 Céline Delestrain1,2,†
Céline Delestrain1,2,† Abdel Aissat1,3,†
Abdel Aissat1,3,† Elodie Nattes1,2,3,†
Elodie Nattes1,2,3,† Isabelle Gibertini4Valérie Lacroze5
Isabelle Gibertini4Valérie Lacroze5 Stéphanie Simon1
Stéphanie Simon1 Xavier Decrouy1Alix de Becdelièvre1,3
Xavier Decrouy1Alix de Becdelièvre1,3 Pascale Fanen1,3*
Pascale Fanen1,3* Ralph Epaud1,2*
Ralph Epaud1,2*
- 1Univ Paris Est Creteil, INSERM, IMRB, Creteil, France
- 2Centre Hospitalier Intercommunal de Créteil, Service de Pédiatrie Générale, Créteil, France
- 3Département de Génétique, AP-HP, Hopital Henri Mondor, DMU de Biologie-Pathologie, Créteil, France
- 4Département de Pédiatrie, Centre Hospitalier Universitaire de Tours, Tours, France
- 5AP-HM, Hôpital de la Conception, Service de Médecine Néonatale, Marseille, France
Background: to perform a functional analysis of a new NK2 homeobox 1 (NKX2-1) variant (c.85_86del denominated NKX2-1DEL) identified in a family presenting with isolated respiratory disease, in comparison to another frameshift variant (c.254dup denominated NKX2-1DUP) identified in a subject with classical brain-lung-thyroid syndrome.
Methods: pathogenic variants were introduced into the pcDNA3-1(+)-wt-TTF1 plasmid. The proteins obtained were analyzed by western blot assay. Subcellular localization was assessed by confocal microscopy in A549 and Nthy cells. Transactivation of SFTPA, SFTPB, SFTPC, and ABCA3 promoters was assessed in A549 cells. Thyroglobulin promoter activity was measured with the paired box gene 8 (PAX8) cofactor in Nthy cells.
Results: The two sequence variants were predicted to produce aberrant proteins identical from the 86th amino acid, with deletion of their functional homeodomain, including the nuclear localization signal. However, 3D conformation prediction of the conformation prediction of the mutant protein assumed the presence of a nuclear localization signal, a bipartite sequence, confirmed by confocal microscopy showing both mutant proteins localized in the nucleus and cytoplasm. Transcriptional activity with SFTPA, SFTPB, SFTPC, ABCA3 and thyroglobulin promoters was significantly decreased with both variants. However, with NKX2-1DEL, thyroglobulin transcriptional activity was maintained with the addition of PAX8.
Conclusion: These results provide novel insights into understanding the molecular mechanism of phenotypes associated with NKX2-1 pathogenic variants.
Introduction
The transcription factor NK2 homeobox 1 (NKX2-1), a member of the NKX2 transcription factor family, partially controls the synthesis of surfactant proteins (1). NKX2-1 was initially identified as a nuclear protein binding the promoter of thyroglobulin gene. However, various studies later showed that NKX2-1 is also expressed in lungs and brain (2). Homozygous mice lacking NKX2-1 die shortly after birth, lack lung parenchyma and the thyroid gland, and exhibit defects in the ventral region of the forebrain, including lack of a pituitary gland (3).
The human gene located in 14q13 is encoded by three exons and is characterized by its functional domain, the homeodomain in exon 3, which also contains the nuclear localization signal (NLS) (4). The most abundant and functional transcript is generated from an open reading frame starting in exon 2 and encoding 371 amino acids (5, 6). In humans, autosomal dominant NKX2-1 pathogenic variants are associated with brain-lung-thyroid syndrome (BLTS), but not always: there is a large variability in the phenotype presentation that may exclude any of the three phenotypes in BLTS (7, 8). Both neurological and pulmonary features are responsible for the disease morbidity, but lung disease is mainly responsible for the mortality (9). Various pulmonary phenotypes have been described, including respiratory distress syndrome in term infants and interstitial lung disease (ILD) in children and more recently in adults owing to abnormal lung development and the lack of pulmonary surfactant (8, 10).
Surfactant is a multi-molecular complex consisting of phospholipids (80%–90%) and proteins (10%–15%); 2% to 3% are specific proteins called surfactant protein A, B, C and D (SP-A, SP-B, SP-C and SP-D, respectively) (11). SP-B and SP-C are synthesized by alveolar type II cells as pro-peptides (pro-SP-B and pro-SP-C), then assembled with lipids into bilayer membranes and secreted into the alveolar airspace, where they form a surface film at the air–liquid interface (12, 13). Adenosine triphosphate-binding cassette transporter A3 (ABCA3) transports phospholipids from the cytoplasm into the lamellar bodies where they combine with surfactant proteins B and C (12, 13).
The thyroid involvement due to NKX2-1 pathogenic variant is defined by thyroid dysgenesis with or without morphological abnormality of the thyroid. Various phenotypes can be observed, from congenital hypothyroidism diagnosed in the first days of life to a compensated hypothyroidism not requiring treatment (14). The NKX2-1 thyroid targets are thyroglobulin (TG), thyroperoxidase (TPO) and TSH receptor (TSHR) promoters (15–17). Paired box gene 8 (PAX8) is another transcription factor involved in thyroglobulin promoter regulation. Functional studies showed the synergism of the association of PAX8 and NKX2-1 on the thyroglobulin promoter (14, 17).
The neurological symptoms associated with NKX2-1 pathogenic variants include hypotonia in the first months of life, with motor delay and benign chorea occurring during childhood. The severity of choreic symptoms varies among families and usually stabilizes in adulthood (18). Other types of abnormal movements such as myoclonus, upper-limb dystonia and verbal tics have been described (18). In the embryonic brain, NKX2-1 controls the specification of the GABAergic interneurons and oligodendrocytes that populate the ventral and dorsal telencephalic region acting on the glial fibrillary acidic protein (GFAP) promoter. Loss of NKX2-1 leads to ventral to dorsal re-specification of the pallidum causing loss of GABAergic interneurons and oligodendrocytes in the dorsal telencephalic region (19). Other neurological targets of NKX2-1 have been described. NKX2-1 promotes GABAergic and cholinergic cell fate by inducing LIM homeobox 6 (LHX6) (20, 21). However, no functional test, such as luciferase assay, is available to clearly assess this function.
Here, we identified and functionally characterized two NKX2-1 variants identified in two children.
The two pathogenic variants were frameshift variants that created the same aberrant protein downstream of the 86th amino acid. The variant c.85_86del (NM_003317.3) was carried by a father and his daughter, who had isolated respiratory symptoms (8). The variant c.254dup (NM_003317.3) was identified in a boy who presented with BLTS. To understand why two frameshift pathogenic variants, produce such different phenotypes, we used functional comparative studies of both variants with thyroid- and lung-specific in vitro functional assays.
