
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Pediatr. , 22 July 2022
Sec. Pediatric Neurology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.960450
 Seungbok Lee1,2
Seungbok Lee1,2 Se Song Jang2
Se Song Jang2 Soojin Park2
Soojin Park2 Jihoon G. Yoon1
Jihoon G. Yoon1 Soo Yeon Kim1,2
Soo Yeon Kim1,2 Byung Chan Lim2Jong Hee Chae1,2*
Byung Chan Lim2Jong Hee Chae1,2*Purpose: Loss-of-function mutations of CTNNB1 have been established as the cause of neurodevelopmental disorder with spastic diplegia and visual defects. Although most patients share key phenotypes such as global developmental delay and intellectual disability, patients with CTNNB1-related neurodevelopmental disorder show a broad spectrum of clinical features.
Methods: We enrolled 13 Korean patients with CTNNB1-related neurodevelopmental disorder who visited Seoul National University Children’s Hospital (5 female and 8 male patients with ages ranging from 4 to 22 years). They were all genetically confirmed as having pathogenic loss-of-function variants in CTNNB1 using trio or singleton whole exome sequencing. Variants called from singleton analyses were confirmed to be de novo through parental Sanger sequencing.
Results: We identified 11 de novo truncating variants in CTNNB1 in 13 patients, and two pathogenic variants, c.1867C > T (p.Gln623Ter) and c.1420C > T (p.Arg474Ter), found in two unrelated patients, respectively. Five of them were novel pathogenic variants not listed in the ClinVar database. While all patients showed varying degrees of intellectual disability, impaired motor performance, and ophthalmologic problems, none of them had structural brain abnormalities or seizure. In addition, there were three female patients who showed autistic features, such as hand stereotypy, bruxism, and abnormal breathing. A literature review revealed a female predominance of autistic features in CTNNB1-related neurodevelopmental disorder.
Conclusion: This is one of the largest single-center cohorts of CTNNB1-related neurodevelopmental disorder. This study investigated variable clinical features of patients and has expanded the clinical and genetic spectrum of the disease.
Since the first discovery of loss-of-function mutations in intellectual disability patients (1), CTNNB1 has been established as a causative gene of neurodevelopmental disorder with spastic diplegia and visual defects (NEDSDV), which is characterized by global developmental delay, intellectual disability, facial dysmorphism, and microcephaly (MIM# 615075) (2). This is an autosomal dominant disorder, mostly caused by loss-of-function mutations resulting in disruption of normal molecular function. Although several studies had reported additional cases of NEDSDV, their clinical presentations are different from each other (1, 3–11).
The CTNNB1 gene encodes the protein β-catenin, a key component of the Wnt signaling pathway. Its roles in carcinogenesis have been well established in many different types of cancers, including colorectal cancer (MIM# 114500), hepatocellular carcinoma (MIM# 114550), medulloblastoma (MIM# 155255), ovarian cancer (MIM# 167000), and pilomatricoma (MIM# 132600) (2). Cancer-causing mutations in CTNNB1 are mostly somatic, gain-of-function mutations. Currently, 28 oncogenic or likely oncogenic CTNNB1 variants are listed in the OncoKB database,1 and all of them are missense or gain-of-function mutations (12). In contrast, most of variants causing NEDSDV are de novo, loss-of-function mutations. There are 33 de novo CTNNB1 variants listed in the ClinVar database to date,2 all of which are predicted to be pathogenic or likely pathogenic (13). Among these, 27 variants are loss-of-function mutations including 14 nonsense, 12 frameshift, and 1 canonical splicing site variants (Figure 1).
