 Huan Xu
Huan Xu Yuxi Wen
Yuxi Wen Runming Jin
Runming Jin Hongbo Chen
Hongbo Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 06 September 2022
Sec. Pediatric Oncology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.975819
Acute myeloid leukemia (AML) is a hematological malignancy resulting from the genetic alterations and epigenetic dysregulations of the hematopoietic progenitor cells. One-third of children with AML remain at risk of relapse even though outcomes have improved in recent decades. Epigenetic dysregulations have been identified to play a significant role during myeloid leukemogenesis. In contrast to genetic changes, epigenetic modifications are typically reversible, opening the door to the development of epigenetic targeted therapy. In this review, we provide an overview of the landscape of epigenetic alterations and describe the current progress that has been made in epigenetic targeted therapy, and pay close attention to the potential value of epigenetic abnormalities in the precision and combinational therapy of pediatric AML.
Acute myeloid leukemia (AML) is a blood cancer resulting from the genetic alterations and epigenetic dysregulations of the hematopoietic stem/progenitor cells (1). Pediatric AML accounts for about 15% of children’s leukemias. One-third of children have relapses and approximately half of childhood leukemia-related deaths are caused by relapsed/refractory AML, even though overall survival (OS) has been improved significantly over the past decades (2). Therefore, more specific drugs and more precise therapeutic strategies are urgently needed to improve the outcomes and prolong the survival time of children with AML.
In recent decades, the rapid advancement of sequencing technologies has led to great progress in clarifying the molecular pathogenesis of AML. These advancements lay the ground work and pave the way for precise therapeutic strategy by uncovering genetic alterations and epigenetic dysregulations in AML. Rather than being the result of changes in DNA sequence itself, epigenetic modifications refer to the changes in gene expression inherited through cell division (3). Epigenetic modifications include histone modification, DNA methylation, and non-coding RNAs (ncRNAs), which contribute to initiating and sustaining epigenetic silencing (4). Recent studies have identified a significant role of epigenetic dysregulation in the pathogenesis of AML, and some recurrent somatic genetic alterations in pediatric AML could interfere with epigenetic regulation (5). Epigenetic modifications are frequently reversible, thus offering potential avenues for epigenetic targeted therapy using specific inhibitors (6). Epigenetic therapy has become a promising therapeutic strategy with many novel inhibitors being applied in adult with AML (7). Therefore, it is timely to consider the important role of epigenetic alterations and the potential targeted therapy in pediatric AML, to promote the development of precision therapy and improve the outcomes of children with AML.
Multiple epigenetic modifications regulate the transition from hematopoietic stem cells to lineage differentiation and maturation at the transcription level, including DNA methylation, histone modifications, and non-coding RNAs. Epigenetic dysfunction is common in most cancers, and extensive studies have also focused on the mechanisms of epigenetic dysregulation in AML, though AML has few mutations compared to other cancer types (5, 8). The current advances of epigenetic modifications in AML are briefly summarized below (Table 1).
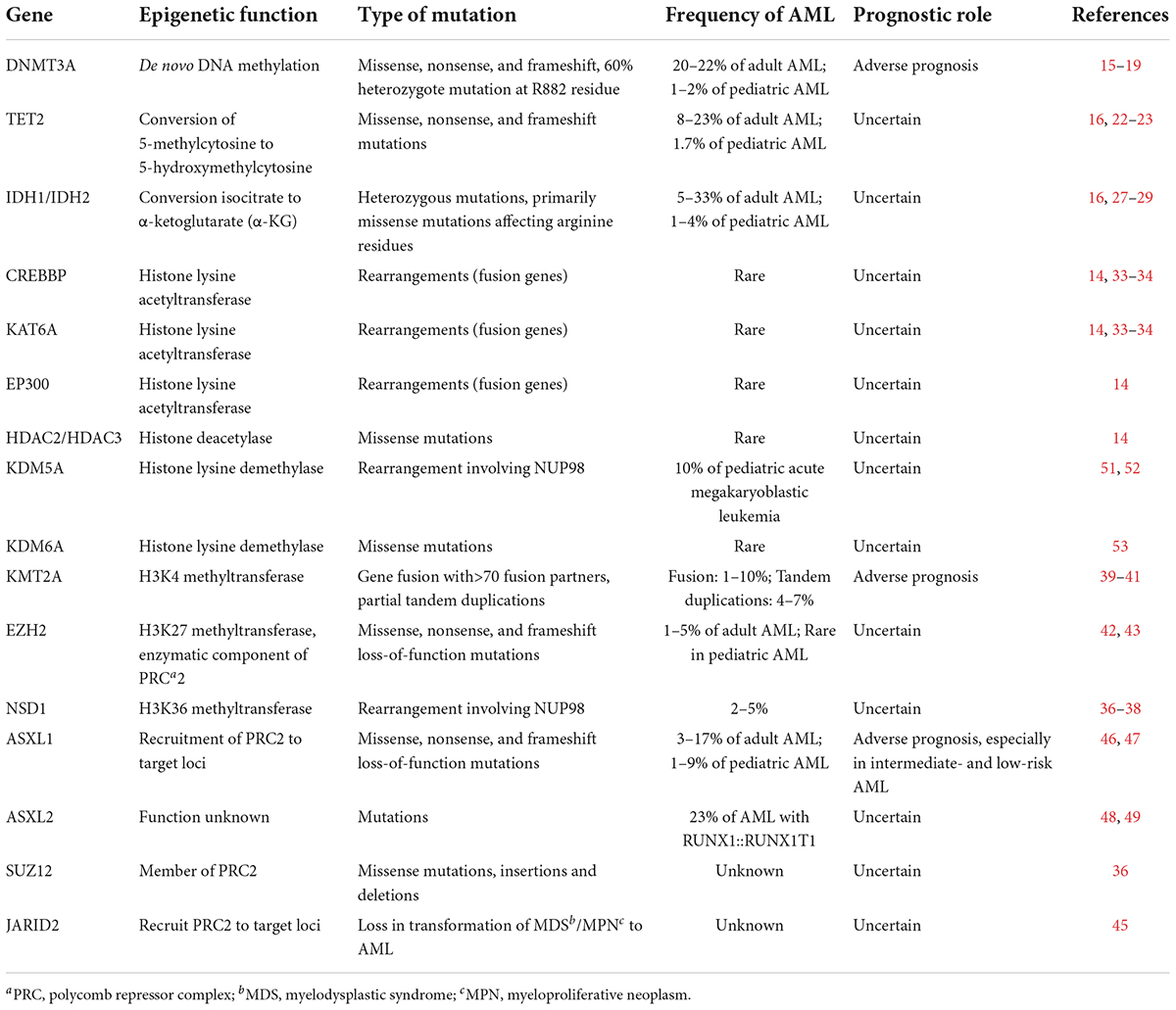
Table 1. Recurrently mutated or translocated genes in epigenetic modification in adult and pediatric AML.