Materials and methods
Genetic analysis
After obtaining informed consent for genetic analysis according to French legislation, NKX2-1 clinical sequencing was performed by the Genetic Department of Henri Mondor Hospital (Creteil, France). Briefly, EDTA samples were collected and used to extract leukocyte genomic DNA. Specific primers were used to amplify the coding regions and the intron–exon boundaries of NKX2-1 (primer sequences available on request) by PCR. The PCR products were subjected to direct Sanger sequencing. For each pathogenic variant, the de novo or inherited status was reported.
Plasmids
The pcDNA3-1(+)-wt-TTF1 plasmid was a gift from David Mu (Addgene plasmid #49989) (22). The plasmid was elongated by introducing an insert encompassing 131 nucleotides (nt) from the last nucleotide of exon 3 (therefore including the stop codon created by the frameshift variant). Then the variants were introduced by site-directed mutagenesis (Stratagene, Waghaeusel-Wiesental, Germany). For transactivation experiments, we used the following plasmids: reporter gene containing NKX2-1 and PAX8 binding sites from the thyroglobulin promoter upstream of luciferase (19), SP-B promoter luciferase (23) for surfactant protein B (SFTPB) promoter, pGL2-SP-C-promoter (24) for surfactant protein C (SFTPC) promoter, pGL3-ABCA3-promoter (25) for ABCA3 promoter, pGL3-SP-A2-promoter (26) for surfactant protein A (SFTPA) promoter, pcDNA3.1-PAX8 (27), and hGFAP-fLuc [gift from Albee Messing, Addgene plasmid # 40589, (28)].
Cell culture
Human lung epithelial cells, A549 cells (ATCC, CCL-185, Manassas, VA), were cultured in DMEM-glutamax (Thermo Fisher Scientific, Waltham, MA) with penicillin-streptomycin (Thermo Fisher Scientific) and 10% FCS (Eurobio, Courtaboeuf, France). Immortalized human thyroid cell lines, Nthy-ori3.1 cells [gift from A. Carré and M. Polak, (29)], were cultured in RPMI-Glutamax (Thermo Fisher Scientific, Waltham, MA) with penicillin-streptomycin (Thermo Fisher Scientific) and 10% FCS (Eurobio, Courtaboeuf, France).
In vitro synthesis of NKX2-1
Proteins were synthesized from the wild-type and mutated NKX2-1 plasmids (1 µg) by in vitro transcription/translation with the TNT-T7–coupled reticulocyte lysate system (Promega, Madison, WI, USA). Western blot analysis was performed with 1 µl TNT reaction as explained below.
Western blot assay
In total, 6.5 × 105 A549 and 1.1 × 106 Nthy cells were plated in 60-mm-diameter culture plates. Cells were transiently transfected with 3 µg wild-type or mutated NKX2-1 by means of lipofectamine 3,000 (Thermo Fisher Scientific) according to the manufacturer's instructions. At 24 h after transfection, proteins were extracted with RIPA lysis and extraction buffer (Thermo Fisher Scientific) and quantified by using Bio-Rad DC protein assay (BioRad, Hercules, CA, USA). A 30-µg amount of protein was fractionated by SDS-PAGE on 12% tris-glycine gels. Proteins were further electrotransferred to a PVDSF 0.45-µM membrane (GE Healthcare, Little Chalfont, UK). Blots were probed overnight at 4°C with NKX2-1 antibody (Abgent, San Diego, CA, USA, 1/500), then incubated with secondary antibody. Bound antibodies were detected by using ECL prime chemiluminescence substrate (Amersham ECL, GE Healthcare) and exposed to a G:Box (Syngene, Cambridge, UK). Images were analyzed by using GeneSys software (Daly City, CA, USA). Experiments were repeated three times.
Immunocytofluorescence
For immunostaining, 2.6 × 105 A549 and 4.5 × 105 Nthy cells per well were grown on coverslips in 6-well plates for 24 h. Then cells were transiently transfected with 2 µg wild-type or mutated NKX2-1 by means of lipofectamine 3,000 (Thermo Fisher Scientific). At 24 h after transfection, cells were washed and fixed with acetone/methanol (3/7) for 5 min at 4 °C and blocked with anti-NKX2-1 antibody (1:250 dilution; Abgent) and Alexa fluor 488 rabbit secondary antibody (1:2000 dilution; Molecular Probes, Eugene, OR, USA). Fluorescence was visualized with a Zeiss confocal microscope. Experiments were repeated three to four times.
Transactivation
For luciferase assay, 1.2 × 105 A549 and 105 Nthy cells per well were plated in 24-well plates. After 24 h, A549 cells were transfected with 400 ng lung reporter plasmid (SP-A2, SP-B, SP-C, or ABCA3 luciferase), and 400 ng wild-type or mutant NKX2-1 plasmid. Nthy cells were transfected with 200 ng thyroglobulin reporter plasmid, 200 ng PAX8 plasmid and 200 ng wild-type or mutant NKX2-1 plasmid. The total amount of plasmids was kept constant by the addition of the respective empty vector (pcDNA3-1). After 24 h, cells were harvested, and underwent firefly luciferase assay (Promega) in a TriStar system (Berthold Technologies GmbH & Co. KG, Germany). The promoter activation was calculated after normalization to the total luminescence of the experiment, in five independent experiments, each condition performed in duplicate.
Statistical analysis and in silico analysis
One-way ANOVA (GraphPad Prism 6, GraphPad Software, La Jolla, CA, USA) was used to analyze differences in transactivation activities. Data are presented as mean ± SEM. The mutated proteins were analyzed by using InterPro (30) and PROSITE (31) software to find new domains and with the cNLS mapper bioinformatic program to find the NLS (32). Multiple sequence alignment was analyzed by using the Clustal Omega online tool (https://www.ebi.ac.uk/Tools/msa/clustalo/) (33).
Results
Subjects
The first index subject was an infant girl born at 38 weeks from a healthy nonconsanguineous couple. At 7 months, she showed failure to thrive and tachypnoea. High-resolution CT (HRCT) revealed diffuse ground-glass opacities suggesting ILD. At 10 months, she required supplemental oxygen support. She received azithromycin, hydroxychloroquine, and corticosteroid pulses. Corticosteroid treatment was stopped at age 3 years (after 27 pulses) after HRCT-evidence of improvement, but she required nocturnal oxygen supplementation until age 4 years. Thyroid function and morphology were within the normal range and remained stable for 8 years' follow up (TSH level = 4 (7 months), 4.9 (2 years 6 months), 3.2 (4 years 7 months) and 3.6 mUI/L (7 years 4 months) for a normal value < 5 mUI/L). During this time, the girl showed normal psychomotor development (with regard to a general pattern of physical, emotional, intellectual and social milestones) with absence of hypotonia, abnormal movements, or motor coordination (gross or fine) abnormalities. She received enteral nutrition by gastrostomy from age 15 months for 6 years. The genetic diagnosis was achieved at 2 years. Her father carried the same variant and had a diagnosis of ILD later in life (age 30 years) during an intensive-care-unit hospitalization for severe viral respiratory infection. He also had normal thyroid function and absence of neurological symptoms. His last HRCT revealed a slight progression of certain fibrotic zones. Finally, her grandmother died without genetic analysis of pulmonary fibrosis complicated by lung cancer.
