
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 06 October 2022
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.982361
 Xuemei Zhao1†
Xuemei Zhao1† Bingbing Wu1†
Bingbing Wu1† Huiyao Chen1Ping Zhang1Yanyan Qian1Xiaomin Peng1Xinran Dong1Yaqiong Wang1Gang Li1
Huiyao Chen1Ping Zhang1Yanyan Qian1Xiaomin Peng1Xinran Dong1Yaqiong Wang1Gang Li1 Chenbin Dong2*
Chenbin Dong2* Huijun Wang1*
Huijun Wang1*Craniosynostosis is a premature fusion of cranial sutures, resulting in abnormally shaped skull and brain development disorder. The description of craniosynostosis in patients with BCL11B mutations is rare. Here, we firstly report a 25-month-old Chinese boy with a novel frameshift variant in BCL11B gene. The patient was identified c.2346_2361del by whole-exome sequencing and was confirmed to be de novo by parental Sanger sequencing. This patient presented clinical phenotype of craniosynostosis as well as global developmental delay. He had a small mouth, thin upper lip, arched eyebrows, a long philtrum, midfacial hypoplasia and craniosynostosis. Brain MRI showed brain extracerebral interval and myelination changes, and brain CT with 3D reconstruction showed multi-craniosynostosis. Our study expands the clinical phenotypes of patients with BCL11B gene mutation, and our findings may help guide clinical treatment and family genetic counseling.
Craniosynostosis is the premature fusion of one or more of the calvarial sutures. The incidence of craniosynostosis is estimated to be 1 in 2000-2500 live births (1). In the cases of syndromic craniosynostosis, a series of clinical phenotypes, such as skull expansion, midfacial hypoplasia, increased intracranial pressure and dysmorphologies, are observed (2). The major etiology of craniosynostosis includes varieties of factors including genetic and environmental factors (3, 4). Moreover, genetic factor has been recognized as a non-negligible etiology of craniosynostosis. In a 13-year birth cohort study, one-quarter of patients with craniosynostosis could be identified as having a genetic cause (4). An early genetic diagnosis can provide valuable information for intervention. For the treatment of craniosynostosis, patients need long-term strategies, including surgery such as open cranial reconstruction, minimally invasive strip craniectomy with spring implantation, cranial distraction and a postoperative molding helmet (1).
The transcription factor BCL11B (B Cell Leukemia 11B), encoding Cys2-His2 zinc-finger protein transcription factor, is located on chromosome 14q32.2. BCL11B is broadly expressed and has critical functions in regulating multiple systems including the central nervous system, immune system, and skin (5, 6). BCL11B contains three types of functional structures: six C2HH zinc-finger binding domains (ZnF1-6), one C2HC zinc-finger binding domain, and a NuRD interacting domain (7). The first report of BCL11B as a potential disease gene was in an infant bearing a de novo BCL11B missense mutation with severe combined immunodeficiency, craniofacial and dermal abnormalities, and the absence of corpus callosum (8). Currently, 20 patients have been reported to harbor BCL11B pathogenic variants, The major clinical features include severe combined immunodeficiency, neurological disorders, dermal defects, congenital diaphragmatic hernia, and craniosynostosis. Recently, Goos et al. reported a de novo missense mutation in a patient with unilateral coronal suture craniosynostosis (9). However, it is rare about the description of craniosynostosis in patients with BCL11B mutations.
Here, we report a patient with a novel de novo frameshift variant exhibiting the combinational phenotype of craniosynostosis and developmental delay. Meanwhile, we review the clinical and genetic features of craniosynostosis patients with BCL11B mutations, which further delineate the phenotype and genotype of such patients.
Genomic DNA was extracted from peripheral blood using a whole blood genomic DNA extraction kit (Qiagen, German) according to the manufacturer’s protocol. DNA fragments were enriched for exome sequences using the IDT xGen Exome Research Panel v2 (Integrated DNA Technologies, Coralville, IA, USA). According to our previous reports, sequencing was performed on NovaSeq 6000 (Illumina, San Diego, USA) according to protocols (10). The data analysis followed the pipeline established by our team (11). PhenoPro, including phenotype-driven analysis and variant-driven analysis, was performed to prioritize the disease-causing genes of Mendelian diseases as described before (12). The pathogenicity was evaluated by the variation frequency, phenotypic coincidence with patients and the type of variation according to ACMG criteria (13). The variant of the BCL11B gene was confirmed by Sanger sequencing on an ABI 3,730 Genetic analyzer (Applied Biosystems, Foster City, CA, USA).