The most well-characterized epigenetic change is DNA methylation, and abnormal methylation patterns have been discovered in gene silencing of tumor suppressor genes and genomic instability (9). The procedure of DNA methylation is the addition of a methyl group to the C5 position of cytosine residues in DNA to form 5-methylcytosine (5-mC). Most CpGs are methylated (70–80%), apart from the CG-dense regions termed CpG islands (CGIs) (10). Malignancy-related aberrant DNA methylation was originally studied in CGIs in gene promoters. Several DNA methyltransferases (DNMTs) and demethylases regulate the methylation modifications of CpGs. The former includes DNMT3A and DNMT3B, while the demethylation procedures are associated with the ten-eleven translocation (TET) family of demethylases (TET1, TET2, and TET3) (11).
Epigenome remodeling is essential for hematopoietic stem cell differentiation and maturation, and there is a direct correlation between DNA methylation patterns and specific cell types during hematopoiesis (12). Epigenetic abnormalities have been identified in AML, and several studies have implicated both hypermethylation and hypomethylation in malignant transformation (13–15). In adult AML, DNMT3A is one of the most commonly mutated genes (16), which occurs in pediatric AML at lower frequencies (20–22% vs. 1–2%, respectively) (17). DNMT3A mutations were reported to be early events in leukemogenesis and are predominately heterozygous R882H in AML (18). Some studies demonstrated that the DNMT3A mutation in hematopoietic stem cells resulted in damaged differentiation, enhanced self-renewal compared to wild-type hematopoietic stem cells, and conferred a poor outcome (19, 20). Meanwhile, DNMT3A mutations have been reported to increase chemotherapy resistance and the risk of relapse (21). It is still not well identified how DNMT3A mutations result in leukemic transformation, but targeting DNMT3A mutations could be a promising therapeutic strategy (22).
TET2 mutations are another pathway to abnormal DNA methylation. TET2 catalyzes the oxidation procedure of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC), resulting in DNA demethylation and reversing the gene silencing driven by DNA methylation (23). Mutations in TET2 occur in 8–23% of patients with AML, but these mutations are observed rarely in children with AML (Table 1) (17). TET2 mutations are associated with reduced levels of 5-hmC, and are related to a poor prognosis in intermediate-risk AML (24, 25). Li et al. found that TET2 knockout HSCs were amplified in vivo and outperformed wild-type HSCs in serial transplantation assays (26). Besides, Rasmussen et al. demonstrated that TET2 deletion led to DNA hypermethylation of the active enhancers, which was related to the upregulation of IGFLR, NOTCH3, and other oncogenes, and the downregulation of tumor suppressor genes, such as LXN and CTDSP1 (27). Subsequently, Shih et al. revealed that TET2 defect and FLT3-ITD mutations remodeled synergistically DNA methylation and gene expression to an extent not seen in either of the mutation alone, including at the GATA2 locus. Then they found that re-expression of GATA2 induced differentiation in AML stem cells and abated leukemogenesis. Consequently, they concluded that the mutations of TET2 and FLT3-ITD induced AML synergistically characterized by site-specific changes in DNA methylation and gene expression (28).
The conversion of isocitrate to α-ketoglutarate (α-KG) is catalyzed by isocitrate dehydrogenase 1/2 (IDH1/2). The conversion of 5-mC to 5-hmC and subsequent DNA demethylation is also the result of combined effect of IDH1/2 and TET2 (29). Mutations in IDH1/2 are frequently observed in adult AML (5–33%), less observed in pediatric AML (1–4%) (17, 30, 31). Mutations in IDH1/2 result in the synthesis of tumor metabolite 2-hydroxyglutarate (2-HG), leading to aberrant DNA methylation (32). Mutational epigenetic profiling revealed that AML cells with IDH1/2 mutations showed global DNA hypermethylation and a specific hypermethylation characteristic, particularly at promoter regions in a large AML patient cohort study (33). Figueroa et al. found that IDH1/2 mutations and TET2 mutations were mutually repellent, and loss-of-function mutation of TET2 was similar to the epigenetic alterations of IDH1/2 mutations. Furthermore, either TET2 depletions or IDH1/2 mutations increased progenitor cell marker expression and damaged hematopoietic differentiation, cooperatively contributing to leukemogenesis (33). Similar to AML with TET2 or IDH1/2 mutations, Rampal et al. identified reduced 5-hmC levels in WT1 mutant AML patients and they found that the overexpression of WT1 increased global levels of 5-hmC, whereas 5-hmC levels were reduced when WT1 was silenced (34). They also demonstrated that WT1 physically interacts with TET2 and TET3, and loss-of-function WT1 caused a hematopoietic differentiation phenotype similar to that observed with TET2 defects (34). Subsequently, Wang et al. also demonstrated that WT1 physically interacts with and recruits TET2 to its target genes, and AML-derived TET2 mutations disrupt the interaction (35). Despite the mechanism by which WT1 silencing or mutations lead to decreased 5-hmC is not completely clarified, the TET2, IDH1/2, and WT1 mutations lead to dysregulated DNA hydroxymethylation, which could be classified as a new subtype of AML.
Histone acetylation is the transformation of acetyl groups to lysine residues in histone proteins, which is modulated by histone lysine acetyltransferases (KATs) and histone deacetylases (HDACs). Histone acetylation plays an important role in gene transcription, chromatin structure, and DNA repair (36). Lysine acetylation leads to open chromatin confirmations and gene activation, whereas lysine deacetylation leads to condensed and closed chromatin, causing gene inactivation (37). KAT rearrangements (as opposed to mutations) occur in AML at extremely low frequencies (15). Although HDACs mutations occur also exceptionally rarely in children with AML, myeloid oncoproteins and leukemia-associated fusions can recruit HDACs abnormally, such as EVI1, RUNX1::RUNX1T1 (previously AML1::ETO), so as to block differentiation and maintain the leukemic phenotype of AML (38).
Histone lysine methylation is regulated by lysine methyltransferases (KMTs), which have several different degrees of methylation, including monomethylation, dimethylation, and trimethylation (39). It has been reported that the methylation of histone lysine could change the affinity of reader proteins to the methylated histone (40). The different target residues and the degree of methylation have distinct effects on transcription levels, and different methylation states could produce different functional effects in the same lysine residue (41). For instance, activation-related marks include methylation of H3K4, H3K79, and H3H36, while the methylation of H3K27, H3K9, and H4K20 contributes to silenced gene transcription (41). In AML, KMTs include components of the polycomb repressor complexes (PRCs) and mixed-lineage leukemia (MLL) proteins, which are frequently involved in translocations or are mutated.
MLL, or called KMT2A belongs to the family of SET domain-containing KMTs. MLL can make transcription activation by targeting H3K4. In AML, a histone methyltransferase DOT1 is recruited by MLL fusion proteins, so as to cause abnormal methylation of H3K79 at MLL gene targets and increase the expression of leukemia-related genes (42). MLL translocations are more common in pediatric than adult AML (30–50% vs. >10%, respectively), which are the most frequent alteration in infant AML (43, 44). Therefore, it is a promising area to develop the inhibitors of this complex and its enzymatic co-factors for infant and pediatric AML.
Enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) is part of the PRC2 polycomb repressor complex, which is an H3K27 methyltransferase to catalyze dimethylation and trimethylation of H3K27, resulting in the suppression of transcription (45, 46). Neff et al. found that EZH2 was necessary for tumor progression rather than leukemogenesis in KMT2A::MLLT3 (previously MLL::AF9) leukemia (47). Another member of the PRC2 complex Jumonji AT-rich interactive domain 2 (JARID2) similarly recruits PRC2 to specific target loci. Puda et al. reported that deletions of PRC2 complex members, especially JARID2, contribute to the leukemic transformation of chronic myeloid disorders (48). Additional sex combs like transcriptional regulator 1/2 (ASXL1/2) mutations occur more frequently in adult than pediatric AML (3–15% vs. 1–9%, respectively) (49, 50). The PRC2-associated protein ASXL1 can recruit PRC2 to its target loci, and mutations of ASXL1 lead to deletion of methylation regulated by PRC2. It has been reported that ASXL1 mutations contributed to the poor prognosis of AML (51). Besides, mutations of ASXL2 are commonly found in patients with the RUNX1::RUNX1T1 fusion gene, and ASXL1 and ASXL2 mutations are mutually exclusive (52).
Histone lysine demethylation is regulated by lysine demethylases (KDMs), which remove the methylation labels on lysine. KDMs include the amine oxidases and Jumonji domain containing proteins (JmjC). Lysine-specific demethylase 1 (LSD1/KDM1A) is the amine oxidase, which has specificity for H3K4 and H3K9 as a transcriptional activator or a transcriptional repressor (53). The demethylase of JmjC lysine, KDM5A (JARID1), is associated with NUP98 fusion protein in about 10% of pediatric acute megakaryoblastic leukemia (54, 55), and the mutation of KDM6A, another member of the JmjC family, occurs rarely in AML (56).
As epigenetic readers, the bromodomain and extra-terminal (BET) protein family members, including BRD2, BRD3, and BRD4, bind to acetylated lysine residues on histone tails (57). The process can initiate chromatin-mediated signal transduction, so as to achieve normal or tumor-dependent functions (58, 59). Some studies have reported that BRD4 could promote the abnormal expression of pivotal oncogenes, including c-Myc and Bcl-2 in AML (60, 61). BET inhibitors can alter the expression of particular genes, despite BRD4 and other BET proteins ubiquitously express at gene promoters and enhancers (62). Zuber et al. indicated that inhibition of BET proteins can block abnormal transcription of some oncogenes associated with leukemia, thus blocking the upregulation of leukemic stem cell (LSC) self-renewal programs and inducing differentiation (63). Furthermore, Dawson et al. demonstrated that BET inhibition could exhibit profound anti-leukemic effects against human and murine MLL-fusion leukemic cell lines, and they also identified the effects in mouse models of murine KMT2A::MLLT3 and human KMT2A::AFF1 (previously MLL::AF4) leukemia (64). Inhibition of tumor cell dependence on high oncogene expression by BET inhibitors is a promising therapeutic strategy, and malignant cells may be eliminated in the therapeutic window that preserves normal hematopoietic cells.
With the rapid advancement of RNA sequencing technology, more and more ncRNA have been discovered that are closely associated with AML leukemogenesis. There is accumulating evidence that ncRNAs play an important role in the pathogenesis of hematological malignancies, especially AML (65). Over the last few decades, the understanding of ncRNAs promote the improvement of the diagnosis, treatment, and prognosis of AML. NcRNA can be classified as housekeeping RNA and regulatory RNA according to their different functions. The regulatory RNA molecules are widely associated with gene transcription and translation, which include microRNAs (miRNAs, 19–24 bp), long non-coding RNAs (lncRNAs, >200 bp), and circular RNAs (circRNAs) (66).
MiRNAs have about 22 nucleotides that bind to the 3’-untranslated regions (3’-UTR) of the target gene and posttranscriptionally suppress the expression level of the target gene (67). Numerous studies have implicated miRNAs in regulating hematopoiesis. Oshima et al. demonstrated that EZH2 cooperated with miRNA let-7 to inhibit HSC function (68). Bolouri et al. performed miRNA sequencing of 152 samples from pediatric AML patients and found a relationship between gene abnormalities and miRNA expression. They found the high expression of miRNA-10a in AML with the mutations of NPM1 and high miRNA-21 expression in Core Binding Factor (CBF)-AMLs (69). Some studies have shown that miRNA-155 was associated with poor prognosis of adult and pediatric AML (70). Zhu et al. analyzed the connection of the miRNA data and clinical data of 229 patients and verified that the high expression of has-miR-542 and has-miR-509 were independent adverse prognostic factors, while has-miR-146a and has-miR-3667 were favorable factors (71).
CircRNAs are ubiquitous, stable, and conserved ncRNAs, which are single-stranded RNA molecules (72). Nicolet et al. demonstrated that the expression of circRNA is cell-type specific and increases during hematopoietic differentiation after analyzing circRNA expression in human hematopoietic progenitors and differentiated lymphoid and myeloid cells (73). Liu et al. utilized a circRNA microarray to analyze the expression pattern of circRNAs in children with AML. Then they verified that circRNF220 was specifically enriched and accumulated in peripheral blood and bone marrow of children with AML (74). Subsequently, they showed that circRNF220 has highly specificity and efficiency in the diagnosis of AML. Meanwhile, they demonstrated that the expression of circRNF220 independently predicted prognosis and high expression of circRNF220 was an adverse prognostic marker for relapse of children with AML. Recently, Wang et al. showed that circ_0040823 sponged miR-516b to inhibit proliferation and induced apoptosis of AML cells (75).
LncRNAs are more than 200 nucleotides in length and lack a meaningful open reading frame (76). Relatively fewer studies have investigated lncRNAs in pediatric AML. It has been reported that urothelial carcinoma-associated 1(UCA1) could maintain the proliferation of AML cells (77, 78). Recently, Liang et al. found that the expression of UCA1 was increased and the expression of miR-204 was inhibited in pediatric patients with AML. Besides, the downregulation of UCA1 suppressed cell proliferation and facilitated apoptosis by upregulating miR-204 in pediatric AML (79). Ma et al. identified that lncRNA LINC00909 promoted cell proliferation and metastasis by miR-625-mediated modulation of the Wnt/β-catenin signal pathway in pediatric AML (80).
The role of miRNAs in the pathogenesis and prognosis of AML is the most studied. However, the mechanism of miRNAs in AML is still complicated and poorly understood. Recently, lncRNAs and circRNAs have been found to participate in the miRNA network and act as competing endogenous RNAs (ceRNAs) and miRNA sponges to regulate the expression of miRNAs in AML (65). It is a promising area to find the crossover of the three ncRNAs to help understand the connection between these three ncRNAs.
The pivotal role of epigenetic modifications in AML has stimulated efforts to study epigenetically targeted drugs. Furthermore, epigenetic targeted therapies provide more chances for patients with AML thanks to the inherent reversibility of epigenetic marks. Numerous clinical trials are ongoing to study the epigenetic targeted therapies in adults with AML, either alone or combinational therapy. Despite mutations of epigenetic modifications are observed less commonly in pediatric AML compared to adult AML, more and more clinical trials are focusing on the important role of epigenetic modifications and targeted therapy in pediatric AML, and a series of small molecules that inhibit epigenetic regulators activity are currently studied in various clinical trial stages (Table 2).