The second index subject was a term-born boy showing respiratory distress syndrome within the first day of life. A chest radiograph showed diffuse ground-glass opacities, compatible with severe hyaline membrane disease. After the first week of life, the boy showed good respiratory condition, with no evidence of respiratory distress recurrence, and a 6-min walk test performed at age 4 years gave normal results. Neonatal screening revealed congenital hypothyroidism [TSH level 420 mU/L and free thyroxine (T4) level 0.008 nmol/L for a normal value < 0,6 mU/L and from 120 to 210 nmol/L before the first week for TSH and free thyroxine respectively], and levothyroxine was started on day 7. Scintigraphy revealed thyroid ectopia with sublingual localization. The boy showed hypotonia at age 3 months and psychomotor delay, with autonomous walking achieved at age 2 years. The choreoathetotic movements appeared at age 3 years.
Identification and characterization of NKX2-1 human variant
For both subjects, screening for surfactant gene SFTPC, SFTPB and ABCA3 was negative by direct sequencing of the subject's DNA. However, we identified an inherited heterozygous variant in NKX2-1 for the girl. The pathogenic variant was a deletion of an adenosine and a thymidine at positions 85 and 86 of the cDNA (transcript NM_003317.3), which generates a frameshift leading to an aberrantly longer protein of 407 amino acids (NKX2-1DEL), retaining only the first 28 amino acids of the wild type (Figure 1A).
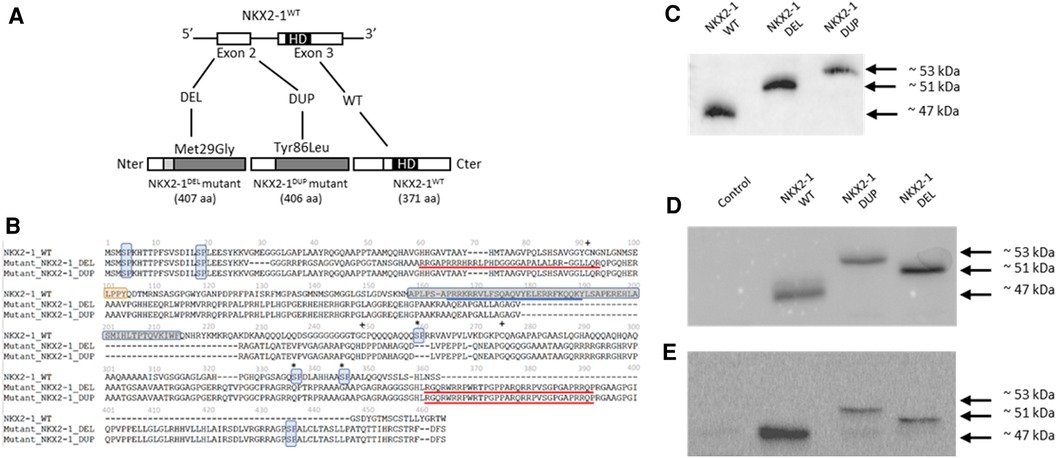
Figure 1. (A) Identification of two NKX2-1 pathogenic variant by direct sequencing and expression of the wild-type and mutated NKX2-1 protein. The diagram of the NKX2-1 transcript variant 2 (NM_003317.3) shows the position of the homeodomain and variants. (B) Deduced amino acid sequences of NKX2-1dup and NKX2-1del as compared with NKX2-1WT. The nuclear localization signal (NLS) is underlined in blue for the NKX2-1WT and the putative NLSs, according to the predictions, are underlined in red for the two mutants. The homeobox domain is framed in gray (position 160 to 220) for the NKX2-1WT and is absent for both mutants. pSP motifs have been highlighted in light blue. The NKX2-1WT harbors five motifs while NKX2-1del and NKX2-1dup have three motifs. LPPY motif, a putative TAZ protein interaction motif, has been highlighted in orange for the NKX2-1-WT. This motif is absent for both mutants. Serines involved in phosphorylation in the wild-type sequence but affected by the mutations are marked by an asterisk. Cysteines involved in the dimerization of the amino-acid chains in the wild-type but affected by the mutations are marked by a plus sign (adapted from Moya et al., 2018). (C) Representative western blot analysis in pcDNA3-1(+)-TTF1 plasmid of translated NKX2-1 wild type and mutant (DEL and DUP). (D) Representative immunoblotting of NKX2-1 in whole cell lysates of A549 cells not transfected (Control) or transiently transfected with NKX2-1WT, NKX2-1DUP and NKX2-1DEL. (E) Representative immunoblotting of NKX2-1 in whole cell lysates of Nthy cells non-transfected (Control) or transiently transfected with NKX2-1WT, NKX2-1DUP and NKX2-1DEL.
We identified a de novo heterozygous variant in the NKX2-1 gene for the boy. The variant was a duplication of guanine at position 254 of the cDNA (transcript NM_003317.3), which generates a frameshift leading to an aberrantly long protein of 406 amino acids (NKX2-DUP)), retaining only the first 85 amino acids of the wild type (Figure 1A).
Amino acid prediction of the mutants by bioinformatic tools anticipated a severe impact on protein function based on reduced conservation of the amino acid sequence with respect to the wild-type protein and the absence of critical domains of this transcription factor, i.e, the homeodomain and the native NLS. Both mutants have lost several serine-proline (SP) residues in the second half of the amino-acid sequence. They also have lost several cysteines which could be involved in dimerization of the NKX2-1 protein (Figure 1B).
Protein expression and subcellular localization of NKX2-1 mutants
To assess the functional consequences of these two pathogenic variants, we used an in vitro transcription and translation system to synthesize the wild-type and NKX2-1DUP and NKX2-1DEL proteins; the identity of the sequences obtained were verified by mass spectrometry (data not shown). As expected, the two mutated proteins had higher molecular weight than the wild type (Figures 1C–E). In silico analysis using a cNLS mapper bioinformatics program, predicted the absence of consensus nuclear localization signal in the two mutants. Considering the 3D conformation of the protein, it also showed the presence of an NLS bipartite that may explain the nuclear localization of both mutants (Supplementary Figure S1). To confirm these predictions, we then performed confocal microscopy analysis of A549 (Figure 2A) and Nthy cells (Figure 2B) transfected with both mutants and the wild-type NKX2-1. As expected with the loss of the NLS, both mutant proteins were localized in cytoplasm, but as predicted by the identification of putative bipartite NLSs, both mutants were also localized in the nucleus. However, they exhibited a very different pattern of expression in the nucleus as compared with the wild-type protein, forming aggregates in the nucleus in each cell line.
