 Aleksandra Szczawińska-Popłonyk
Aleksandra Szczawińska-Popłonyk Katarzyna Tąpolska-Jóźwiak
Katarzyna Tąpolska-Jóźwiak Eyal Schwartzmann
Eyal Schwartzmann Barbara Pietrucha3
Barbara Pietrucha3

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 20 October 2022
Sec. Pediatric Immunology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.972952
Ataxia-telangiectasia (A-T) is a severe syndromic neurodegenerative inborn error of immunity characterized by DNA reparation defect, chromosomal instability, and hypersensitivity to ionizing radiation, thereby predisposing affected individuals to malignant transformation. While the leading disease symptomatology is associated with progressively debilitating cerebellar ataxia accompanied by central and peripheral nervous system dysfunctions, A-T is a multisystemic disorder manifesting with the heterogeneity of phenotypic features. These include airway and interstitial lung disease, chronic liver disease, endocrine abnormalities, and cutaneous and deep-organ granulomatosis. The impaired thymic T cell production, defective B cell development and antibody production, as well as bone marrow failure, contribute to a combined immunodeficiency predisposing to infectious complications, immune dysregulation, and organ-specific immunopathology, with the A-T hyper-IgM (HIGM) phenotype determining the more severe disease course. This study aimed to clarify the immunodeficiency and associated immune dysregulation as well as organ-specific immunopathology in children with A-T. We also sought to determine whether the hyper-IgM and non-hyper-IgM phenotypes play a discriminatory role and have prognostic significance in anticipating the clinical course and outcome of the disease. We retrospectively reviewed the medical records of twelve A-T patients, aged from two to eighteen years. The patients' infectious history, organ-specific symptomatology, and immunological workup including serum alpha-fetoprotein, immunoglobulin isotypes, IgG subclasses, and lymphocyte compartments were examined. For further comparative analysis, all the subjects were divided into two groups, HIGM A-T and non-HIGM A-T. The clinical evaluation of the study group showed that recurrent respiratory tract infections due to viral and bacterial pathogens and a chronic obstructive airway disease along with impaired humoral immunity, in particular complete IgA deficiency, were noted in all the A-T patients, with both HIGM and non-HIGM phenotypes. The most important features with the discriminatory role between groups, were autoimmune disorders, observable four times more frequently in HIGM than in non-HIGM A-T. Two patients with the HIGM A-T phenotype were deceased due to liver failure and chronic Epstein-Barr virus (EBV) infection. It may therefore be assumed that the HIGM form of A-T is associated with more profound T cell dysfunction, defective immunoglobulin class switching, chronic EBV expansion, and poorer prognosis.
Ataxia-telangiectasia (A-T) is an autosomal recessive syndromic inborn error of immunity (IEI) (1) characterized by the DNA reparation defect, genomic instability, and sensitivity to ionizing radiation with multisystemic involvement and unfavorable outcome. The disease pathophysiology results from mutations in the Ataxia-Telangiectasia Mutated (ATM) gene encoding for a protein kinase which is a member of the large phosphoinositidil 3-kinase related protein kinase (PIKK) family. The nuclear and cytoplasmic enzymatic ATM kinase activity is pleiotropic and heterogeneous. The enzyme plays an important role in controlling the cell-cycle checkpoints and coordinating the cellular signaling pathways in response to DNA double-strand breaks (DSBs), genotoxic factors, and oxidative stress. It is also involved in cytoplasmic processes, phosphorylating numerous substrates important in mitochondrial respiration and energy metabolism. In A-T, the disruption of the multiplicity of ATM kinase functions in DNA damage response, transcription and translation regulation, protein aggregation, and autophagy, maintaining cellular homeostasis (2–6) is underpinning the multisystemic involvement with the multiplicity of phenotypic features (7–9). The leading A-T symptomatology is characterized by neurodegeneration and progressively debilitating cerebellar ataxia with postural instability, oculomotor apraxia, dysarthria and orolingual insufficiency, as well as extrapyramidal dysfunctions with choreoathetotic movements, dystonia and muscle tremor (4, 10–12). The extended disease phenotype also includes chronic obstructive airway and interstitial lung disease (13–16), chronic inflammatory non-alcoholic liver disease (17–20), cutaneous and systemic granulomatosis (21–24), and hormonal dysfunctions with growth retardation, gonadal insufficiency, and diabetogenic insulin resistance (25–27). The impaired thymic T cell production, defective B cell development accompanied by hypogammaglobulinemia, IgG subclass and antigen-specific antibody generation, and bone marrow failure contribute to a combined immunodeficiency (28–30). The more severe clinical disease course with infectious complications, immune dysregulation, organ-specific immunopathology, and a high risk of lymphoid malignancy has been assigned to the phenotype characterized by low serum IgG and/or IgA and normal to elevated IgM immunoglobulin isotype levels corresponding with the hyper-IgM (HIGM) A-T variant (31–34).
This study aimed to clarify the immunodeficiency and associated infectious complications, immune dysregulation as well as organ-specific immunopathology in children with A-T. We also sought to determine whether the hyper-IgM (HIGM) and non-hyper-IgM (non-HIGM) phenotypes play a discriminatory role and have prognostic significance in anticipating the clinical course of the disease.