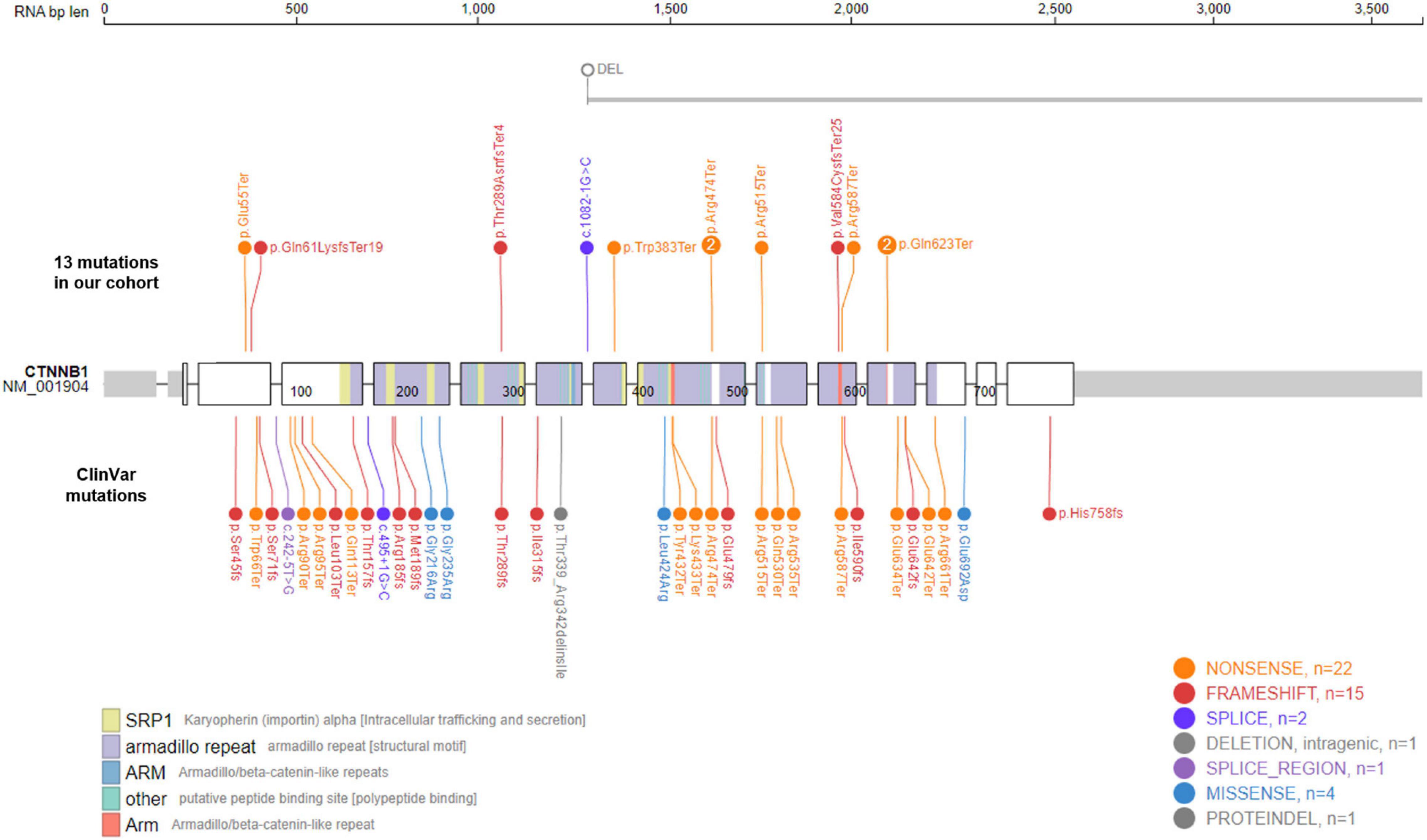
Figure 1. Mutational spectrum of CTNNB1-related neurodevelopmental disorder. Eleven de novo CTNNB1 variants were identified in in 13 patients. Two mutations, c.1867C > T (p.Gln623Ter) and c.1420C > T (p.Arg474Ter), were found in two unrelated patients, respectively. The large deletion of Case 4 spans at least 685 kb including exons 8–15 of CTNNB1 (NM_001904). We also visualized 33 de novo ClinVar mutations in CTNNB1 together, all of which are predicted to be pathogenic or likely pathogenic.
In addition to NEDSDV and several kinds of cancers mentioned above, the defects in CTNNB1 can also result in the ophthalmologic disorder, exudative vitreoretinopathy 7 (MIM# 617572). Panagiotou et al. reported that Mendelian inherited CTNNB1 mutations can cause non-syndromic familial exudative vitreoretinopathy (14). They reported one missense mutation (p.Arg710Cys) and one truncating mutation (p.His720Ter) located in the carboxy-terminal domain of the β-catenin protein. Recent studies also associated missense or truncating variants of CTNNB1 with vitreoretinopathy and suggested that ophthalmologic examination should be performed in every patient with CTNNB1-related disorders (15, 16).
Advances in sequencing technology have greatly improved genetic diagnosis in clinical practice. Whole exome sequencing (WES) or targeted gene panel sequencing is generally used, and multiple lines of evidence have already demonstrated its efficacy as a first-tier or second-tier genetic test in various kinds of diseases (17–19). Trio WES is an especially efficient diagnostic strategy for patients with neurodevelopmental disorders but without known etiologies because it enables detection of de novo or compound heterozygous variants (20).
In this study, we report clinical presentations of 13 Korean NEDSDV patients, whose CTNNB1 loss-of-function mutations were identified using singleton or trio WES analyses. This is one of the largest single-center cohorts of NEDSDV, expanding the clinical and genetic spectrum of CTNNB1-related neurodevelopmental disorders.