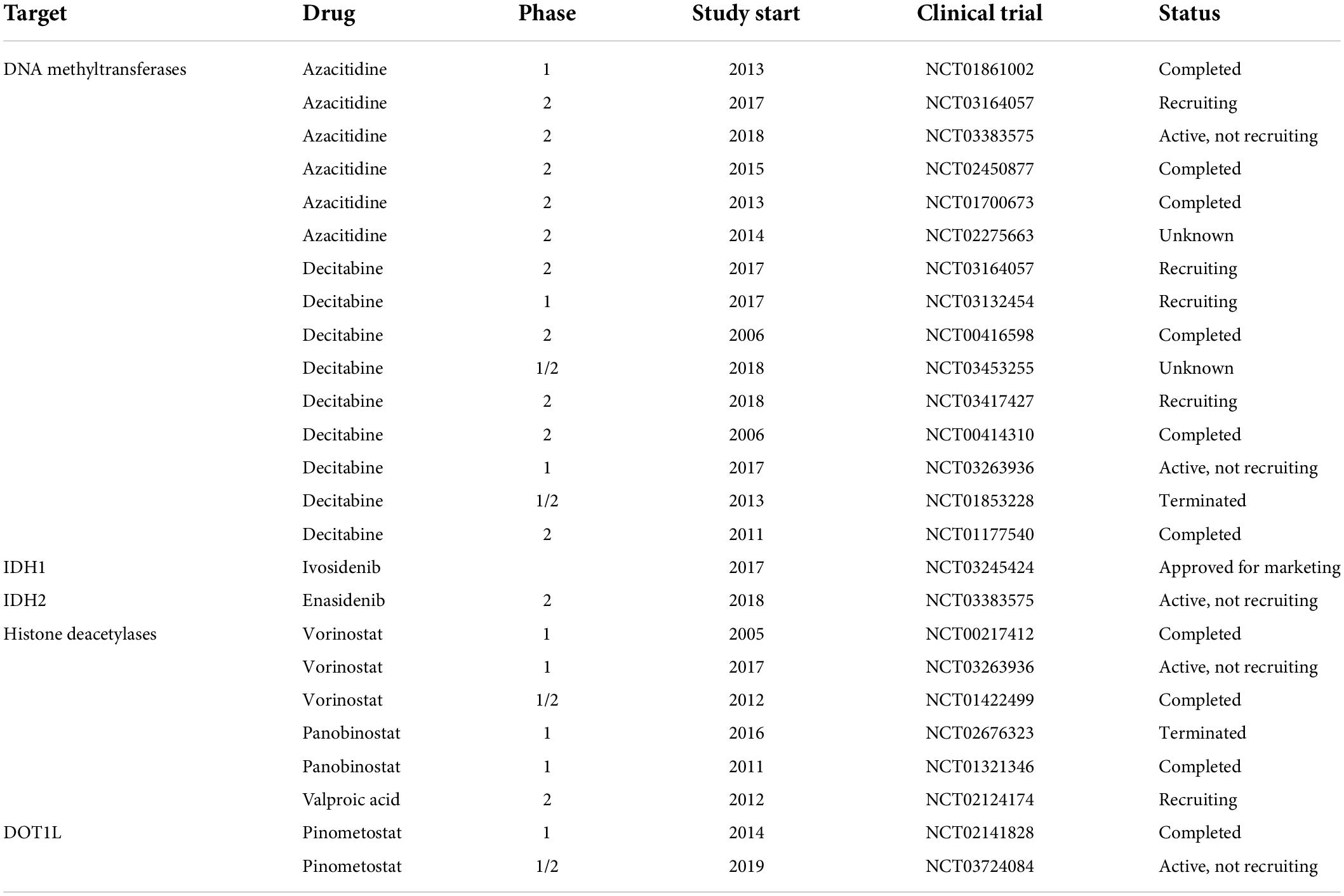
Table 2. Clinical trials of epigenetic targeted therapies in pediatric AML.
DNA methyltransferase inhibitors (DNMTi) or called hypomethylating agents (HMAs) include 5-azacytidine (azacitidine) and 5-aza-2’-deoxycytidine (decitabine). HMAs can alter DNA methylation patterns, promote the expression of tumor suppressors, and increase apoptosis (81). HMAs are the best-established epigenetic therapies in adult AML, which have shown efficacy and safety in older patients with AML. HMAs covalently bond with DNMTs irreversibly, which leads to proteasomal degradation of DNMTs, resulting in hypomethylation and transcriptional repression, and direct cytotoxic effects through DNA damage (Figure 1) (82). Several studies have demonstrated that the action of azacitidine is not limited to DNA demethylation (83). Schaefer et al. found that azacitidine inhibited the RNA methyltransferase DNMT2, which is variably expressed in human cancer cell lines (84). Subsequently, some studies have identified the novel targets of azacitidine in RNA methylation, which provides a new insight into its more widespread clinical use either or in combination in AML (85, 86). HMAs have been demonstrated to prolong overall survival (OS) compared to standard treatment in adult patients (87), and Stahl et al. identified the important role of HMAs in relapsed/refractory AML in a large patient cohort study (88). Despite the low frequency of mutations in DNMTs in pediatric AML, some studies have shown that DNMTi might be efficient in pediatric AML. Gore et al. demonstrated that decitabine is feasible and well- tolerated in children with newly diagnosed AML before standard combination chemotherapy (89). Subsequently, Sun et al. identified that azacitidine was safe when utilized in sequence with intensive chemotherapy in pediatric relapsed/refractory AML (90). Some studies have indicated that HMAs only have limited efficacy and difficulty leading to sustained remission when used as a single agent in adult AML (91), thus further preclinical and clinical studies should focus on the combination therapy with other chemotherapy agents, targeted drugs, and immunotherapy.