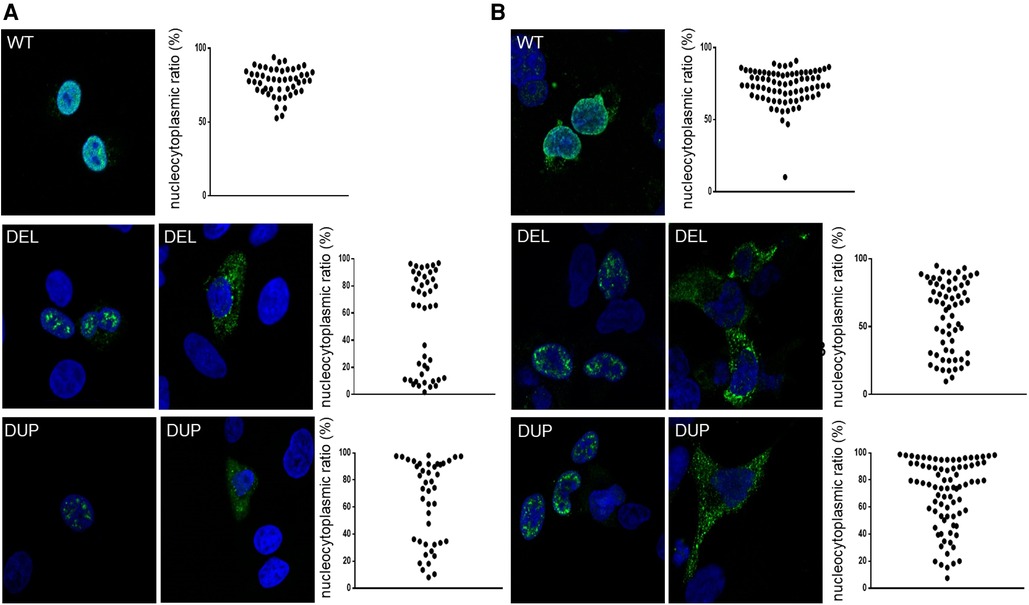
Figure 2. Representative confocal images of A549 cells (A) and nthy cells (B) after 24-h expression of NKX2-1WT, NKX2-1DEL and NKX2-1DUP. Immunostaining involved incubation with rabbit polyclonal anti-NKX2-1 antibody (green). Nuclei were detected by ToPro-3 iodide (blue). Scatter graphs show the nucleocytoplasmic ratio of each protein (≥10 cells per experiment, n = 3 or 4).
Transcriptional activity of NKX2-1wt, NKX2-1DUP and NKX2-1DEL on tissue-specific promoters
Given the nuclear localization of the two mutants, we then aimed to study their transcriptional activity. We measured the transcriptional activity of the mutant proteins by using representative target promoters specific for the tissues affected in BLTS: SFTPA, SFTPB, SFTPC, and ABCA3 for lung and thyroglobulin for thyroid. As compared with the wild-type protein, the two mutant proteins added to the four lung and thyroglobulin promoters showed significantly decreased activity (Figure 3). Transcriptional activity of thyroglobulin promoter was also assessed with the addition of the co-factor PAX8. Thyroglobulin promoter activation was significantly decreased by NKX2-1DUP as compared with the wild type (Figure 4). In contrast, in agreement with the clinical phenotype and despite the absence of homeodomain, thyroglobulin promoter activity was significantly increased by NKX2-1DEL, similar to the wild type (Figure 4).
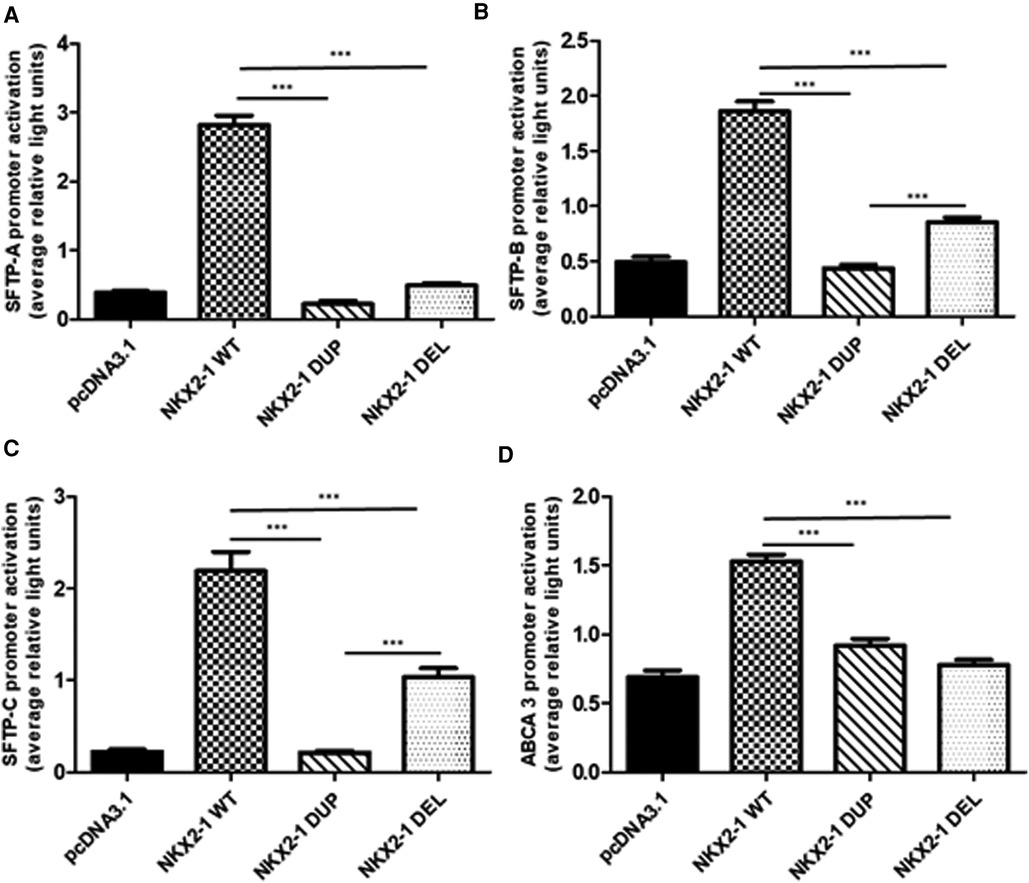
Figure 3. Functional activity of wild-type vs. mutant NKX2-1 on promoters of lung-specific genes (SFTPC, SFTPB, ABCA3 and SFTPA). Transactivation of SFTPA (A), SFTPB (B), SFTPC (C), and ABCA3 (D) by wild-type NKX2-1, NKX2-1DUP or NKX2-1DEL. Promoter constructs were co-transfected in A549 cells (A–D) with the empty pcDNA3.1 vector or expression vector for the wild-type or mutated NKX2-1 cDNAs. Data are mean ± SEM of five independent experiments performed in duplicate. *p < 0.05, **p < 0.01, ***p < 0.001.