Medical records of twelve A-T children, six boys and six girls, aged from two to eighteen years who have been followed and treated in our tertiary care university department of pediatric immunology, have been reviewed. The clinical analysis comprised the individual patients' infectious history with particular emphasis on respiratory complications, autoimmune disorders, and organ-specific symptomatology. The immunological laboratory work-up included serum immunoglobulin G, M, and A isotypes, IgG subclass levels, and peripheral blood B and T lymph cell flow cytometric immunophenotyping. The alpha-fetoprotein (AFP) activity was also examined in all the children studied.
Referring to the individual patients' IgM levels, further comparative analysis and dividing all the subjects into two groups, HIGM A-T and non-HIGM A-T was done. Herein, we stratified patients as HIGM A-T when their serum IgG and/or IgA levels were decreased with IgM levels above the reference values and non-HIGM A-T when their serum IgM levels were normal or below the reference ranges.
Peripheral venous blood samples anticoagulated with ethylenediaminetetracetic acid (EDTA-K2) were stored at a temperature of between 4 and 8°C and processed within 24 h. Cells were labelled with the following murine fluorochrome-stained monoclonal antibodies: anti-CD45 FITC (fluoresceine isothiocyanate), anti-CD14 PE (phycoerithrin), anti-CD19 PE, anti-CD19 PerCP (peridinin chlorophyll protein), anti-IgM FITC, anti-IgD FITC, anti-CD38 APC (allophycocyanin), anti-CD27 PE, anti-CD21 FITC, as well as anti-CD3 FITC, anti-CD4 FITC, CD45RA FITC, CD127 FITC, CD185 FITC, anti-CD8 PE, anti-CD16 + CD56 PE, CD25 PE, CD31 PE, CD45RO PE, anti-CD3 PerCP, CD197 PerCP, anti-CD4 APC and anti-CD8 APC (all Beckton-Dickinson Biosciences, USA).
Blood samples were mixed with antibodies, incubated in a lysing solution (FACS Lysing Solution, Beckton-Dickinson, USA), centrifuged twice, and suspended in a phosphate buffered saline (PBS, Roche, Germany). The acquisition of cells and analysis were carried out with the use of the flow cytometer FACSCanto and FACSDiva software (Beckton-Dickinson, USA). On biparametric scattering CD45 + CD14- lymphocytes, the following sequential gating strategies for the characterization of lymphocyte subpopulations were applied:
B cells were identified as CD19-expressing cells in the lymphocyte population, and CD19+ B cells were then analyzed either for the expression of CD27 and IgD, or CD21 and CD38 and IgM. The following B cell subsets were delineated: immature CD19 + CD21lo, immature activated CD19 + CD38loCD21lo, transitional CD19 + CD38hisIgMhi, non-switched memory CD19 + CD27 + sIgD+, switched memory CD19 + CD27 + IgD- B cells, and CD19 + CD38hisIgM- plasmablasts.
T cells were identified as CD3-expressing cells in the lymphocyte population and analyzed either for the expression of CD4 and CD8. Subsequently, CD3 + CD4+ T helper cells, and CD3 + CD8+ T cytotoxic cells were delineated.
T helper cells were then analyzed for either the expression of CD27 and CD45RO or CD45RA, or CD31, CD127 and CD25, or CD185. This approach enabled to identify the following T helper cell subsets: CD3 + CD4 + CD31 + CD45RA + recent thymic emigrants, naïve CD3 + CD4 + CD27 + CD45RA+, regulatory CD3 + CD4 + CD25 ++ CD27-, central memory CD3 + CD4 + CD27 + CD45RO+, effector memory CD3 + CD4 + CD27-CD45RO+, terminally differentiated CD3 + CD4 + CD27-CD45RA+, follicular CD3 + CD4 + CD185 + CD45RO+, and regulatory CD3 + CD4 + CD45RO + CD127-CD25++ T helper cells.
Among CD3 + CD8 + cytotoxic T cells, analyzed for the expression of CD197, CD27 and CD45RO or CD45RA, the following subsets were distinguished: naïve CD3 + CD8 + CD197 + CD27 + CD45RA+, central memory CD3 + CD8 + CD197 + CD27 + CD45RO+, effector memory CD3 + CD8 + CD197-CD27-CD45RO+, and terminally differentiated CD3 + CD8 + CD197-CD27-CD45RA + cells.
NK cells were defined as CD3- and CD16 + and/or CD56 + cells.
The relative values of PB lymphocytes, the B, T, and NK cells of the total lymphocyte population as well as B and T cell subsets were calculated. The absolute counts of all cell subsets were calculated from the PB leukocyte counts. A comparative analysis was done with the reference cut-off values of B and T cell subsets for pediatric populations of different age groups.
Due to the small patient sample size, resulting from the rarity of the disease, statistical methods were not employed and a descriptive showcasing of the study group was done.