We enrolled 13 Korean NEDSDV patients who visited the pediatric neurology clinic of Seoul National University Children’s Hospital. They were genetically confirmed as having de novo loss-of-function mutations in CTNNB1, which were not found in their parents. The medical records of the patients were retrospectively reviewed by a pediatric neurologist. The study was performed in accordance with the ethical standards of the Declaration of Helsinki and was approved by the Institutional Review Board of Seoul National University Hospital (#2003-192-1112 and #1406-081-588).
CTNNB1 mutations were detected through next generation sequencing, seven patients by trio WES and six patients by singleton WES analyses. Except Cases 1, 2, 12, and 13 who were sequenced in other hospitals or laboratories, we conducted WES using the Illumina technology and the detailed WES methods were described in our previous study (21). WES data were aligned to the reference genome hg19 and processed according to the best practice of Genome Analysis Toolkit (22). We used the ANNOVAR program for variant annotation, such as the RefSeq gene set and gnomAD (23, 24), and focused on rare protein-altering variants (< 0.001% frequency in gnomAD). The Human Gene Mutation Database3 and ClinVar databases were screened to check whether identified variants were previously reported (13, 25).
We further selected de novo variants in trio WES analyses, and candidate variants called from singleton WES were confirmed to be de novo through parental Sanger sequencing. Genomic DNA was extracted from blood using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany) and PCR was performed using Solg™ 2X h-Taq PCR Smart mix (Solgent, Daejeon, Korea) according to the manufacturers’ instructions. All primers were synthesized by the Bioneer Company (Daejeon, Korea) and PCR products were sequenced by Macrogen (Seoul, Korea).
We used the Moralizer tool4 to check the description and predicted protein change for each candidate variant (26), and visualized them using the PeCan protein viewer5 (27). Identified variants were described in accordance with the Human Genome Variation Society guidelines (28).
We identified 11 different CTNNB1 variants in 13 patients, 8 nonsense, 3 frameshift, 1 canonical splicing site, and 1 large deletion mutation (Table 1 and Figure 1). All of them were confirmed as de novo variants by parental analysis and predicted to be pathogenic according to the American College of Medical Genetics and Genomics criteria. They were not reported in gnomAD (allele frequency = 0), and five of them were novel pathogenic variants not listed in ClinVar or HGMD (Supplementary Table 1 and Supplementary Figure 1).
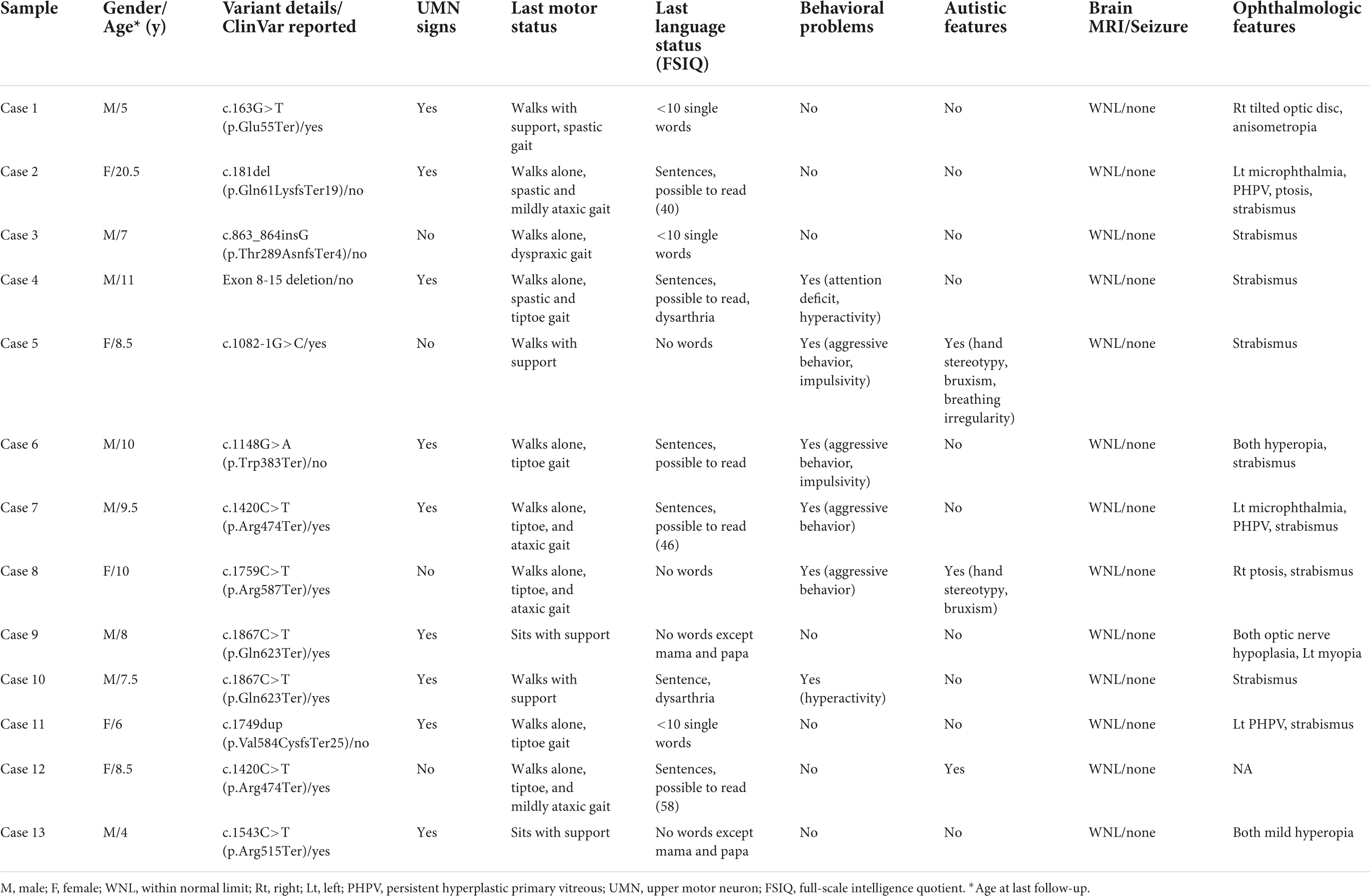
Table 1. Neurodevelopmental disorder patients with de novo loss-of-function mutations in CTNNB1 (NM_001904.4).