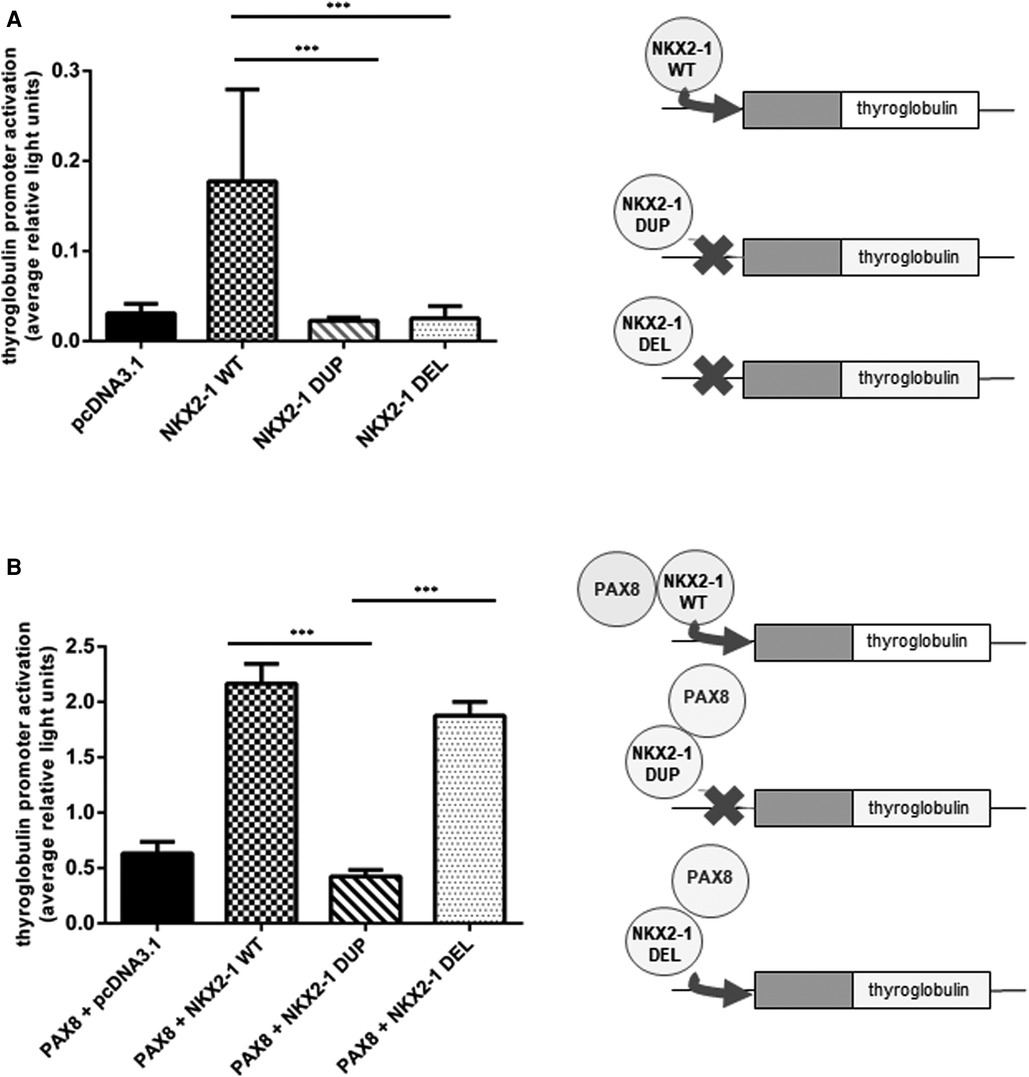
Figure 4. Functional activity of wild-type vs. mutants NKX2-1 on thyroid-specific (thyroglobulin) gene. Transactivation of the thyroglobulin promoter by NKX2-1WT, NKX2-1DUP or NKX2-1DEL. Promoter constructs were co-transfected without (A) or with (B) PAX8 in Nthy cells with the empty pcDNA3.1 vector or the expression vector for wild-type or mutated NKX2-1 cDNAs. Data are mean ± SEM of five independent experiments performed in duplicate. ***p < 0.001.
Discussion
In the present report, we describe two heterozygous pathogenic variants in the NKX2-1 transcription factor. The first frameshift pathogenic variant, c.85_86del, was carried by a girl who showed ILD during the first year of life. This girl and her father, who carried the same variant, exhibited only lung disease. The second variant, c.254dup, previously described (19), was carried by a boy who had BLTS. The boy had respiratory distress syndrome at birth, but the condition improved progressively without treatment. Congenital hypothyroidism was diagnosed at birth, and the boy also showed choreic movements and psychomotor delay.
These two variants shift the reading frame in the same way after the 86th amino acid, leading to aberrant proteins associated with different BLTS phenotypes. Variants (n = 10) that shift the reading frame in the same way leading to an aberrant protein of 406 amino acids with deletion of the functional homeodomain were previously described (10, 34–36). Seven of the 9 subjects described had BLTS; two of them had thyroid and neurological symptoms, and one presented thyroid and respiratory symptoms.
To improve our understanding of the molecular basis of this heterogenous phenotype, we first performed an in vitro study to compare the NKX2-1DUP and NKX2-1DEL mutant proteins. Indeed, western blots showed that both had higher molecular weight than NKX2-1WT, the weight of NKX2-1DUP being much higher than NKX2-1DEL in both Nthy and A549 cells. These discrepancies could be explained by the functional motifs within the amino acid sequence of the NKX2-1wt protein that were affected by the pathogenic variants (Figure 1B). NKX2-1 posttranslational modifications, such as phosphorylations, acetylations, and redox states could alter their molecular weight, mobility in electrophoresis assays and transcriptional activity (37). Among the five SP motifs present in the NKX2-1wt protein, three were absent in mutants, these latter being important in the phosphorylation state of the protein and its cellular-specific transcriptional activity (38). One putative new SP motif appeared at the Cter of both mutants and could also impact their phosphorylation state and thus their activity. Additionally, three cysteines involved in the dimerization of the wild-type protein (39) disappeared in both mutants, whereas four other cysteines appeared which could disturb the dimerization of the mutants and/or their redox state (40), and consequently affect the molecular weight. We also showed that despite the loss of the native NLS, the two mutant proteins could reach the nucleus, according to putative NLSs that could be present in both mutant sequences (Supplementary Figure S1). Indeed, in both cell lines, the mutants were in cytoplasm but also in the nucleus, where they formed aggregates instead of the diffuse appearance seen with the wild-type protein. The cell nucleus contains distinct classes of subnuclear structures (cajal bodies, splicing speckles, paraspeckles). Many nuclear proteins are known to interact dynamically with one or another of these bodies, and disruption of the specific organization of nuclear proteins can result in defects in cell functions and may cause molecular disease (41). Further experiments to explain the subnuclear re-localization of the two mutants in one or several of these specific nuclear structures are needed and would help refine the cartography of functional domains within the NKX2-1 protein sequence.