In all A-T affected patients, humoral immunodeficiency was observable. Whereas the elevated IgM levels were reflecting a defective class switch recombination (CSR) process discriminatory for the two HIGM and non-HIGM patient groups, IgA deficiency was revealed in all but one patient in each group. While serum IgG, IgG1 and IgG2 subclass levels were decreased below the reference values in all the HIGM A-T patients, in three out of six non-HIGM patients, IgG and IgG1 subclass levels were within the reference values. In the latter group, in one patient only, a low IgG3 subclass level was shown, whereas IgG4 levels were normal in all six patients. In flow cytometric PB lymphocyte analysis, impaired B and T lymph cell differentiation and maturation were revealed. Persistent lymphopenia, abnormal B cell neogenesis with low B cell numbers, and defective development of memory B cells were observable in all patients. T cell lymphopenia with low numbers of recent thymic emigrants (RTE) reflecting thymic dysfunction in generating naive T cells, with low CD4 + CD45RA: CD4 + CD45RO + ratio alike was the universal feature of both HIGM and non-HIGM A-T phenotypes. In all the A-T patients studied, both HIGM and non-HIGM, serum AFP activity was remarkably elevated (reference serum level <8 ng/ml). The results of the immunological workup in the A-T children studied are shown in Table 1.
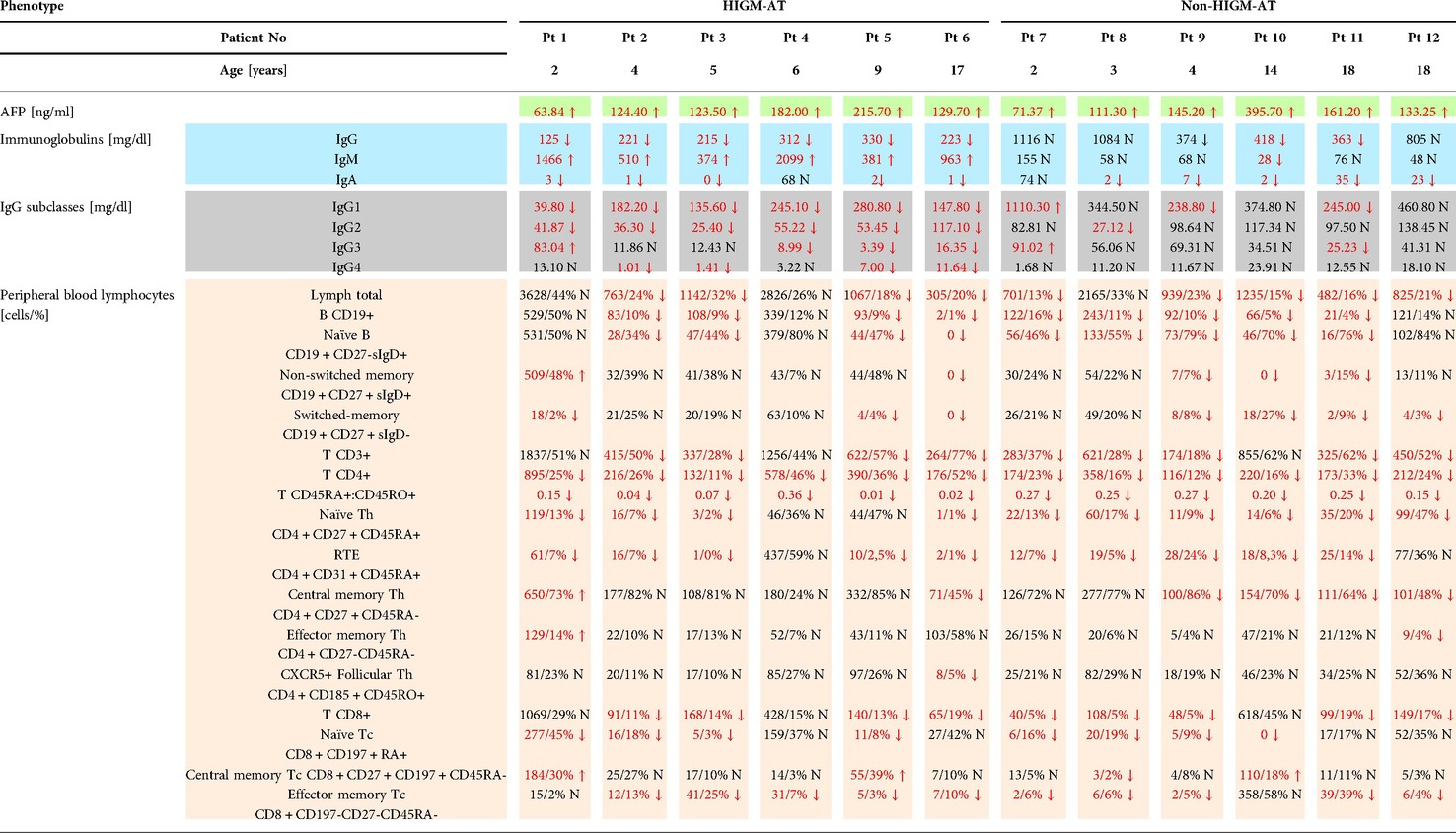
Table 1. The immunological workup of twelve A-T patients studied with discrimination between HIGM and non-HIGM phenotypes.
