10.1002/sctm.17-0074
181. Cotten CM, Murtha AP, Goldberg RN, Grotegut CA, Smith PB, Goldstein RF, et al. Feasibility of autologous cord blood cells for infants with hypoxic-ischemic encephalopathy. J Pediatr. (2014) 164(5):973–9.e1. doi: 10.1016/j.jpeds.2013.11.036
182. Cotten CM, Fisher K, Kurtzberg J, Simmons R. Phase I trial of allogeneic umbilical cord tissue-derived mesenchymal stromal cells in neonates with hypoxic-ischemic encephalopathy. Cytotherapy. (2020) 22(5):S192. doi: 10.1016/j.jcyt.2020.04.052
183. Tsuji M, Sawada M, Watabe S, Sano H, Kanai M, Tanaka E, et al. Autologous cord blood cell therapy for neonatal hypoxic-ischaemic encephalopathy: a pilot study for feasibility and safety. Sci Rep. (2020) 10(1):4603. doi: 10.1038/s41598-020-61311-9
184. Bruschettini M, Romantsik O, Moreira A, Ley D, Thebaud B. Stem cell-based interventions for the prevention of morbidity and mortality following hypoxic-ischaemic encephalopathy in newborn infants. Cochrane Database Syst Rev. (2020) 8(8):CD013202. doi: 10.1002/14651858.CD013202.pub2
185. Serrenho I, Rosado M, Dinis A, Cardoso CM, Graos M, Manadas B, et al. Stem cell therapy for neonatal hypoxic-ischemic encephalopathy: a systematic review of preclinical studies. Int J Mol Sci. (2021) 22(6):3142. doi: 10.3390/ijms22063142
186. Matsuyama N, Shimizu S, Ueda K, Suzuki T, Suzuki S, Miura R, et al. Safety and tolerability of a multilineage-differentiating stress-enduring cell-based product in neonatal hypoxic-ischaemic encephalopathy with therapeutic hypothermia (SHIELD trial): a clinical trial protocol open-label, non-randomised, dose-escalation trial. BMJ Open. (2022) 12(4):e057073. doi: 10.1136/bmjopen-2021-057073
Keywords: asphyxia, ROS, HMGB1, NF-κB, cytokines, neutrophils, TLRs, neonate
Citation: Okazaki K, Nakamura S, Koyano K, Konishi Y, Kondo M and Kusaka T (2023) Neonatal asphyxia as an inflammatory disease: Reactive oxygen species and cytokines. Front. Pediatr. 11:1070743. doi: 10.3389/fped.2023.1070743
Received: 15 October 2022; Accepted: 10 January 2023;
Published: 27 January 2023.
Edited by:
Robert Galinsky, Hudson Institute of Medical Research, AustraliaReviewed by:
Jiang-Qin Liu, Shanghai First Maternity and Infant Hospital, ChinaTayla Penny, Hudson Institute of Medical Research, Australia
© 2023 Okazaki, Nakamura, Koyano, Konishi, Kondo and Kusaka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takashi Kusaka a3VzYWthLnRha2FzaGlAa2FnYXdhLXUuYWMuanA=
Specialty Section: This article was submitted to Neonatology, a section of the journal Frontiers in Pediatrics