Because the mutant proteins can reach the nucleus, they could activate tissue-specific promoters such as thyroglobulin, a specific thyroid gene. Of note, unlike NKX2-1DUP, NKX2-1DEL was able to transactivate thyroglobulin but required a PAX8 cofactor for its activity. PAX8 and NKX2-1 are both expressed only in the thyroid, and PAX8 has a synergic effect with NKX2-1 to transactivate the thyroglobulin promoter (14, 17). Despite its highly modified amino acid, NKX2-1DEL protein could transactivate the thyroglobulin promoter with PAX8, which suggests that PAX8 is essential for the thyrocyte-specific promoter activation of thyroglobulin (42–44). Both mutants have lost their homeodomain and have equivalent sequence sizes. However, they differ significantly from amino-acids 30 to 88 in their sequences, suggesting that this part of the Nterm region could be of interest for a selective binding of the PAX8 cofactor and putative other factors, such as TAZ. Although the two mutants we studied have both lost the LPPY motif (illustrated in Figure 1B), which may be involved in the binding of TAZ protein (45), a mechanism involving both TAZ and PAX8 within this Nterm region of NKX2-1 could be possible. Interestingly, Moya et al. (36) showed that the TAZ cofactor could bind to the 74 first amino-acids of the NKX2-1 protein, maybe through pS-P in the Nterm region. Thus, further experiments focusing on binding of both TAZ and PAX8 on the 30-to-74 first amino acids of NKX2-1 could help to decipher their roles in the tissue-specificity of NKX2-1. Synergy with PAX8 can overcome the functional defect of a specific NKX2-1 variant on the thyroglobulin promoter (14). PAX8 may recruit NKX2-1 via protein–protein interaction without NKX2-1 binding to DNA (46, 47). For the present variants, we hypothesize that the loss of the NKX2-1DEL homeodomain was associated with the creation of a new interaction domain with PAX8, thus allowing for thyroglobulin promoter transactivation.
To gain insight into the mechanism of the pulmonary phenotype of our two subjects, we assessed transactivation of specific surfactant gene promoters, including SFTPA, SFTPB, SFTPC and ABCA3, in two cell lines. All these proteins are essential for surfactant homeostasis and normal lung development (48–53). Both mutant proteins resulted in decreased luciferase activity suggesting reduced transcriptional activity of the SFTPA, SFTPB, SFTPC and ABCA3 promoters, which is consistent with the pulmonary phenotype of our two subjects. In the 2 index children, the lung phenotype differed as one child presented with severe respiratory distress with rapid improvement with the first weeks, while the other experienced a progressive respiratory failure at 7 months evolving to chILD. Moya et al. have studied 2 children with BLTS, but one with absence of lung involvement. The authors demonstrated that the coactivator TAZ/WWTR1 (transcriptional coactivator with PDZ-binding motif/WW domain-containing transcription regulator protein (1) was able to restore NKX2-1 transactivation suggesting a potential role of TAZ in the development of the lung disease (36). Alternatively, we and others have shown that decreased surfactant protein levels may be associated with a less severe phenotype as compared with complete deficiency (54). The distinct respiratory phenotypes (chILD vs. resolved neonatal RDS) might be related to the differences in the level transcription of SP-B and SP-C which were significantly decreased in NKX2-1DUP compared to NKX2-1DEL. Indeed SP-B and SP-C protein levels are crucial in the adaptation of the newborn at extrauterine conditions (55) as illustrated by SP-B deficiency causing severe neonatal respiratory distress and death within the first few months of life (56) whereas chILD is mainly triggered by surfactant metabolism disruption due to abnormal proSP-C protein together with decrease ABCA3, SP-C and SP-B levels (57).
In conclusion, here we explain the difference in phenotype in two subjects carrying two frameshifts NKX2-1 pathogenic variants, c.85_86del and c.254dup. The two variants (dup + 1nt/del-2nt) produce an aberrant protein of 407 and 406 amino acids identical from the 86th amino acid lacking the functional homeodomain and the nuclear localization signal. Unexpectedly, 3D conformation prediction of the mutant protein assumed the presence of a nuclear localization signal, a bipartite sequence, which was experimentally confirmed by confocal microscopy showing both mutant proteins localized in the nucleus and cytoplasm. However, only the c.175_176del NKX2-1 mutant could transactivate the thyroglobulin promoter because of the PAX8 cofactor. Other functional studies are warranted to further elucidate the molecular mechanisms involved in NKX2-1 variants associated with heterogeneous phenotypes.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: 10.6084/m9.figshare.21898911.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
CD, AA, EN, PF, RE: designed the study. RE and PF: coordinated the study. CD, AA, EN, AB, SS, PF and RE: conceived of the experiments and drafted the manuscript. CD, AA, EN, VL, XD: performed experiments and analyzed data. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by public grants from the Institut National de la Santé et de la Recherche Médicale.
Acknowledgments
We are grateful to Laura Smales for English editing assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.978598/full#supplementary-material.
References
1. Hamdan H, Liu H, Li C, Jones C, Lee M, deLemos R, et al. Structure of the human Nkx2.1 gene. Biochim Biophys Acta. (1998) 1396(3):336–48. doi: 10.1016/S0167-4781(97)00210-8
2. Trueba SS, Auge J, Mattei G, Etchevers H, Martinovic J, Czernichow P, et al. Pax8, Titf1, and Foxe1 gene expression patterns during human development: new insights into human thyroid development and thyroid dysgenesis-associated malformations. J Clin Endocrinol Metab. (2005) 90(1):455–62. doi: 10.1210/jc.2004-1358
3. Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, et al. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. (1996) 10(1):60–9. doi: 10.1101/gad.10.1.60
4. Ghaffari S, Dougherty GJ, Eaves AC, Eaves CJ. Diverse effects of anti-Cd44 antibodies on the stromal cell-mediated support of normal but not leukaemic (cml) haemopoiesis in vitro. Br J Haematol. (1997) 97(1):22–8. doi: 10.1046/j.1365-2141.1997.d01-2139.x