An in-depth analysis of individual patients' infectious history showed that all the patients studied suffered from respiratory tract infections yet the course of the lung and airway disease was variable. In two out of twelve patients, one with the HIGM A-T and another one with the non-HIGM A-T phenotypes, aged 17 and 18 years, respectively, an interstitial lung disease (ILD) with concomitant chronic obstructive airway disease was noted. While the tracheal aspirate cultures were positive in all the six HIGM A-T patients, in two out of six non-HIGM A-T children the cultures did not show bacterial pathogens. The most frequently cultured pathogen was Streptococcus pneumoniae, which was identified in five and two HIGM and non-HIGM A-T patients, respectively. It is worth noting that an opportunistic pathogen, Pseudomonas aeruginosa was identified in cultures exclusively from children with the HIGM A-T variant. Chronic infections with herpes viruses, such as cytomegalovirus (CMV), Epstein-Barr virus (EBV), and human herpes virus 6 (HHV6) assessed as peripheral blood viral loads were found in four HIGM A-T patients and only two out of six non-HIGM A-T patients. The results of microbiologic investigations in all the A-T children studied and a comparison between HIG and non-HIGM patients are displayed in Table 2.
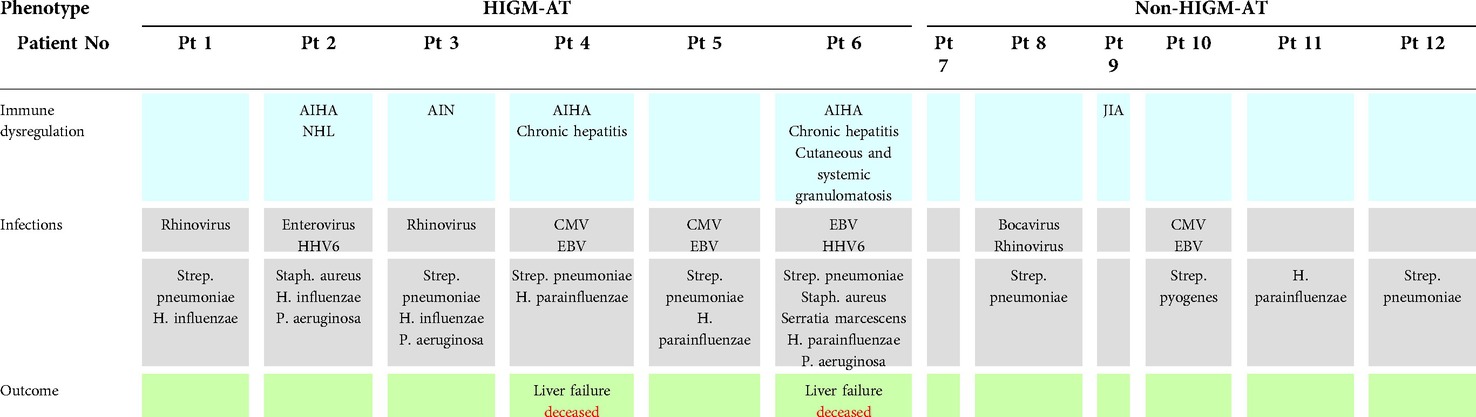
Table 2. The summary of infections and immune dysregulation disorders in HIGM A-T and non-HIGM A-T patients.
Immune dysregulation phenotypes were two-fold more frequently observable in HIGM A-T than in non-HIGM A-T patients. Autoimmune hemolytic anemia (AIHA) was the most common autoimmune disorder as it was seen in three out of six HIGM A-T individuals. Other autoimmune diseases such as autoimmune neutropenia (AIN), and juvenile idiopathic arthritis (JIA), were seen in single cases (the first occurred in HIGM A-T, whereas the latter was the only autoimmune disorder diagnosed in a non-HIGM A-T patient). Inflammatory chronic liver disease was the organ-specific immunopathology in two patients with the HIGM A-T variant who deceased due to liver failure. Non-Hodgkin B-cell lymphoma (NHL) was a sequela in one HIGM A-T patient. Two children with the HIGM A-T variant died, aged 6 and 17 years old, both due to chronic liver failure, pancytopenia, and chronic EBV infection. In one of these patients, chronic hepatitis was accompanied by cutaneous and systemic granulomatosis, involving the palate, pharynx, larynx, and lungs. The summary of immune dysregulation disorders with discrimination between HIGM and non-HIGM A-T patients is displayed in Table 2.