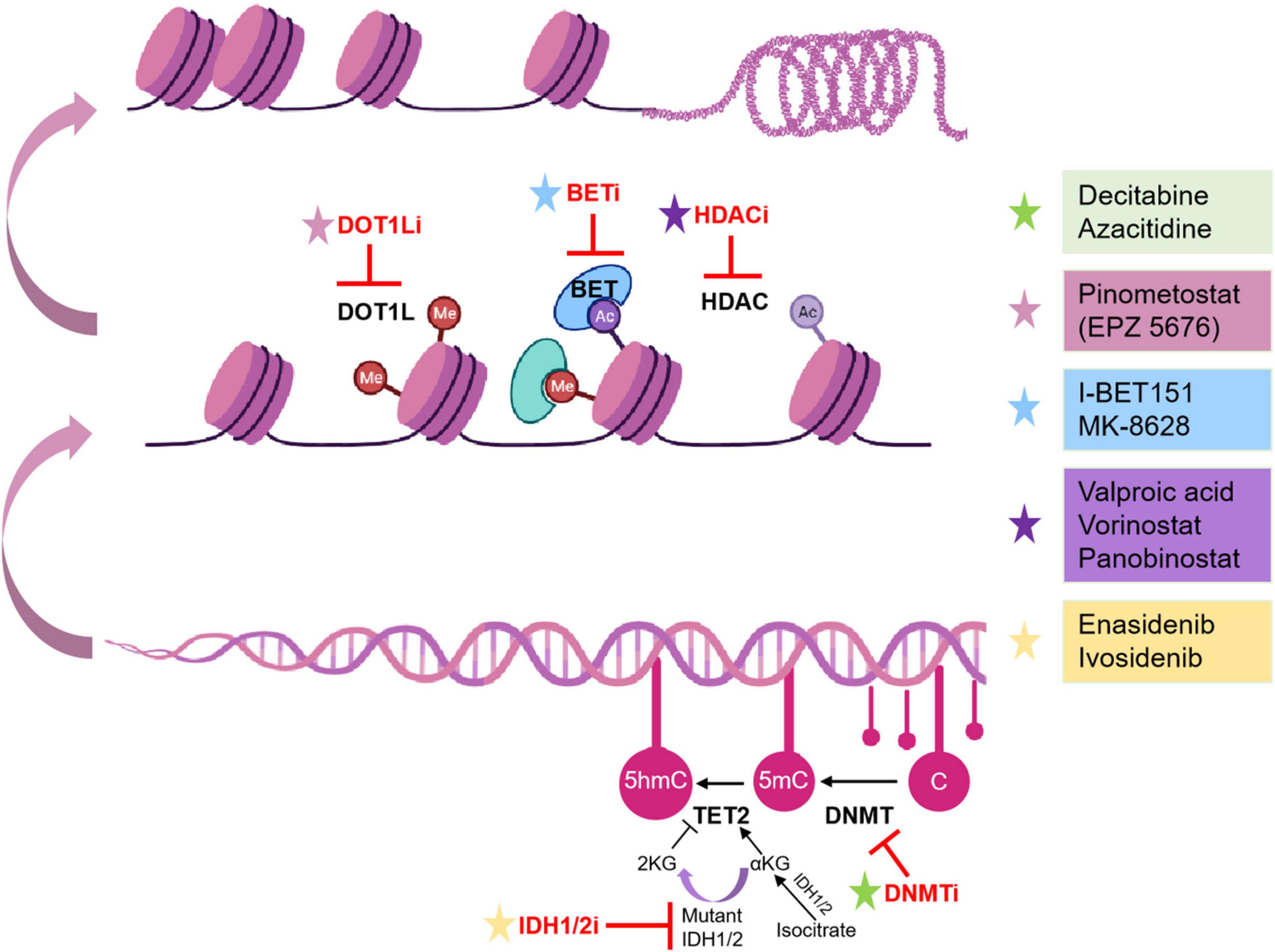
Figure 1. Epigenetic inhibitors and mechanisms in pediatric AML. Epigenetic inhibitors are highlighted in red. C, cytosine; Ac, acetylation.
IDH mutations have been identified to block cell differentiation by competitively inhibiting α-KG-dependent dioxygenases involved in histone and DNA demethylation (92, 93). Current IDH inhibitors selectively inhibit mutant IDH protein and promote the terminal differentiation of abnormal myeloid cells in vitro and in vivo models (94). Stein et al. firstly reported the IDH inhibitor, enasidenib, could be used in relapsed/refractory IDH2-mutant AML in a phase 1/2 clinical trial (NCT01915498) (95). Then DiNardo et al. reported that ivosidenib, a small-molecule inhibitor of IDH1, promoted durable remission and molecular remission in patients with CR in a phase 1 clinical trial of patients with advanced IDH1-mutant relapsed/refractory AML (96). However, few clinical trials accept children with AML at present. The only clinical trial currently that accepts pediatric AML patients is investigating the synergistic effect of azacytidine and enasidenib in myelodysplastic syndrome (MDS) with IDH2 mutation (NCT03383575), whereas there is no ongoing trial in pediatric AML yet.
HDAC inhibitors (HDACi) can activate tumor suppressor genes and promote tumor cell killing, which have been evaluated in clinical trials for adult AML patients with limited efficacy, either alone or in combination with chemotherapy (97). Leukemia-associated fusion proteins have been reported to block gene expression by recruitment of HDACs, which could be alleviated by inhibiting HDACs, causing differentiation of leukemic blasts. Meanwhile, these fusion proteins are more frequent in pediatric AML, raising the possibility that children with AML may benefit more from HDAC inhibitors than adult patients. Karol et al. reported that panobinostat could be safely administrated with chemotherapy and increased histone acetylation in a phase 1 clinical trial (98). Recently, Pommert et al. demonstrated that the combination therapy of decitabine and vorinostat with fludarabine, cytarabine, and G-CSF (FLAG) was well-tolerated and effective in pediatric patients with relapsed/refractory AML in phase 1 clinical trial (NCT02412475) (99). The results of these clinical trials will promote the development of HDAC inhibitors in pediatric patients with AML.
Owing to more frequent MLL translocations of pediatric AML than adult AML (30–50% vs. >10%, respectively), highly promising therapeutic approaches can be applied in MLL-associated leukemias. Inhibition of DOT1L blocks MLL target gene expression by regulating the aberrant methylation of H3K79. Pinometostat (EPZ-5676) is a DOT1L inhibitor that has been applied in relapsed/refractory AML patients with MLL rearrangements in a phase 1 clinical trial (100). However, the study reported only temporary reductions in leukemic blasts (NCT02141828). Despite the lack of clinical benefit of EPZ-5676 as a single agent, preclinical and clinical studies are warranted to evaluate the combinational efficacy of DOT1L inhibitors and conventional regimens in relapsed/refractory AML.
BET inhibitors have an important role in inhibiting histone acetylation. Dawson et al. showed that I-BET151, a small-molecule inhibitor of the BET family, was effective against murine and human leukemic cell lines with MLL-fusion by inducing early cell cycle arrest and apoptosis (64). Then they indicated that IBET151 significantly promoted survival in two distinct mouse models of murine KMT2A:MLLT3 and human KMT2A::AFF1leukemia (64). These results provide a promising epigenetic therapy target for pediatric AML. Other BET inhibitors, including MK-8628 (OTX015) and CPI-0610, also are now being evaluated in phase 1 and 2 trials in adult AML (NCT02698189, NCT02158858) (101).
Although the prognosis for children with AML has improved in recent decades, it remains poor due to the high risk of relapse and few therapeutic choices available when initial treatment fails. There remains an urgent need for better and more precise therapeutic approaches for patients with AML, especially for pediatric AML. Rapid advancements of next-generation sequencing technologies have contributed to understanding genetic alterations and epigenetic abnormalities of AML, and promoting the development of precise therapeutic strategies. Based on the clinical efficacy of epigenetic therapies in adult AML, the preclinical and clinical study of epigenetic targeted therapy needs more attention, despite the low frequency of epigenetic alterations in children with AML. Some inhibitors of epigenetic targeted therapies have benefited many children with the appropriate patient stratification. Meanwhile, it is critical that more personalized medicine will need more precise and appropriate patient stratification with different genetic and epigenetic alterations. One drug is unlikely to be curative in AML, either epigenetic targeted drugs or other inhibitors, which give opportunities for drug resistance and increase the risk of relapse due to the clonal heterogeneity. Several clinical trials have demonstrated that the Bcl-2 inhibitor, venetoclax plus HMAs dramatically improved CR rates in elderly AML patients (102). And venetoclax plus azacitidine was approved by FDA in newly diagnosed AML ineligible for induction chemotherapy in 2018. Recently, IDH1 inhibitor, ivosidenib in combination with azacitidine was also approved for newly diagnosed AML (103). Further preclinical and clinical studies should focus on the combination therapeutic strategies of epigenetic targeted drugs with other inhibitors and immunotherapy to promote cell killing and improve the prognosis for children with AML.