Nonsense-mediated mRNA decay (NMD) efficacy of the BCL11B frameshift was evaluated using online NMDective resource (14). The wildtype 3-D protein structure of BCL11B was analyzed by the neural network AlphaFold, a new online source to predict the protein domain (15). The mutated amino acid sequence was acquired on the Mutalyzer website. Then we used SWISS-MODEL to predict the truncated protein structure according to the protein template from AlphaFold. All the protein structures were visualized by PyMOL 2.3.0.
The boy is the first child of healthy non-consanguineous parents. He visited our hospital at 12 months of age with global developmental delay and recurrent allergy. He was born after uneventful full-term pregnancy with birth weight of 3,650 g (0.60 SD), and birth length of 52.0 cm (1.49 SD). He was observed with plagiocephaly, his birth head circumference is 33.2 cm (−0.99 SD) (Supplementary Figure 1). He had midfacial hypoplasia, hypertelorism, arched eyebrows, a long philtrum and a small mouth (Table 1). His motor development milestones were delayed: he could not sit steadily at 14 months, he could walk with help at 18 months, and he could not clap his hands until 24 months, he could only say “baba” and “mama”. Brain MRI at the age of 13 months showed widening of the extracerebral interval, dysmyelination of the bilateral periventricular white matter, abnormal signals of the cornu occipitale and a reduced right-brain cerebral volume (Figure 1A and Table 1).
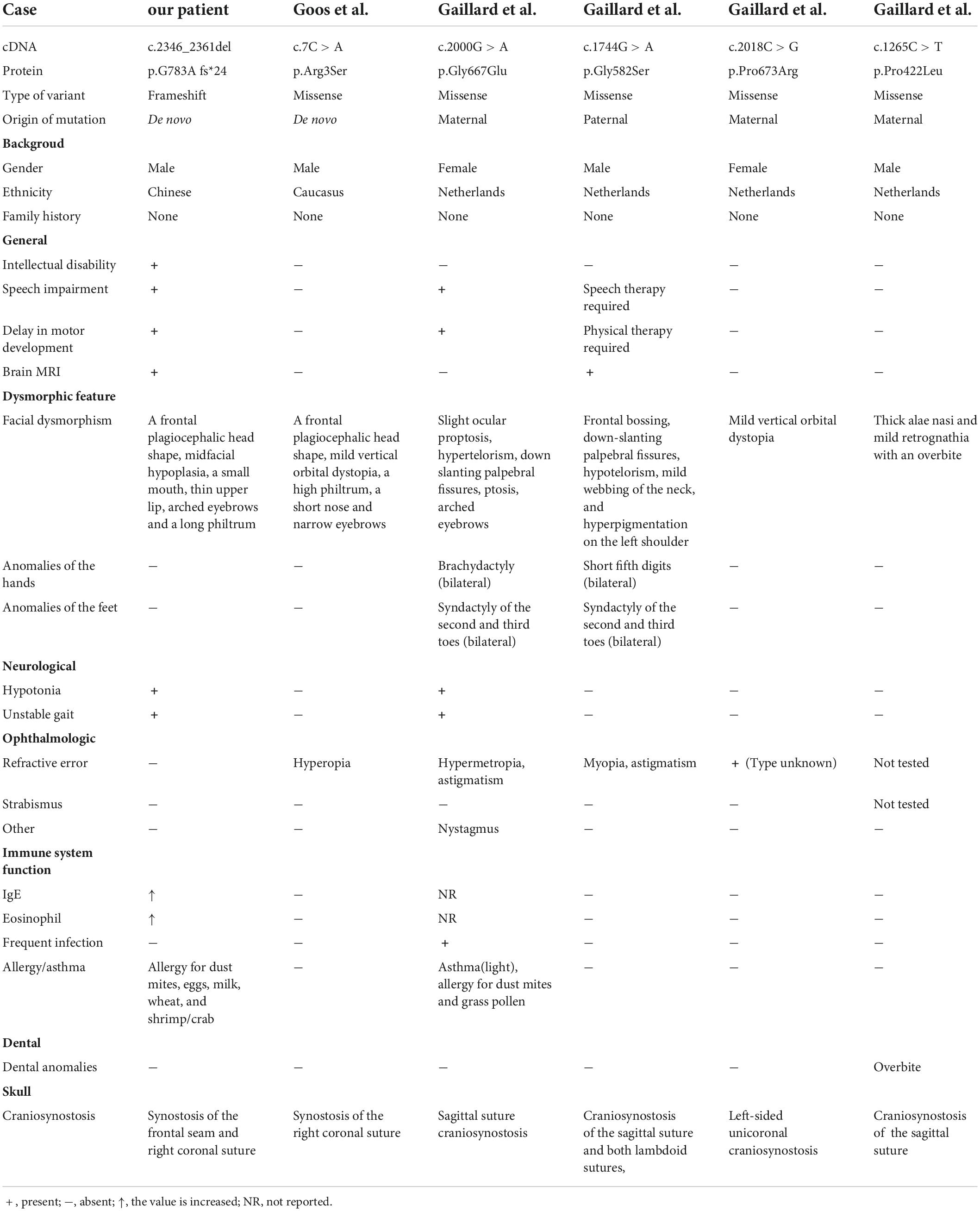
Table 1. Clinical features of six craniosynostosis patients with mutations in BCL11B.
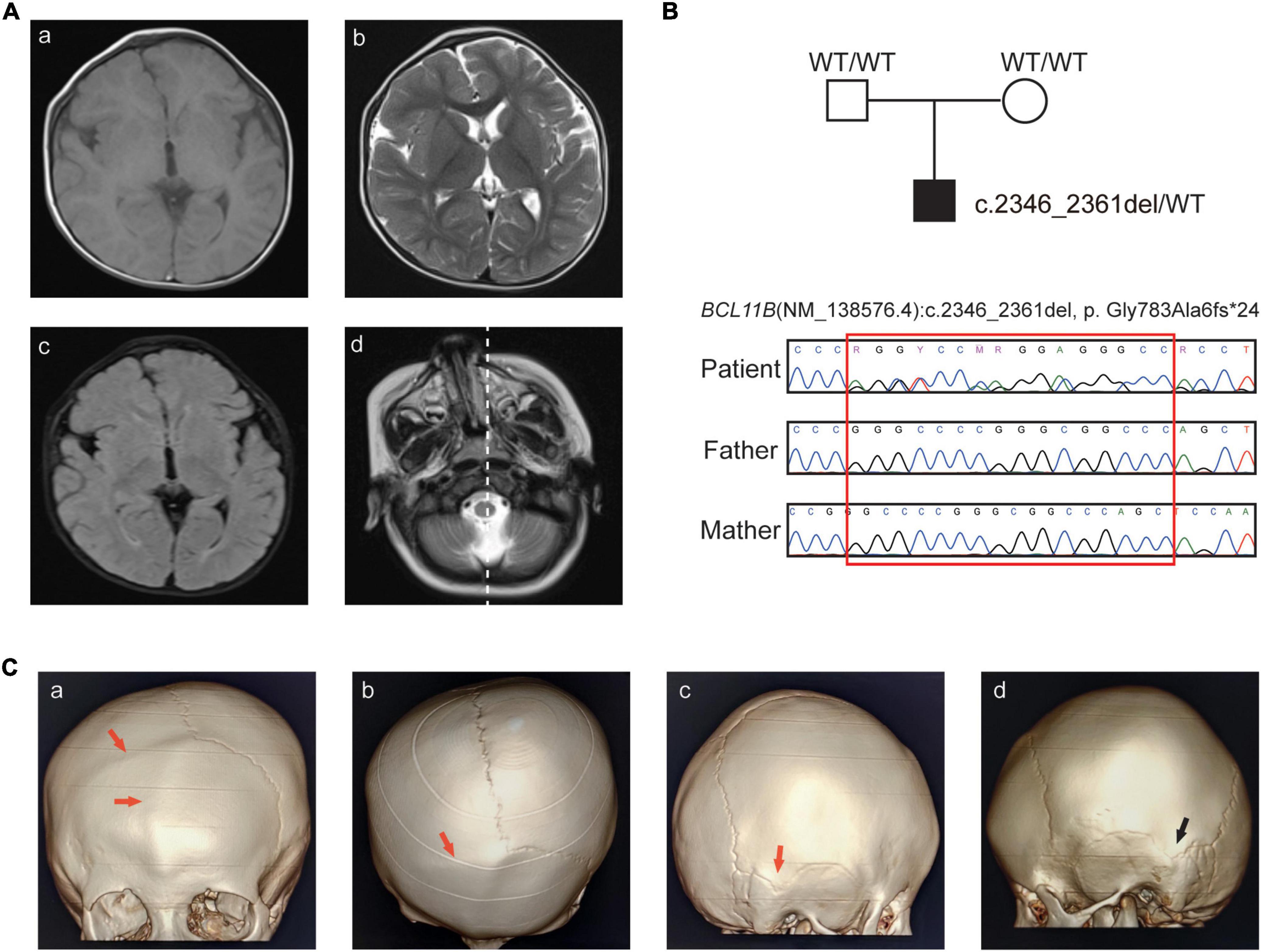
Figure 1. Clinical features and sequence analysis. (A) Brain MR images. Abnormal signals of the cornu occipitale. (a) T1WI hypodensities and the smaller right brain volume; (b) T2WI hyperintensity and the widening of the extracerebral space; (c) increased FLAIR signals; (d) right-skewed midfacial hypoplasia. (B) Pedigree of the family and Sanger sequencing analysis. (C) CT images with 3D reconstruction. (a–b) Frontal seam and right coronal seam, (c) right apex seam, (d) left apex seam. The abnormal seam fusions are indicated by the red arrow, and the normal seam is indicated by the black arrow.
Further examinations, including brain CT and blood immunophenotyping analysis, were undertaken at the age of 25 months. Skull radiographs and three-dimensional computed tomography (3D-CT) showed craniosynostosis of the frontal and right-coronal suture fusion, and skull asymmetry was observed, with the eye axis deviating to the right side (Figures 1A,C).
His immunophenotyping analysis results are shown in Supplementary Table 1 and Supplementary Figure 2. Notably, the number and percentage of eosinophil cells were increased to 910 cells/μl (normal range: 60-300 cells/μl) and 13.7% (normal range: 0.5-5%), respectively. The value of IgE in this patient was markedly increased to 944.91 IU/ml (normal range < 100 IU/ml).
WES was performed for the patient, and a novel heterozygous variant (NM_138576.4: c.2346_2361del, p.Gly783Ala6fs*24) of the BCL11B gene was detected. The variant was located on the exon4, just before the ZnF4 domain (Figure 2A) and was predicted to generate a truncated protein with a lack of ZnF4-6 (Figure 2B). The BCL11B gene variant was highly conserved and partly located in the ZnF4 domain (Figure 2C).