5. Li C, Cai J, Pan Q, Minoo P. Two functionally distinct forms of Nkx2.1 protein are expressed in the pulmonary epithelium. Biochem Biophys Res Commun. (2000) 270(2):462–8. doi: 10.1006/bbrc.2000.2443
6. Oguchi H, Kimura S. Multiple transcripts encoded by the thyroid-specific enhancer-binding protein (T/ebp)/thyroid-specific transcription factor-1 (ttf-1) gene: evidence of autoregulation. Endocrinology. (1998) 139(4):1999–2006. doi: 10.1210/endo.139.4.5933
7. Nettore IC, Mirra P, Ferrara AM, Sibilio A, Pagliara V, Kay CS, et al. Identification and functional characterization of a novel mutation in the Nkx2-1 gene: comparison with the data in the literature. Thyroid. (2013) 23(6):675–82. doi: 10.1089/thy.2012.0267
8. Nattes E, Lejeune S, Carsin A, Borie R, Gibertini I, Balinotti J, et al. Heterogeneity of lung disease associated with Nk2 homeobox 1 mutations. Respir Med. (2017) 129:16–23. doi: 10.1016/j.rmed.2017.05.014
9. Maquet E, Costagliola S, Parma J, Christophe-Hobertus C, Oligny LL, Fournet JC, et al. Lethal respiratory failure and mild primary hypothyroidism in a term girl with a De Novo heterozygous mutation in the Titf1/Nkx2.1 gene. J Clin Endocrinol Metab. (2009) 94(1):197–203. doi: 10.1210/jc.2008-1402
10. Hamvas A, Deterding RR, Wert SE, White FV, Dishop MK, Alfano DN, et al. Heterogeneous pulmonary phenotypes associated with mutations in the thyroid transcription factor gene Nkx2-1. Chest. (2013) 144(3):794–804. doi: 10.1378/chest.12-2502
11. Whitsett JA, Weaver TE. Alveolar development and disease. Am J Respir Cell Mol Biol. (2015) 53(1):1–7. doi: 10.1165/rcmb.2015-0128PS
12. Wert SE, Whitsett JA, Nogee LM. Genetic disorders of surfactant dysfunction. Pediatr Dev Pathol. (2009) 12(4):253–74. doi: 10.2350/09-01-0586.1
13. Perez-Gil J. Structure of pulmonary surfactant membranes and films: the role of proteins and lipid-protein interactions. Biochim Biophys Acta. (2008) 1778(7-8):1676–95. doi: 10.1016/j.bbamem.2008.05.003
14. Carre A, Szinnai G, Castanet M, Sura-Trueba S, Tron E, Broutin-L'Hermite I, et al. Five new Ttf1/Nkx2.1 mutations in brain-lung-thyroid syndrome: rescue by Pax8 synergism in one case. Hum Mol Genet. (2009) 18(12):2266–76. doi: 10.1093/hmg/ddp162
15. Civitareale D, Lonigro R, Sinclair AJ, Di Lauro R. A thyroid-specific nuclear protein essential for tissue-specific expression of the thyroglobulin promoter. Embo J. (1989) 8(9):2537–42. doi: 10.1002/j.1460-2075.1989.tb08391.x
16. Mizuno K, Gonzalez FJ, Kimura S. Thyroid-Specific enhancer-binding protein (T/Ebp): cdna cloning, functional characterization, and structural identity with thyroid transcription factor ttf-1. Mol Cell Biol. (1991) 11(10):4927–33. doi: 10.1128/mcb.11.10.4927-4933.1991
17. Di Palma T, Nitsch R, Mascia A, Nitsch L, Di Lauro R, Zannini M. The paired domain-containing factor Pax8 and the homeodomain-containing factor ttf-1 directly interact and synergistically activate transcription. J Biol Chem. (2003) 278(5):3395–402. doi: 10.1074/jbc.M205977200
18. Gras D, Jonard L, Roze E, Chantot-Bastaraud S, Koht J, Motte J, et al. Benign hereditary chorea: phenotype, prognosis, therapeutic outcome and long term follow-up in a large series with new mutations in the Titf1/Nkx2-1 gene. J Neurol Neurosurg Psychiatry. (2012) 83(10):956–62. doi: 10.1136/jnnp-2012-302505
19. Pohlenz J, Dumitrescu A, Zundel D, Martine U, Schonberger W, Koo E, et al. Partial deficiency of thyroid transcription factor 1 produces predominantly neurological defects in humans and mice. J Clin Invest. (2002) 109(4):469–73. doi: 10.1172/JCI0214192
20. Sandberg M, Flandin P, Silberberg S, Su-Feher L, Price JD, Hu JS, et al. Transcriptional networks controlled by Nkx2-1 in the development of forebrain gabaergic neurons. Neuron. (2016) 91(6):1260–75. doi: 10.1016/j.neuron.2016.08.020
21. Du T, Xu Q, Ocbina PJ, Anderson SA. Nkx2.1 specifies cortical interneuron fate by activating Lhx6. Development. (2008) 135(8):1559–67. doi: 10.1242/dev.015123
22. Runkle EA, Rice SJ, Qi J, Masser D, Antonetti DA, Winslow MM, et al. Occludin is a direct target of thyroid transcription factor-1 (ttf-1/Nkx2-1). J Biol Chem. (2012) 287(34):28790–801. doi: 10.1074/jbc.M112.367987
23. Yan C, Sever Z, Whitsett JA. Upstream enhancer activity in the human surfactant protein B gene is mediated by thyroid transcription factor 1. J Biol Chem. (1995) 270(42):24852–7. doi: 10.1074/jbc.270.42.24852
24. Kelly SE, Bachurski CJ, Burhans MS, Glasser SW. Transcription of the lung-specific surfactant protein C gene is mediated by thyroid transcription factor 1. J Biol Chem. (1996) 271(12):6881–8. doi: 10.1074/jbc.271.12.6881
25. Matsuzaki Y, Besnard V, Clark JC, Xu Y, Wert SE, Ikegami M, et al. Stat3 regulates Abca3 expression and influences lamellar body formation in alveolar type ii cells. Am J Respir Cell Mol Biol. (2008) 38(5):551–8. doi: 10.1165/rcmb.2007-0311OC
26. Liu D, Hinshelwood MM, Giguere V, Mendelson CR. Estrogen related receptor-alpha enhances surfactant protein-a gene expression in fetal lung type ii cells. Endocrinology. (2006) 147(11):5187–95. doi: 10.1210/en.2006-0664
27. Vilain C, Rydlewski C, Duprez L, Heinrichs C, Abramowicz M, Malvaux P, et al. Autosomal dominant transmission of congenital thyroid hypoplasia due to loss-of-function mutation of Pax8. J Clin Endocrinol Metab. (2001) 86(1):234–8. doi: 10.1210/jcem.86.1.7140
28. Cho W, Hagemann TL, Johnson DA, Johnson JA, Messing A. Dual transgenic reporter mice as a tool for monitoring expression of glial fibrillary acidic protein. J Neurochem. (2009) 110(1):343–51. doi: 10.1111/j.1471-4159.2009.06146.x
29. Carre A, Stoupa A, Kariyawasam D, Gueriouz M, Ramond C, Monus T, et al. Mutations in borealin cause thyroid dysgenesis. Hum Mol Genet. (2017) 26(3):599–610. doi: 10.1093/hmg/ddw419
30. Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ, et al. Interpro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. (2017) 45(D1):D190–D9. doi: 10.1093/nar/gkw1107
31. Sigrist CJ, de Castro E, Cerutti L, Cuche BA, Hulo N, Bridge A, et al. New and continuing developments at prosite. Nucleic Acids Res. (2013) 41(Database issue):D344–7. doi: 10.1093/nar/gks1067
32. Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A. (2009) 106(25):10171–6. doi: 10.1073/pnas.0900604106