HX and YW collected the data and wrote the manuscript. HC and RJ reviewed and finalized the manuscript. All authors have read and agreed to the published version of the manuscript.
This work was supported by the National Natural Science Foundation of China (No. 31701207).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tarlock K, Meshinchi S. Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am. (2015) 62:75–93. doi: 10.1016/j.pcl.2014.09.007
2. Lonetti A, Pession A, Masetti R. Targeted therapies for pediatric AML: gaps and perspective. Front Pediatr. (2019) 7:463. doi: 10.3389/fped.2019.00463
3. Chen Y, Hong T, Wang S, Mo J, Tian T, Zhou X. Epigenetic modification of nucleic acids: from basic studies to medical applications. Chem Soc Rev. (2017) 46:2844–72. doi: 10.1039/C6CS00599C
4. Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. (2019) 571:489–99. doi: 10.1038/s41586-019-1411-0
5. Jones L, McCarthy P, Bond J. Epigenetics of paediatric acute myeloid leukaemia. Br J Haematol. (2020) 188:63–76. doi: 10.1111/bjh.16361
6. Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. (2016) 127:42–52. doi: 10.1182/blood-2015-07-604512
7. Döhner H, Wei AH, Löwenberg B. Towards precision medicine for AML. Nat Rev Clin Oncol. (2021) 18:577–90. doi: 10.1038/s41571-021-00509-w
8. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. (2012) 150:12–27. doi: 10.1016/j.cell.2012.06.013
9. Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. (1975) 187:226–32. doi: 10.1126/science.187.4173.226
10. Nishiyama A, Nakanishi M. Navigating the DNA methylation landscape of cancer. Trends Genet. (2021) 37:1012–27. doi: 10.1016/j.tig.2021.05.002
11. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. (2016) 30:733–50. doi: 10.1101/gad.276568.115
12. Farlik M, Halbritter F, Müller F, Choudry FA, Ebert P, Klughammer J, et al. DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell. (2016) 19:808–22. doi: 10.1016/j.stem.2016.10.019
13. Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. (2012) 8:e1002781. doi: 10.1371/journal.pgen.1002781
14. Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. (2010) 17:13–27. doi: 10.1016/j.ccr.2009.11.020
15. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
16. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. (2010) 363:2424–33. doi: 10.1056/NEJMoa1005143
17. Liang DC, Liu HC, Yang CP, Jaing TH, Hung IJ, Yeh TC, et al. Cooperating gene mutations in childhood acute myeloid leukemia with special reference on mutations of ASXL1, TET2, IDH1, IDH2, and DNMT3A. Blood. (2013) 121:2988–95. doi: 10.1182/blood-2012-06-436782
18. Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. (2014) 25:442–54. doi: 10.1016/j.ccr.2014.02.010
19. Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. (2011) 44:23–31. doi: 10.1038/ng.1009
20. Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. (2015) 125:629–38. doi: 10.1182/blood-2014-08-594648
21. Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med. (2016) 22:1488–95. doi: 10.1038/nm.4210
22. Brunetti L, Gundry MC, Goodell MA. DNMT3A in leukemia. Cold Spring Harb Perspect Med. (2017) 7:a030320. doi: 10.1101/cshperspect.a030320
23. Bowman RL, Levine RL. TET2 in normal and malignant hematopoiesis. Cold Spring Harb Perspect Med. (2017) 7:a026518. doi: 10.1101/cshperspect.a026518
24. Metzeler KH, Maharry K, Radmacher MD, Mrózek K, Margeson D, Becker H, et al. TET2 mutations improve the new European leukemianet risk classification of acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. (2011) 29:1373–81. doi: 10.1200/JCO.2010.32.7742
25. Zhang T, Zhao Y, Zhao Y, Zhou J. Expression and prognosis analysis of TET family in acute myeloid leukemia. Aging (Albany N Y). (2020) 12:5031–47. doi: 10.18632/aging.102928
26. Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. (2011) 118:4509–18. doi: 10.1182/blood-2010-12-325241
27. Rasmussen KD, Jia G, Johansen JV, Pedersen MT, Rapin N, Bagger FO, et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. (2015) 29:910–22. doi: 10.1101/gad.260174.115
28. Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. (2015) 27:502–15. doi: 10.1016/j.ccell.2015.03.009
29. Montalban-Bravo G, DiNardo CD. The role of IDH mutations in acute myeloid leukemia. Future Oncol. (2018) 14:979–93. doi: 10.2217/fon-2017-0523
30. Damm F, Thol F, Hollink I, Zimmermann M, Reinhardt K, van den Heuvel-Eibrink MM, et al. Prevalence and prognostic value of IDH1 and IDH2 mutations in childhood AML: a study of the AML-BFM and DCOG study groups. Leukemia. (2011) 25:1704–10. doi: 10.1038/leu.2011.142
31. Valerio DG, Katsman-Kuipers JE, Jansen JH, Verboon LJ, de Haas V, Stary J, et al. Mapping epigenetic regulator gene mutations in cytogenetically normal pediatric acute myeloid leukemia. Haematologica. (2014) 99:e130–2. doi: 10.3324/haematol.2013.094565
32. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. (2010) 17:225–34. doi: 10.1016/j.ccr.2010.01.020
33. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. (2010) 18:553–67. doi: 10.1016/j.ccr.2010.11.015
34. Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. (2014) 9:1841–55. doi: 10.1016/j.celrep.2014.11.004
35. Wang Y, Xiao M, Chen X, Chen L, Xu Y, Lv L, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell. (2015) 57:662–73. doi: 10.1016/j.molcel.2014.12.023
36. Shvedunova M, Akhtar A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat Rev Mol Cell Biol. (2022) 23:329–49. doi: 10.1038/s41580-021-00441-y
37. Zhao S, Zhang X, Li H. Beyond histone acetylation-writing and erasing histone acylations. Curr Opin Struct Biol. (2018) 53:169–77. doi: 10.1016/j.sbi.2018.10.001
38. Fong CY, Morison J, Dawson MA. Epigenetics in the hematologic malignancies. Haematologica. (2014) 99:1772–83. doi: 10.3324/haematol.2013.092007
39. Husmann D, Gozani O. Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol. (2019) 26:880–9. doi: 10.1038/s41594-019-0298-7
40. Bhat KP, Ümit Kaniskan H, Jin J, Gozani O. Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat Rev Drug Discov. (2021) 20:265–86. doi: 10.1038/s41573-020-00108-x
41. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. (2007) 129:823–37. doi: 10.1016/j.cell.2007.05.009