The combined immunodeficiency belongs to the complex and heterogeneous individual phenotype in ataxia telangiectasia patients. The impaired T cell compartment manifesting as thymic dysfunction in generation recent thymic emigrants and naive T CD4 + helper cells and also suppressor/ cytotoxic T CD8 + cells is a universal feature of A-T, observable in both HIGM and non-HIGM variants. A decrease in CD4 + and CD8 + naive, central memory, and terminally differentiated effector memory CD4+ T cells was shown in the peripheral T cell compartment in all the A-T affected individuals. This impaired generation of naive CD4+ T helper cells with a low number of RTE and a CD4 + CD45RA+: CD4 + CD45RO + ratio has, therefore, no discriminatory significance between our HIGM and non-HIGM A-T patients. Consequently, disturbed T cell neogenesis and reduced T cell repertoire diversity contribute to the increased predisposition to infections (28–30, 35). The pathogenesis of defective T cell homeostasis in A-T patients is complex and thymus dysfunction with low thymic output (36), disorders of the cell cycle checkpoints regulation, oxidative stress responses (37, 38), premature immune aging process (39, 40) as well as increased CD95-mediated apoptosis (41) have been postulated as candidate contributory pathways. Among B cell subsets, a reduced number of CD19 + CD27- cells representing naive B cells and corresponding with the impairment of the bone marrow output has been observed in our A-T children studied, consistently with other reports (42, 43). The switched-memory B cells have been depleted in three out of six HIGM and four out of six non-HIGM children, which is an unexpected finding in individuals with elevated serum IgM levels reflecting an impaired class switch recombination (CSR) (31–34) being the major criterion for stratifying our patients with A-T. We have also noted that IgA serum levels were admittedly higher in the non-HIGM group with normal or partially deficient IgA in as many as four patients. Referring to the recent report (44) on the predictive role of IgA as a simple, surrogate marker in anticipating the poorer prognosis in A-T patients, our five HIGM A-T presented with a complete IgA deficiency. The HIGM group also more frequently presented a deficit in IgG subclass generation and this parameter has also been shown to correlate with shorter survival in A-T (45). These findings mirror a deeply impaired process of immunoglobulin class switching and correspond with a higher risk of respiratory tract viral, most frequently due to Rhinovirus, and bacterial, such as Streptococcus pneumoniae and Haemophilus influenzae infections (46–48). Interestingly, the heterogeneity of IgG subclass distribution had an important clinical significance also in patients with non-HIGM A-T. Whereas the IgG2 deficiency was associated with recurrent infections due to Streptococcus pneumoniae, in patients with IgG3 deficiency no bacterial pathogens or Haemophilus parainfluenzae were cultured in the airways. Furthermore, the opportunistic pathogen, Pseudomonas aeruginosa which was cultured exclusively in patients with the HIGM A-T variant, is associated with a more severe course of lung disease and a higher risk of chronic respiratory failure (16, 49). In our group of HIGM A-T children, impaired CSR was associated with more severe clinical phenotypes, including autoimmune phenomena, granulomatous disorders, inflammatory organ-specific immunopathology, lymphoproliferation, and malignancy, consistently with previous reports (18, 24, 33, 34). Whereas in three out of all the six HIGM A-T patients, Epstein-Barr virus (EBV)-DNA was persistently identified in peripheral blood pointing to the EBV reactivation, important concerns are raised regarding the role of the dysfunctional ATM kinase activity in favoring the EBV life cycle and promoting lymphoproliferation and malignancy in immunocompromized A-T individuals (50, 51). Interestingly, our six-year-old A-T patient with HIGM variant, in whom non-Hodgkin lymphoma (NHL) was diagnosed, showed reactivation of human herpes virus (HHV)-6 but not EBV, which DNA was undetectable. This observation might be supported by studies of the immune response to HHV-6 in T-cell immunodeficient humans failing protection against the reactivation of the virus causing infectious, inflammatory, and malignant life-threatening complications (52, 53).
In conclusion, due to the rarity of A-T, our single-center group of pediatric patients is relatively small, a fact that presents as a major limitation of this study. Whereas we were able to stratify our A-T children into HIGM and non-HIGM groups according to the serum IgM levels, we observed B cell developmental impairment in both phenotypes. Accordingly, IgA deficiency was a universal feature in all but one A-T patient studied and did not play any discriminatory role between HIGM and non-HIGM groups. Likewise, low thymic RTE output and low naive T helper to memory T helper lymph cell ratio was the cardinal feature of both HIGM and non-HIGM A-T immunophenotypes. However, immune dysregulation associated with autoimmune disorders showed a striking predilection to the HIGM phenotype. Non-effective antibody maturation, expansion of IgM autoantibodies during immune response to infection, and an increased germinal center autoreactive B cell population have been proposed as contributing factors to autoimmune disorders in HIGM-AT (34). Furthermore, a fatality due to EBV expansion and liver failure was observable solely in HIGM A-T patients. It may therefore be assumed that more profound T cell dysfunction may be assigned to the defective immunoglobulin class switching and HIGM A-T phenotype with a poorer prognosis.