33. Madeira F, Pearce M, Tivey ARN, Basutkar P, Lee J, Edbali O, et al. Search and sequence analysis tools services from embl-ebi in 2022. Nucleic Acids Res. (2022) 50(W1):W276–9. doi: 10.1093/nar/gkac240
34. Thorwarth A, Schnittert-Hubener S, Schrumpf P, Muller I, Jyrch S, Dame C, et al. Comprehensive genotyping and clinical characterisation reveal 27 novel Nkx2-1 mutations and expand the phenotypic Spectrum. J Med Genet. (2014) 51(6):375–87. doi: 10.1136/jmedgenet-2013-102248
35. de Filippis T, Marelli F, Vigone MC, Di Frenna M, Weber G, Persani L. Novel Nkx2-1 frameshift mutations in patients with atypical phenotypes of the brain-lung-thyroid syndrome. Eur Thyroid J. (2014) 3(4):227–33. doi: 10.1159/000366274
36. Moya CM, Zaballos MA, Garzon L, Luna C, Simon R, Yaffe MB, et al. Taz/Wwtr1 mediates the pulmonary effects of Nkx2-1 mutations in brain-lung-thyroid syndrome. J Clin Endocrinol Metab. (2018) 103(3):839–52. doi: 10.1210/jc.2017-01241
37. Tagne JB, Gupta S, Gower AC, Shen SS, Varma S, Lakshminarayanan M, et al. Genome-Wide analyses of Nkx2-1 binding to transcriptional target genes uncover novel regulatory patterns conserved in lung development and tumors. PloS one. (2012) 7(1):e29907. doi: 10.1371/journal.pone.0029907
38. Zannini M, Acebron A, De Felice M, Arnone MI, Martin-Perez J, Santisteban P, et al. Mapping and functional role of phosphorylation sites in the thyroid transcription factor-1 (ttf-1). J Biol Chem. (1996) 271(4):2249–54. doi: 10.1074/jbc.271.4.2249
39. Arnone MI, Zannini M, Di Lauro R. The DNA binding activity and the dimerization ability of the thyroid transcription factor I are redox regulated. J Biol Chem. (1995) 270(20):12048–55. doi: 10.1074/jbc.270.20.12048
40. Tell G, Pines A, Paron I, D'Elia A, Bisca A, Kelley MR, et al. Redox effector factor-1 regulates the activity of thyroid transcription factor 1 by controlling the redox state of the N transcriptional activation domain. J Biol Chem. (2002) 277(17):14564–74. doi: 10.1074/jbc.M200582200
41. Fox AH, Lam YW, Leung AK, Lyon CE, Andersen J, Mann M, et al. Paraspeckles: a novel nuclear domain. Curr Biol. (2002) 12(1):13–25. doi: 10.1016/S0960-9822(01)00632-7
42. Mansouri A, Chowdhury K, Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet. (1998) 19(1):87–90. doi: 10.1038/ng0598-87
43. Pasca di Magliano M, Di Lauro R, Zannini M. Pax8 has a key role in thyroid cell differentiation. Proc Natl Acad Sci U S A. (2000) 97(24):13144–9. doi: 10.1073/pnas.240336397
44. Zannini M, Francis-Lang H, Plachov D, Di Lauro R. Pax-8, a paired domain-containing protein, binds to a sequence overlapping the recognition site of a homeodomain and activates transcription from two thyroid-specific promoters. Mol Cell Biol. (1992) 12(9):4230–41. doi: 10.1128/mcb.12.9.4230-4241.1992
45. Park KS, Whitsett JA, Di Palma T, Hong JH, Yaffe MB, Zannini M. Taz interacts with ttf-1 and regulates expression of surfactant protein-C. J Biol Chem. (2004) 279(17):17384–90. doi: 10.1074/jbc.M312569200
46. De Leo R, Miccadei S, Zammarchi E, Civitareale D. Role for P300 in pax 8 induction of thyroperoxidase gene expression. J Biol Chem. (2000) 275(44):34100–5. doi: 10.1074/jbc.M003043200
47. Grasberger H, Ringkananont U, Lefrancois P, Abramowicz M, Vassart G, Refetoff S. Thyroid transcription factor 1 rescues Pax8/P300 synergism impaired by a natural Pax8 paired domain mutation with dominant negative activity. Mol Endocrinol. (2005) 19(7):1779–91. doi: 10.1210/me.2004-0426
48. Kroner C, Wittmann T, Reu S, Teusch V, Klemme M, Rauch D, et al. Lung disease caused by Abca3 mutations. Thorax. (2017) 72(3):213–20. doi: 10.1136/thoraxjnl-2016-208649
49. Nogee LM, Dunbar AE 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. (2001) 344(8):573–9. doi: 10.1056/NEJM200102223440805
50. Delestrain C, Simon S, Aissat A, Medina R, Decrouy X, Nattes E, et al. Deciphering the mechanism of Q145h sftpc mutation unmasks a splicing defect and explains the severity of the phenotype. Eur J Hum Genet. (2017) 25(6):779–82. doi: 10.1038/ejhg.2017.36
51. Nogee LM, Wert SE, Proffit SA, Hull WM, Whitsett JA. Allelic heterogeneity in hereditary surfactant protein B (Sp-B) deficiency. Am J Respir Crit Care Med. (2000) 161(3 Pt 1):973–81. doi: 10.1164/ajrccm.161.3.9903153
52. Garcia-Laorden MI, Rodriguez de Castro F, Sole-Violan J, Rajas O, Blanquer J, Borderias L, et al. Influence of genetic variability at the surfactant proteins a and D in community-acquired pneumonia: a prospective, observational, genetic study. Crit Care. (2011) 15(1):R57. doi: 10.1186/cc10030
53. Giannoni E, Sawa T, Allen L, Wiener-Kronish J, Hawgood S. Surfactant proteins a and D enhance pulmonary clearance of pseudomonas aeruginosa. Am J Respir Cell Mol Biol. (2006) 34(6):704–10. doi: 10.1165/rcmb.2005-0461OC
54. Flamein F, Riffault L, Muselet-Charlier C, Pernelle J, Feldmann D, Jonard L, et al. Molecular and cellular characteristics of Abca3 mutations associated with diffuse parenchymal lung diseases in children. Hum Mol Genet. (2011) 21(4):765–75. doi: 10.1093/hmg/ddr508
55. Hillman NH, Kallapur SG, Jobe AH. Physiology of transition from intrauterine to extrauterine life. Clin Perinatol. (2012) 39(4):769–83. doi: 10.1016/j.clp.2012.09.009
56. Nogee LM, de Mello DE, Dehner LP, Colten HR. Brief report: deficiency of pulmonary surfactant protein B in congenital alveolar proteinosis. N Engl J Med. (1993) 328(6):406–10. doi: 10.1056/NEJM199302113280606
Keywords: child, NKX2-1, lung, surfactant protein, thyroid
Citation: Delestrain C, Aissat A, Nattes E, Gibertini I, Lacroze V, Simon S, Decrouy X, de Becdelièvre A, Fanen P and Epaud R (2023) Deciphering an isolated lung phenotype of NKX2-1 frameshift pathogenic variant. Front. Pediatr. 10:978598. doi: 10.3389/fped.2022.978598
Received: 26 June 2022; Accepted: 20 December 2022;
Published: 17 January 2023.
Edited by:
Ron Rubenstein, Washington University in St. Louis, United StatesReviewed by:
Jennifer Wambach, Washington University in St. Louis, United StatesF. Sessions Cole, Washington University in St. Louis, United States
© 2023 Delestrain, Aissat, Nattes, Gibertini, Lacroze, Simon, Decrouy, de Becdelièvre, Fanen and Epaud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pascale Fanen cGFzY2FsZS5mYW5lbkBpbnNlcm0uZnI= Ralph Epaud cmFscGguZXBhdWRAaW5zZXJtLmZy
†These authors have contributed equally to this work
Specialty Section: This article was submitted to Pediatric Pulmonology, a section of the journal Frontiers in Pediatrics