42. Slany RK. The molecular biology of mixed lineage leukemia. Haematologica. (2009) 94:984–93. doi: 10.3324/haematol.2008.002436
43. Harrison CJ, Hills RK, Moorman AV, Grimwade DJ, Hann I, Webb DK, et al. Cytogenetics of childhood acute myeloid leukemia: united kingdom medical research council treatment trials AML 10 and 12. J Clin Oncol. (2010) 28:2674–81. doi: 10.1200/JCO.2009.24.8997
44. Chaudhury SS, Morison JK, Gibson BE, Keeshan K. Insights into cell ontogeny, age, and acute myeloid leukemia. Exp Hematol. (2015) 43:745–55. doi: 10.1016/j.exphem.2015.05.008
45. Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. (2020) 13:104. doi: 10.1186/s13045-020-00937-8
46. Kempf JM, Weser S, Bartoschek MD, Metzeler KH, Vick B, Herold T, et al. Loss-of-function mutations in the histone methyltransferase EZH2 promote chemotherapy resistance in AML. Sci Rep. (2021) 11:5838. doi: 10.1038/s41598-021-84708-6
47. Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci USA. (2012) 109:5028–33. doi: 10.1073/pnas.1202258109
48. Puda A, Milosevic JD, Berg T, Klampfl T, Harutyunyan AS, Gisslinger B, et al. Frequent deletions of JARID2 in leukemic transformation of chronic myeloid malignancies. Am J Hematol. (2012) 87:245–50. doi: 10.1002/ajh.22257
49. Shiba N, Yoshida K, Shiraishi Y, Okuno Y, Yamato G, Hara Y, et al. Whole-exome sequencing reveals the spectrum of gene mutations and the clonal evolution patterns in paediatric acute myeloid leukaemia. Br J Haematol. (2016) 175:476–89. doi: 10.1111/bjh.14247
50. Schnittger S, Eder C, Jeromin S, Alpermann T, Fasan A, Grossmann V, et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia. (2013) 27:82–91. doi: 10.1038/leu.2012.262
51. Paschka P, Schlenk RF, Gaidzik VI, Herzig JK, Aulitzky T, Bullinger L, et al. ASXL1 mutations in younger adult patients with acute myeloid leukemia: a study by the German-Austrian acute myeloid leukemia study group. Haematologica. (2015) 100:324–30. doi: 10.3324/haematol.2014.114157
52. Yamato G, Shiba N, Yoshida K, Shiraishi Y, Hara Y, Ohki K, et al. ASXL2 mutations are frequently found in pediatric AML patients with t(8;21)/ RUNX1-RUNX1T1 and associated with a better prognosis. Genes Chromosomes Cancer. (2017) 56:382–93. doi: 10.1002/gcc.22443
53. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. (2004) 119:941–53. doi: 10.1016/j.cell.2004.12.012
54. Gruber TA, Larson Gedman A, Zhang J, Koss CS, Marada S, Ta HQ, et al. An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell. (2012) 22:683–97. doi: 10.1016/j.ccr.2012.10.007
55. de Rooij JD, Hollink IH, Arentsen-Peters ST, van Galen JF, Berna Beverloo H, Baruchel A, et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia. (2013) 27:2280–8. doi: 10.1038/leu.2013.87
56. Stief SM, Hanneforth AL, Weser S, Mattes R, Carlet M, Liu WH, et al. Loss of KDM6A confers drug resistance in acute myeloid leukemia. Leukemia. (2020) 34:50–62. doi: 10.1038/s41375-019-0497-6
57. Newcombe AA, Gibson BES, Keeshan K. Harnessing the potential of epigenetic therapies for childhood acute myeloid leukemia. Exp Hematol. (2018) 63:1–11. doi: 10.1016/j.exphem.2018.03.008
58. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. (2014) 13:337–56. doi: 10.1038/nrd4286
59. Kulikowski E, Rakai BD, Wong NCW. Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Med Res Rev. (2021) 41:223–45. doi: 10.1002/med.21730
60. Donati B, Lorenzini E, Ciarrocchi A. BRD4 and cancer: going beyond transcriptional regulation. Mol Cancer. (2018) 17:164. doi: 10.1186/s12943-018-0915-9
61. Dey A, Yang W, Gegonne A, Nishiyama A, Pan R, Yagi R, et al. BRD4 directs hematopoietic stem cell development and modulates macrophage inflammatory responses. EMBO J. (2019) 38:e100293. doi: 10.15252/embj.2018100293
62. Ramsey HE, Greenwood D, Zhang S, Childress M, Arrate MP, Gorska AE, et al. BET inhibition enhances the antileukemic activity of low-dose venetoclax in acute myeloid leukemia. Clin Cancer Res. (2021) 27:598–607. doi: 10.1158/1078-0432.CCR-20-1346
63. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. (2011) 478:524–8. doi: 10.1038/nature10334
64. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. (2011) 478:529–33. doi: 10.1038/nature10509
65. Liu Y, Cheng Z, Pang Y, Cui L, Qian T, Quan L, et al. Role of microRNAs, circRNAs and long noncoding RNAs in acute myeloid leukemia. J Hematol Oncol. (2019) 12:51. doi: 10.1186/s13045-019-0734-5
66. Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. (2017) 16:167–79. doi: 10.1038/nrd.2016.117
67. Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. (2007) 318:1931–4. doi: 10.1126/science.1149460
68. Oshima M, Hasegawa N, Mochizuki-Kashio M, Muto T, Miyagi S, Koide S, et al. Ezh2 regulates the Lin28/let-7 pathway to restrict activation of fetal gene signature in adult hematopoietic stem cells. Exp Hematol. (2016) 44:282–96.e3. doi: 10.1016/j.exphem.2015.12.009
69. Bolouri H, Farrar JE, Triche T Jr., Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. (2018) 24:103–12. doi: 10.1038/nm.4439
70. Narayan N, Bracken CP, Ekert PG. MicroRNA-155 expression and function in AML: an evolving paradigm. Exp Hematol. (2018) 62:1–6. doi: 10.1016/j.exphem.2018.03.007
71. Zhu R, Lin W, Zhao W, Fan F, Tang L, Hu Y. A 4-microRNA signature for survival prognosis in pediatric and adolescent acute myeloid leukemia. J Cell Biochem. (2019) 120:3958–68. doi: 10.1002/jcb.27679
72. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. (2019) 20:675–91. doi: 10.1038/s41576-019-0158-7
73. Nicolet BP, Engels S, Aglialoro F, van den Akker E, von Lindern M, Wolkers MC. Circular RNA expression in human hematopoietic cells is widespread and cell-type specific. Nucleic Acids Res. (2018) 46:8168–80. doi: 10.1093/nar/gky721
74. Liu X, Liu X, Cai M, Luo A, He Y, Liu S, et al. CircRNF220, not its linear cognate gene RNF220, regulates cell growth and is associated with relapse in pediatric acute myeloid leukemia. Mol Cancer. (2021) 20:139. doi: 10.1186/s12943-021-01395-7
75. Wang N, Yang B, Jin J, He Y, Wu X, Yang Y, et al. Circular RNA circ_0040823 inhibits the proliferation of acute myeloid leukemia cells and induces apoptosis by regulating miR-516b/PTEN. J Gene Med. (2022) 24:e3404. doi: 10.1002/jgm.3404