Our observations highlight the complexity of pathophysiology and symptomatology in A-T, a disease at the interface of immune deficiency, autoinflammation, organ-specific immunopathology, and malignancy with a more severe and unfavorable course associated with the HIGM A-T variant. Therefore, affected children require pediatricians' awareness, careful and attentive evaluation, and multidisciplinary care under the pediatric immunologist's supervision to improve prognosis.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
ASP was responsible for the conception and design of the study, acquisition, and interpretation of data, and drafted the manuscript, KTJ contributed to the acquisition, and interpretation of data, and drafting the manuscript, ES helped in the acquisition and analysis of data and drafting the manuscript, BP critically revised the manuscript All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypic classifications. J Clin Immunol. (2020) 40:66–81. doi: 10.1007/s10875-020-00758-x
2. Guleria A, Chandna S. ATM Kinase: much more than a DNA damage responsive protein. DNA Repair. (2016) 39:1–20. doi: 10.1016/j.dnarep.2015.12.009
3. Lee JH, Paull TT. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat Rev Mol Cell Biol. (2021) 22:796–814. doi: 10.1038/s41580-021-00394-2
4. Choy KR, Watters DJ. Neurodegeneration in ataxia-telangiectasia: multiple roles of ATM kinase in cellular homeostasis. Dev Dyn. (2018) 247:33–46. doi: 10.1002/dvdy.24522
5. Lee JH, Paull TT. Mitochondria at the crossroads of ATM-mediated stress signaling and regulation of reactive oxygen species. Redox Biol. (2020) 32:101511. doi: 10.1016/j.redox.2020.101511
6. Subramanian GN, Yeo AJ, Gatei MH, Coman DJ, Lavin MF. Metabolic stress and mitochondrial dysfunctions in ataxia-telangiectasia. Antioxidants. (2022) 11:653. doi: 10.3390/antiox11040653
7. Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman H. Ataxia-telangiectasia: a review. Orphanet J Rare Dis. (2016) 11:159. doi: 10.1186/s13023-016-0543-7
8. McGrath-Morrow SA, Rothblum-Oviatt CC, Wright J, Schlechter H, Lefton-Greif MA, Natale VA, et al. Multidisciplinary management of ataxia-telangiectasia: current perspectives. J Mulidiscip Healthc. (2021) 14:1637–44. doi: 10.2147/JMDH.S295486
9. Van Os NJH, Haaxma CA, van der Flier M, Merkus PJFM, van Deuren M, de Groot IJM, et al. Ataxia-telangiectasia: recommendations for multidisciplinary treatment. Dev Med Child Neurol. (2017) 59:680–9. doi: 10.1111/dmcn.13424
10. Hoche F, Seidel K, Theis M, Vlaho S, Schubert R, Zielen S, et al. Neurodegeneration in ataxia-telangiectasia: what is new? What is evident? Neuropediatrics. (2012) 43:119–29. doi: 10.1055/s-0032-1313915
11. Teive HAG, Camargo CHF, Munhoz RP. More than ataxia: movement disorders in ataxia-telangiectasia. Parkinsonism Relat Disord. (2018) 45:3–8. doi: 10.1016/j.parkreldis.2017.12.009
12. Petley E, Yule A, Alexander S, Ojha S, Whitehouse WP. The natural history of ataxia-telangiectasia (A-T): a systematic review. PLoS One. (2022) 17:e0264177. doi: 10.1371/journal.pone.0264177
13. Berkun Y, Vilozni D, Levi Y, Borik S, Waldman D, Somech R, et al. Reversible airway obstruction in children with ataxia telangiectasia. Pediatr Pulmonol. (2010) 45:230–5. doi: 10.1002/ppul.21095
14. Schroeder SA, Swift M, Sandoval C, Langston C. Interstitial lung disease in patients with ataxia-telangiectasia. Pediatr Pulmonol. (2005) 39:537–43. doi: 10.1002/ppul.20209
15. McGrath-Morrow SA, Lederman HM, Aherrera AD, Lefton-Greif MA, Crawford TO, Ryan T, et al. Pulmonary function in children and young adults with ataxia telangiectasia. Pediatr Pulmonol. (2014) 49:84–90. doi: 10.1002/ppul.22760
16. Bhatt JM, Bush A, van Gerven M, Nissenkorn A, Renke M, Yarlett L, et al. ERS statement on the multidisciplinary respiratory management of ataxia-telangiectasia. Eur Respir Rev. (2015) 24:565–81. doi: 10.1183/16000617.0066-2015
17. Donath H, Woelke S, Theis M, Hess U, Knop V, Herrmann E. Progressive liver disease in patients with ataxia telangiectasia. Front Pediatr. (2019) 7:458. doi: 10.3389/fped.2019.00458
18. Szczawińska-Popłonyk A, Ossowska L, Jończyk-Potoczna K. Granulomatous liver disease in ataxia-telangiectasia with the hype-IgM phenotype: a case report. Front Pediatr. (2020) 8:570330. doi: 10.3389/fped.2020.570330