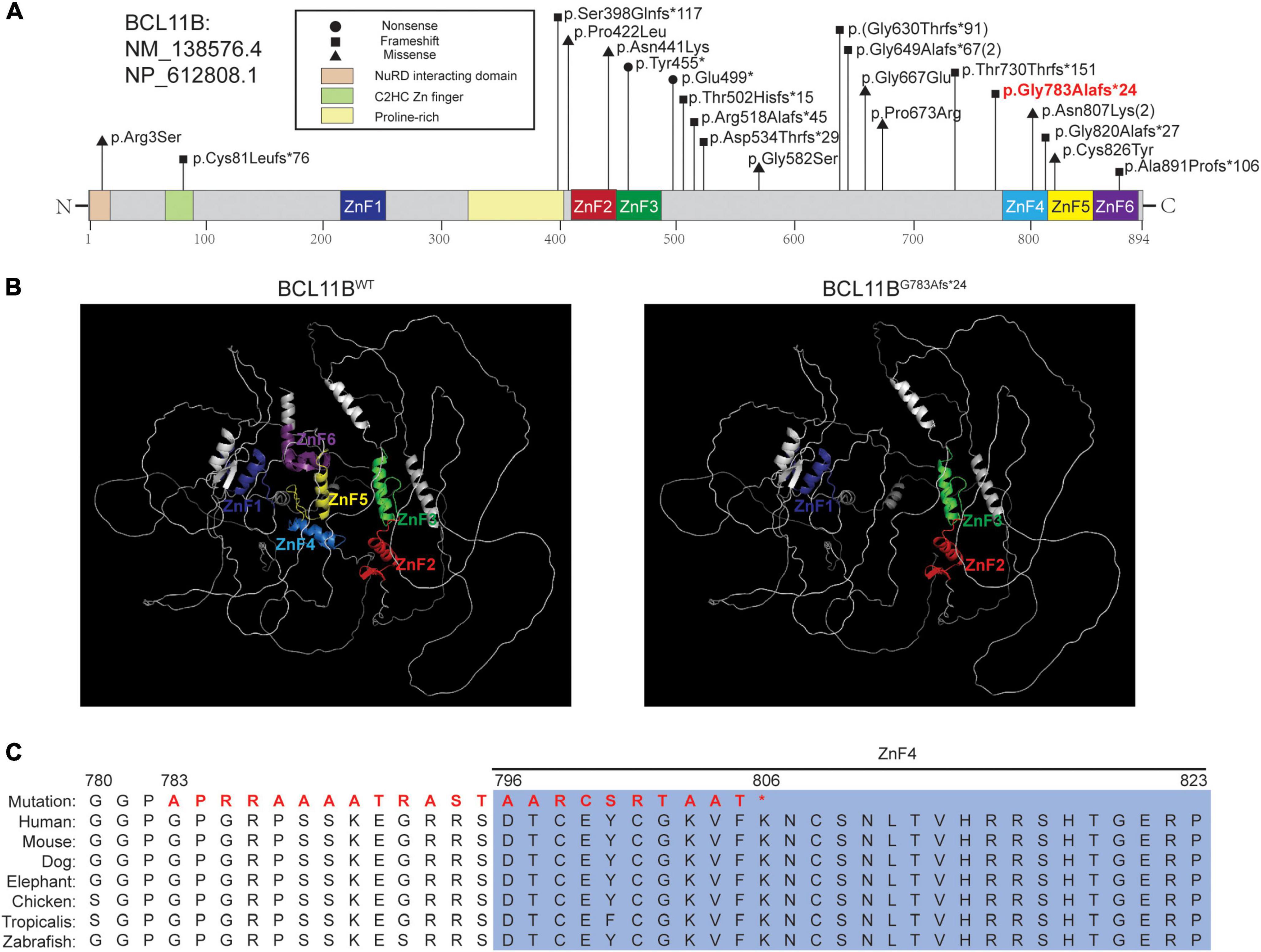
Figure 2. The pathogenic variant of BCL11B and the in silico prediction results. (A) The localization of variants reported in the literature and the variant in our patient. The variant in bold red font is that of the patient in this report. (B) Structures of BCL11BWT analyzed by the AlphaFold network and BCL11BG783Afs*24 predicted by SWISS-MODEL. The ZnF1-ZnF6 domains are marked in the corresponding colors of the schematic representations. (C) Sequence alignment across species with amino acid residues was performed, and our patient’s mutated sequence is shown in bold red font.
This frameshift variant was curated according to the ACMG criteria base on the following evidence: (i) NMDetective-A (score: 0.12) and NMDetective-B (score: 0) predicted the variant escaping NMD; the pLI (probability of being loss-of-function intolerance) score of BCL11B was 0.99, the observed/expected (o/e) ratio was 0.09, which suggests that BCL11B is highly possible of loss-of-function, PVS1_Strong; (ii) Sanger sequencing of parental samples confirmed that the variant was de novo (Figure 1B), and the phenotype was consistent with the BCL11B gene, PM6; (iii) the variant had not been recorded in gnomAD, the 1,000 Genomes database or our in-house database (43,000 individuals), PM2. (iv) we had submitted this variant to ClinVar database (VCV001184138.4) as pathogenic, and the lab of Institute of Human Genetics, University Hospital Muenster also submitted to ClinVar, with the evidence of PVS1 + PS2 + PS4, this same variant identified from a different patient. Therefore, the class of this variant was curated as pathogenetic.