76. Lin W, Zhou Q, Wang CQ, Zhu L, Bi C, Zhang S, et al. LncRNAs regulate metabolism in cancer. Int J Biol Sci. (2020) 16:1194–206. doi: 10.7150/ijbs.40769
77. Li J, Li Z, Bai X, Chen X, Wang M, Wu Y, et al. LncRNA UCA1 promotes the progression of AML by upregulating the expression of CXCR4 and CYP1B1 by affecting the stability of METTL14. J Oncol. (2022) 2022:2756986. doi: 10.1155/2022/2756986
78. Li J, Wang M, Chen X. Long non-coding RNA UCA1 modulates cell proliferation and apoptosis by regulating miR-296-3p/Myc axis in acute myeloid leukemia. Cell Cycle. (2020) 19:1454–65. doi: 10.1080/15384101.2020.1750814
79. Liang Y, Li E, Zhang H, Zhang L, Tang Y, Wanyan Y. Silencing of lncRNA UCA1 curbs proliferation and accelerates apoptosis by repressing SIRT1 signals by targeting miR-204 in pediatric AML. J Biochem Mol Toxicol. (2020) 34:e22435. doi: 10.1002/jbt.22435
80. Ma L, Wang YY, Jiang P. LncRNA LINC00909 promotes cell proliferation and metastasis in pediatric acute myeloid leukemia via miR-625-mediated modulation of Wnt/β-catenin signaling. Biochem Biophys Res Commun. (2020) 527:654–61. doi: 10.1016/j.bbrc.2020.05.001
81. Gardin C, Dombret H. Hypomethylating agents as a therapy for AML. Curr Hematol Malig Rep. (2017) 12:1–10. doi: 10.1007/s11899-017-0363-4
82. Flotho C, Claus R, Batz C, Schneider M, Sandrock I, Ihde S, et al. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. (2009) 23:1019–28. doi: 10.1038/leu.2008.397
83. Lu LW, Chiang GH, Medina D, Randerath K. Drug effects on nucleic acid modification. I. A specific effect of 5-azacytidine on mammalian transfer RNA methylation in vivo. Biochem Biophys Res Commun. (1976) 68:1094–101. doi: 10.1016/0006-291X(76)90308-9
84. Schaefer M, Hagemann S, Hanna K, Lyko F. Azacytidine inhibits RNA methylation at DNMT2 target sites in human cancer cell lines. Cancer Res. (2009) 69:8127–32. doi: 10.1158/0008-5472.CAN-09-0458
85. Aimiuwu J, Wang H, Chen P, Xie Z, Wang J, Liu S, et al. RNA-dependent inhibition of ribonucleotide reductase is a major pathway for 5-azacytidine activity in acute myeloid leukemia. Blood. (2012) 119:5229–38. doi: 10.1182/blood-2011-11-382226
86. Cheng JX, Chen L, Li Y, Cloe A, Yue M, Wei J, et al. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat Commun. (2018) 9:1163. doi: 10.1038/s41467-018-03513-4
87. Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. (2015) 126:291–9. doi: 10.1182/blood-2015-01-621664
88. Stahl M, DeVeaux M, Montesinos P, Itzykson R, Ritchie EK, Sekeres MA, et al. Hypomethylating agents in relapsed and refractory AML: outcomes and their predictors in a large international patient cohort. Blood Adv. (2018) 2:923–32. doi: 10.1182/bloodadvances.2018016121
89. Gore L, Triche TJ Jr., Farrar JE, Wai D, Legendre C, Gooden GC, et al. A multicenter, randomized study of decitabine as epigenetic priming with induction chemotherapy in children with AML. Clin Epigenetics. (2017) 9:108. doi: 10.1186/s13148-017-0411-x
90. Sun W, Triche T Jr., Malvar J, Gaynon P, Sposto R, Yang X, et al. A phase 1 study of azacitidine combined with chemotherapy in childhood leukemia: a report from the TACL consortium. Blood. (2018) 131:1145–8. doi: 10.1182/blood-2017-09-803809
91. Zeidan AM, Wang R, Wang X, Shallis RM, Podoltsev NA, Bewersdorf JP, et al. Clinical outcomes of older patients with AML receiving hypomethylating agents: a large population-based study in the United States. Blood Adv. (2020) 4:2192–201. doi: 10.1182/bloodadvances.2020001779
92. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. (2016) 21:373–80.
93. Waitkus MS, Diplas BH, Yan H. Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell. (2018) 34:186–95. doi: 10.1016/j.ccell.2018.04.011
94. Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. (2013) 340:622–6. doi: 10.1126/science.1234769
95. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. (2017) 130:722–31. doi: 10.1182/blood-2017-04-779405
96. DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. (2018) 378:2386–98. doi: 10.1056/NEJMoa1716984
97. Stahl M, Gore SD, Vey N, Prebet T. Lost in translation? Ten years of development of histone deacetylase inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Expert Opin Investig Drugs. (2016) 25:307–17. doi: 10.1517/13543784.2016.1146251
98. Karol SE, Cooper TM, Mead PE, Crews KR, Panetta JC, Alexander TB, et al. Safety, pharmacokinetics, and pharmacodynamics of panobinostat in children, adolescents, and young adults with relapsed acute myeloid leukemia. Cancer. (2020) 126:4800–5. doi: 10.1002/cncr.33156
99. Pommert L, Schafer ES, Malvar J, Gossai N, Florendo E, Pulakanti K, et al. Decitabine and vorinostat with FLAG chemotherapy in pediatric relapsed/refractory AML: report from the therapeutic advances in childhood leukemia and lymphoma (TACL) consortium. Am J Hematol. (2022) 97:613–22. doi: 10.1002/ajh.26510
100. Shukla N, Wetmore C, O’Brien MM, Silverman LB, Brown P, Cooper TM, et al. Final report of phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in children with relapsed or refractory MLL-r acute leukemia. Blood. (2016) 128:2780. doi: 10.1182/blood.V128.22.2780.2780
101. Djamai H, Berrou J, Dupont M, Coudé MM, Delord M, Clappier E, et al. Biological effects of BET inhibition by OTX015 (MK-8628) and JQ1 in NPM1-mutated (NPM1c) acute myeloid leukemia (AML). Biomedicines. (2021) 9:1704. doi: 10.3390/biomedicines9111704
102. DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. (2019) 133:7–17. doi: 10.1182/blood-2018-08-868752
Keywords: acute myeloid leukemia, pediatric, epigenetics, DNA methylation, histone modification, non-coding RNAs, therapy
Citation: Xu H, Wen Y, Jin R and Chen H (2022) Epigenetic modifications and targeted therapy in pediatric acute myeloid leukemia. Front. Pediatr. 10:975819. doi: 10.3389/fped.2022.975819
Received: 06 July 2022; Accepted: 10 August 2022;
Published: 06 September 2022.
Edited by:
Sergi Aranda, National Center for Genomic Analysis (CRG), SpainReviewed by:
Jan Stary, University Hospital in Motol, CzechiaCopyright © 2022 Xu, Wen, Jin and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Runming Jin, amlucnVubUBxcS5jb20=; Hongbo Chen, aGJjaGVuQGh1c3QuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.