19. Weiss B, Krauthammer A, Soudack M, Lahad A, Sarouk I, Somech R, et al. Liver disease in pediatric patients with ataxia telangiectasia: a novel report. J Pediatr Gastroenterol Nutr. (2016) 62:550–5. doi: 10.1097/MPG.0000000000001036
20. Viswanathan P, Sharma Y, Maisuradze L, Tchaikovskaya T, Gupta S. Ataxia telangiectasia mutated pathway disruption affects hepatic DNA and tissue damage in nonalcoholic fatty liver disease. Exp Mol Pathol. (2020) 113:104369. doi: 10.1016/j.yexmp.2020.104369
21. Folgori L, Scarselli A, Angelino G, Ferrari F, Antoccia A, Chessa L, et al. Cutaneous granulomatosis and combined immunodeficiency revealing ataxia-telangiectasia: a case report. Ital J Pediatr. (2010) 36:29. doi: 10.1186/1824-7288-36-29
22. Woelke S, Valesky E, Bakhtiar S, Pommerening H, Pfeffermann LM, Schubert R, et al. Treatment of granulomas in patients with ataxia telangiectasia. Front Immunol. (2018) 9:2000. doi: 10.3389/fimmu.2018.02000
23. Szczawińska-Popłonyk A, Olejniczak K, Tąpolska-Jóźwiak K, Boruczkowski M, Jończyk-Potoczna K, Małdyk J, et al. Cutaneous and systemic granulomatosis in ataxia-telangiectasia: a clinico-pathological study. Adv Dermatol Allergol. (2020) 27:760–5. doi: 10.5114/ada.2020.100485
24. Meyer AK, Banks M, Nadasdy T, Clark JJ, Zheng R, Gelfand EW, et al. Vasculitis in a child with the hiper-IgM variant of ataxia telangiectasia. Front Pediatr. (2019) 7:390. doi: 10.3389/fped.2019.00390
25. Nissenkorn A, Levy-Shraga Y, Banet-Levi Y, Lahad A, Sarouk I, Modan-Moses D. Endocrine abnormalities in ataxia telangiectasia: findings from a national cohort. Pediatr Res. (2016) 79:889–94. doi: 10.1038/pr.2016.19
26. Voss S, Pietzner J, Hoche F, Taylor AMR, Last JI, Schubert R, et al. Growth retardation and growth hormone deficiency in patients with ataxia telangiectasia. Growth Factors. (2014) 32:123–9. doi: 10.3109/08977194.2014.939805
27. Donath H, Hess U, Kieslich M, Theis M, Ohlenschlager U, Schubert R, et al. Diabetes in patients with ataxia telangiectasia: a national cohort study. Front Pediatr. (2020) 8:317. doi: 10.3389/fped.2020.00317
28. Driessen GJ, Ijspert H, Weemaes CMR, Haraldsson A, Trip M, Warris A, et al. Antibody deficiency in patients with ataxia telangiectasia is caused by disturbed B- and T-cell homeostatsis and reduced immune repertoire diversity. J Allergy Clin Immunol. (2013) 131:1367–75. doi: 10.1016/j.jaci.2013.01.053
29. Moeini Shad T, Yousefi B, Amirifar P, Delavari S, Rae W, Kokhaei P, et al. Variable abnormalities in T and B cell subsets in ataxia telangiectasia. J Clin Immunol. (2021) 41:76–88. doi: 10.1007/s10875-020-00881-9
30. Cirillo E, Polizzi A, Soresina A, Prencipe R, Giardino G, Cancrini C, et al. Progressive depletion of B and T lymphocytes in patients with ataxia telangiectasia: results of the Italian primary immunodeficiency network. J Clin Immunol. (2022) 42:783–97. doi: 10.1007/s10875-022-01234-4
31. Mohammadinejad P, Abolhassani H, Aghamohammadi A, Pourhamidi S, Sadeghi B, Nasiri Kalmarzi R, et al. Class switch recombination proces in ataxia-telangiectasia patients with elevated serum levels of IgM. J Immunoassay Immunochem. (2015) 36:16–26. doi: 10.1080/15321819.2014.891525
32. Krauthammer A, Lahad A, Goldberg L, Sarouk I, Weiss B, Somech R, et al. Elevated IgM levels as a marker for a unique phenotype in patients with ataxia telangiectasia. BMC Pediatr. (2018) 18:185. doi: 10.1186/s12887-018-1156-1
33. Ghiasy S, Parvaneh I, Azizi G, Sadri G, Zaki Dizaji M, Abolhassani H, et al. The clinical significance of complete class switching defect in ataxia telangiectasia patients. Expert Rev Clin Immunol. (2017) 13:499–505. doi: 10.1080/1744666X.2017.1292131
34. Amirifar P, Mozdarami H, Yazdani R, Kiaei F, Moeini Shad T, Shahkarami S, et al. Effect of class switch recombination defect on the phenotype of ataxia-telangiectasia patients. Immunol Invest. (2021) 50:201–15. doi: 10.1080/08820139.2020.1723104
35. Kraus M, Lev A, Simon AJ, Levran I, Nissenkorn A, Levi YB, et al. Disturbed B and T cell homeostasis and neogenesis in patients with ataxia-telangiectasia. J Clin Immunol. (2014) 34:561–72. doi: 10.1007/s10875-014-0044-1