We also reviewed currently reported 20 patients in literature. 18 patients carried 16 variations of the BCL11B gene, including ten frameshifts, four missense mutations and two nonsense mutations. The other two had chromosomal rearrangements resulting in diminished BCL11B expression (data not shown) (Figure 2A).
In this study, we report a novel de novo frameshift pathogenic variant (Gly783Ala fs*24) in the BCL11B gene associated with craniosynostosis and developmental delay. Using the newly reported AlphaFold algorithm, a computational method that predicts protein structures with atomic accuracy (15), we predicted that this truncated protein would lack the last three C2HH zinc-finger domains (ZnF4-6). As BCL11B is a transcriptional activator, the premature stop-codon sequence of BCL11B may affect the protein’s function in binding to its target DNA and its interactions with target proteins (7, 16). We curated the variant as pathogenic based on the ACMG criteria (PVS1_Strong + PS2 + PM6 + PM2). Although we predicted this frameshift variant escape NMD, whether the pathogenic mechanism of the variant is haploinsufficiency or gain of function remains unclear, and further experiment would be needed to address this. In the literature (17), the Cys81Leufs*76 variant was predicted to activate NMD, while the Gly820Alafs*27 variant was predicted to result in a protein with loss of DNA binding to the last C-terminus. Therefore, further studies are needed to elucidate the pathogenic mechanism of BCL11B gene.
In addition, this frameshift maybe a pathogenic hotpot of BCL11B gene as a second patient who carried the same frameshift mutation was recently submitted in the ClinVar database. Like our patient, this patient presented global developmental delay and the dysmorphic feature like trigonocephaly. However, no more detail clinical information was recorded in this patient. In our patient, we noticed that the number and percentage of eosinophils and the IgE values of our patient significantly increased. Interestingly, five cases with a high level of eosinophils were also reported by Lessel et al and Lu et al. (17, 18). Lu and associates proposed this germline BCL11B-related atopic disease as a novel primary atopic disorder (PAD). The immunophenotyping of the patient revealed intact T-cell numbers as our patient presented (Supplementary Table 1). However, zebrafish experiment demonstrated that the BCL11B variant (p.Cys826Tyr) was unable to rescue T-cell development. Therefore, it would be important to explore how immune deficiency arose despite intact thymic development.
In a summary of the neurodevelopment disorder phenotype of all reported patients (8, 9, 17–23), four were reported to present with abnormal MRI, exhibiting moderate ectopia of the amygdala (17), hypoplasia of the globus pallidus (17), callosal agenesis (8), or an abnormal myelination pattern of white matter (23). Our patient had brain morphological abnormalities with brain asymmetry and affected growth of the right brain.
In this study, we reviewed the clinical phenotype of the reported patients with craniosynostosis and BCL11B variants (9, 24) and our patient (Table1). One de novo missense variant (c.7C > A, p. Arg3Ser) with craniosynostosis in BCL11B gene has been reported (9). The patient presented with sever craniosynostosis and received three corrective surgeries before the age of 13 years. Fortunately, the boy was otherwise healthy. Recently, another literature has reported four BCL11B missense mutations (p.Gly667Glu, p.Gly582Ser, p.Pro673Arg, p.Pro422Leu) with craniosynostosis or combined congenital diaphragmatic hernia (CDH) and craniosynostosis (24). All the BCL11B variants were inherited from their normal parents. The variant of c.1744G > A (p.Gly582Ser) was recorded in three individuals without craniosynostosis in our internal database and in one individual in gnomAD database. In this literature, clinical exome sequencing was performed in one patient and targeted sequencing of single gene (BCL11B) in other three patients, these sequence methods may miss real pathogenic gene variants. Therefore, the pathogenicity of these inherited missense BCL11B variants in craniosynostosis is questionable as the paper’s title mentioned (24).
In this study, our data revealed disruptions of BCL11B as a monogenic cause of craniosynostosis combined with a neurodevelopmental disorder. Our findings expand the genetic and phenotypic spectrum of the BCL11B gene in pediatric patients.
The datasets presented in this study can be found in online repositories. The names of the repository and accession number(s) can be found here: the GSA repository, accession number: subHRA004434.
The studies involving human participants were reviewed and approved by the ethics committees of the Children’s Hospital, Fudan University (2015-130). Written informed consent to participate in this study and for the publication of any potentially identifiable images or data included in this article was provided by the participants’ legal guardian/next of kin.
XZ and BW contributed to the conception and design, data acquisition and analysis, and drafting of the manuscript. HC and YW performed the in silico analysis. PZ and YQ performed the data analysis of next-generation sequencing. GL performed the genetic experiment and sanger sequence analysis. XP collected clinical information and recorded the data. XD confirmed the clinical diagnosis and silico analysis. HW and CD contributed to the conception, design of this study, and revised the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (grant no. 81471483).
We sincerely thank the members of the patient’s family for their kind participation in this study. We thank Xiangtong Ji from Huashan Hospital, affiliated with Fudan University, for clinical assistance. We also thank Professor Merisa Robbertze for reviewing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.982361/full#supplementary-material
1. Governale LS. Craniosynostosis. Pediatr Neurol. (2015) 53:394–401. doi: 10.1016/j.pediatrneurol.2015.07.006
2. Kyrylkova K, Iwaniec UT, Philbrick KA, Leid M. Bcl11b regulates sutural patency in the mouse craniofacial skeleton. Dev Biol. (2016) 415:251–60. doi: 10.1016/j.ydbio.2015.10.010
3. Wu X, Gu Y. Signaling mechanisms underlying genetic pathophysiology of craniosynostosis. Int J Biol Sci. (2019) 15:298–311. doi: 10.7150/ijbs.29183
4. Wilkie AOM, Johnson D, Wall SA. Clinical genetics of craniosynostosis. Curr Opin Pediatr. (2017) 29:622–8. doi: 10.1097/MOP.0000000000000542
5. Daher MT, Bausero P, Agbulut O, Li Z, Parlakian A. Bcl11b/Ctip2 in skin, tooth, and craniofacial system. Front Cell Dev Biol. (2020) 8:581674. doi: 10.3389/fcell.2020.581674
6. Drashansky TT, Helm E, Huo Z, Curkovic N, Kumar P, Luo X, et al. Bcl11b prevents fatal autoimmunity by promoting Treg cell program and constraining innate lineages in Treg cells. Sci Adv. (2019) 5:eaaw0480. doi: 10.1126/sciadv.aaw0480
7. Simon R, Wiegreffe C, Britsch S. Bcl11 transcription factors regulate cortical development and function. Front Mol Neurosci. (2020) 13:51. doi: 10.3389/fnmol.2020.00051
8. Punwani D, Zhang Y, Yu J, Cowan MJ, Rana S, Kwan A, et al. Multisystem anomalies in severe combined immunodeficiency with mutant Bcl11b. N Engl J Med. (2016) 375:2165–76. doi: 10.1056/NEJMoa1509164
9. Goos JAC, Vogel WK, Mlcochova H, Millard CJ, Esfandiari E, Selman WH, et al. A de novo substitution in Bcl11b leads to loss of interaction with transcriptional complexes and craniosynostosis. Hum Mol Genet. (2019) 28:2501–13. doi: 10.1093/hmg/ddz072
10. Wang H, Xiao F, Dong X, Lu Y, Cheng G, Wang L, et al. Diagnostic and clinical utility of next-generation sequencing in children born with multiple congenital anomalies in the china neonatal genomes project. Hum Mutat. (2021) 42:434–44. doi: 10.1002/humu.24170