36. Micheli R, Pirovano S, Calandra G, Valotti M, Plebani A, Albertini A, et al. Low thymic output and reduced heterogeneity of alpha/beta, but not gamma/delta, T lymphocytes in infants with ataxia telangiectasia. Neuropediatrics. (2003) 34:165–7. doi: 10.1055/s-2003-41280
37. Shibata A, Jeggo PA. ATM’s role in the repair of DNA double-strand breaks. Genes. (2021) 12:1370. doi: 10.3390/genes12091370
38. Maciejczyk M, Heropolitańska-Pliszka E, Pietrucha B, Sawicka-Powierza J, Bernatowska E, Wolska-Kuśnierz B, et al. Antioxidant defence, redox homeostasis, and oxidative damage in children with ataxia telangiectasia and Nijmegen breakage syndrome. Front Immunol. (2019) 10:2322. doi: 10.3389/fimmu.2019.02322
39. Exley AR, Buckenham S, Hodges F, Hallam R, Byrd P, Last J, et al. Premature aging of the immune system underlies immunodeficiency in ataxia telangiectasia. Clin Immunol. (2011) 140:26–36. doi: 10.1016/j.clim.2011.03.007
40. Shiloh Y, Lederman HM. Ataxia-telangiectasia (A-T): an emerging dimension of premature aging. Aging Res Rev. (2017) 33:76–88. doi: 10.1016/j.arr.2016.05.002
41. Carney EF, Srinivasan V, Moss PA, Taylor AM. Classical ataxia telangiectasia patients have a congenitally aged immune system with high expression of CD95. J Immunol. (2012) 189:261–8. doi: 10.4049/jimmunol.1101909
42. Boyarchuk O, Makukh H, Kostyuchenko L, Yarema N, Haibonuk I, Kravets V, et al. TREC/KREC levels in children with ataxia-telangiectasia. Immunol Res. (2021) 69:436–44. doi: 10.1007/s12026-021-09216-1
43. Dasouki M, Jabr A, Al Dakheed G, Elbadaoui F, Alazami AM, Al-Saud B, et al. TREC and KREC profiling as a representative of thymus and bone marrow output in patients with various inborn errors of immunity. Clin Exp Immunol. (2020) 202:60–71. doi: 10.1111/cei.13484
44. Zielen S, Duecker RP, Woelke S, Donath H, Bakhtiar S, Buecker A, et al. Simple measurement of IgA predicts immunity and mortality in ataxia-telangiectasia. J Clin Immunol. (2021) 41:1878–82. doi: 10.1007/s10875-021-01090-8
45. Van Os NJH, Jansen AFM, van Deuren M, Haraldsson A, van Driel NTM, Etzioni A, et al. Ataxia-telangiectasia: immunodeficiency and survival. Clin Immunol. (2017) 178:45–55. doi: 10.1016/j.clim.2017.01.009
46. Mendez-Echevarria A, Caminoa M, del Rosal T, Casas I, Pozo F, Pascual-Pascual SI, et al. The role of respiratory viruses in children with ataxia-telangiectasia. Viruses. (2021) 13:867. doi: 10.3390/v13050867
47. Schroeder SA, Zielen S. Ifections of the respiratory system in patients with ataxia-telangiectasia. Pediatr Pulmonol. (2014) 49:389–99. doi: 10.1002/ppul.22817
48. Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr. (2004) 144:505–11. doi: 10.1016/j.jpeds.2003.12.046
49. Bhatt JM, Bush A, van Gerven M, Nissenkorn A, Renke M, Yarlett L, et al. Ataxia telangiectasia: why should the ERS care? Eur Resp J. (2015) 46:1557–60. doi: 10.1183/13993003.01456-2015
50. Hau PM, Tsao SW. Epstein-Barr virus hijacks DNA damage response transducers to orchestrate its life cycle. Viruses. (2017) 9:341. doi: 10.3390/v9110341
51. Tafti M, Hermine O, Suarez F. Epstein-Barr (EBV)-related lymphoproliferative disorders in ataxia-telangiectasia: does ATM regulate EBV life cycle? Front Immunol. (2019) 9:3060. doi: 10.3389/fimmu.2018.03060
52. Hanson D, Hill JA, Koelle DM. Advances in the characterization of the T-cell response to human herpesvirus-6. Front Immunol. (2018) 9:1454. doi: 10.3389/fimmu.2018.01454
Keywords: ataxia-telangiectasia, inborn errors of immunity, syndromic immunodeficiency, infection, immune dysregulation, malignancy
Citation: Szczawińska-Popłonyk A, Tąpolska-Jóźwiak K, Schwartzmann E and Pietrucha B (2022) Infections and immune dysregulation in ataxia-telangiectasia children with hyper-IgM and non-hyper-IgM phenotypes: A single-center experience. Front. Pediatr. 10:972952. doi: 10.3389/fped.2022.972952
Received: 19 June 2022; Accepted: 28 September 2022;
Published: 20 October 2022.
Edited by:
Jutte Van Der Werff Ten Bosch, ZNA Queen Paola Children's Hospital, BelgiumReviewed by:
Mirjam van der Burg, Leiden University Medical Center (LUMC), Netherlands© 2022 Szczawińska-Popłonyk, Tąpolska-Jóźwiak, Schwartzmann and Pietrucha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleksandra Szczawińska-Popłonyk YXN6Y3phd2luc2thQHVtcC5lZHUucGw=
Specialty Section: This article was submitted to Pediatric Immunology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.