11. Yang L, Kong Y, Dong X, Hu L, Lin Y, Chen X, et al. Clinical and genetic spectrum of a large cohort of children with epilepsy in China. Genet Med. (2019) 21:564–71. doi: 10.1038/s41436-018-0091-8
12. Li Z, Zhang F, Wang Y, Qiu Y, Wu Y, Lu Y, et al. Phenopro: a novel toolkit for assisting in the diagnosis of mendelian disease. Bioinformatics. (2019) 35:3559–66. doi: 10.1093/bioinformatics/btz100
13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
14. Lindeboom RGH, Vermeulen M, Lehner B, Supek F. The impact of nonsense-mediated MRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat Genet. (2019) 51:1645–51. doi: 10.1038/s41588-019-0517-5
15. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with Alphafold. Nature. (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
16. Lennon MJ, Jones SP, Lovelace MD, Guillemin GJ, Brew BJ. Bcl11b: a new piece to the complex puzzle of amyotrophic lateral sclerosis neuropathogenesis? Neurotox Res. (2016) 29:201–7. doi: 10.1007/s12640-015-9573-5
17. Lessel D, Gehbauer C, Bramswig NC, Schluth-Bolard C, Venkataramanappa S, van Gassen KLI, et al. Bcl11b mutations in patients affected by a neurodevelopmental disorder with reduced type 2 innate lymphoid cells. Brain. (2018) 141:2299–311. doi: 10.1093/brain/awy173
18. Lu HY, Sertori R, Contreras AV, Hamer M, Messing M, Del Bel KL, et al. A novel germline heterozygous Bcl11b variant causing severe atopic disease and immune dysregulation. Front Immunol. (2021) 12:788278. doi: 10.3389/fimmu.2021.788278
19. Homma TK, Freire BL, Honjo Kawahira RS, Dauber A, Funari MFA, Lerario AM, et al. Genetic disorders in prenatal onset syndromic short stature identified by exome sequencing. J Pediatr. (2019) 215:192–8. doi: 10.1016/j.jpeds.2019.08.024
20. Prasad M, Balci TB, Prasad C, Andrews JD, Lee R, Jurkiewicz MT, et al. Bcl11b-related disorder in two Canadian children: expanding the clinical phenotype. Eur J Med Genet. (2020) 63:104007. doi: 10.1016/j.ejmg.2020.104007
21. Qiao F, Wang C, Luo C, Wang Y, Shao B, Tan J, et al. A De novo heterozygous frameshift mutation identified in Bcl11b causes neurodevelopmental disorder by whole exome sequencing. Mol Genet Genomic Med. (2019) 7:e897. doi: 10.1002/mgg3.897
22. Yan S, Wei YS, Yang QY, Yang L, Zeng T, Tang XM, et al. [A case report of Bcl11b mutation induced neurodevelopmental disorder and literature review]. Zhonghua Er Ke Za Zhi. (2020) 58:223–7. doi: 10.3760/cma.j.issn.0578-1310.2020.03.012
23. Yang S, Kang Q, Hou Y, Wang L, Li L, Liu S, et al. Mutant Bcl11b in a patient with a neurodevelopmental disorder and T-cell abnormalities. Front Pediatr. (2020) 8:544894. doi: 10.3389/fped.2020.544894
Keywords: BCL11B, truncating variation, pediatrics, craniosynostosis, developmental delay
Citation: Zhao X, Wu B, Chen H, Zhang P, Qian Y, Peng X, Dong X, Wang Y, Li G, Dong C and Wang H (2022) Case report: A novel truncating variant of BCL11B associated with rare feature of craniosynostosis and global developmental delay. Front. Pediatr. 10:982361. doi: 10.3389/fped.2022.982361
Received: 30 June 2022; Accepted: 14 September 2022;
Published: 06 October 2022.
Edited by:
Ruen Yao, Shanghai Children’s Medical Center, ChinaReviewed by:
Muhammad Naeem, Hebei Normal University, ChinaCopyright © 2022 Zhao, Wu, Chen, Zhang, Qian, Peng, Dong, Wang, Li, Dong and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chenbin Dong, ZGNiNDI2QDE2My5jb20=; Huijun Wang, aHVpanVud2FuZ0BmdWRhbi5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.