The proportion of nonsense variants (61.5%, 8 out of 13) was higher than that of ClinVar (42.4%, 14 out of 33). All identified mutations in our cohort were loss-of-function mutations, and the large deletion of Case 4, spanning at least 685 kb from exon 8 to exon 15, is also predicted to result in an out-of-frame mutation. Like other kinds of mutations causing diseases by loss-of-function mechanisms, CTNNB1 mutations in our patients were located throughout the gene without any identifiable hot spots. Two mutations, c.1867C > T (p.Gln623Ter) and c.1420C > T (p.Arg474Ter), were found in two unrelated patients, respectively.
We have summarized the clinical features of our patients with CTNNB1 mutations in Table 1. While most patients shared the key phenotypes of NEDSDV, such as developmental delay and intellectual disability, they also showed a broad spectrum of clinical presentations particular to individual patients. All patients in our cohort were 4 years of age or older and diagnosed with neurodevelopmental disorder by pediatric neurologists. While Cases 5 and 8 could not speak any meaningful words until nearly 10 years of age, almost half of the patients could speak sentences and read some words or sentences. Among them, full-scale intelligence quotients were evaluated in three patients, with results ranging from 40 (Case 2) to 58 (Case 12).
Motor development also differed from patient to patient. While Cases 9 and 13 could not sit alone at their last follow-up, others could walk with or without support. Although more than half of the patients (61.5%, 8 out of 13) were able to walk alone without any assistance, all of them showed abnormal gait patterns such as spastic, tiptoe, and ataxic gaits. Similarly, upper motor neuron signs such as increased muscle tone, increased deep tendon reflexes, and Babinski signs were evident in 9 of the 13 patients (69.2%).
Twelve patients were evaluated by ophthalmologists and they all showed various kinds of ocular abnormalities. Strabismus, shown in 9 patients (69.2%), was the most common ophthalmologic problem in our cohort. Unilateral persistent hyperplastic primary vitreous (PHPV) was shown in 3 patients, and two of them also had microphthalmia on the affected side.
Although every patient in our cohort underwent brain MRI and their images were reviewed by pediatric radiologists, there were no remarkable abnormalities noted in their brains. In addition, none of our patients had any seizure history during follow-up. As shown in Figure 2A, we reviewed a total of 69 patients with CTNNB1-related neurodevelopmental disorder reported in the literature, including our patients, and found that only one (1.4%) and three (5.4%) patients had seizure history and brain abnormalities, respectively (1, 3–11, 13–15). The patient with seizure history was suspected as having absence seizure in early childhood. However, her electroencephalogram was normal, and it was uncertain that such events were true clinical seizures. In addition, while one patient had definite brain abnormalities including dysgenesis of the corpus callosum, absence of the right fornix, and hypoplastic brainstem, the brain MRI findings of the other two were just left temporal lobe atrophy and hypoplasia of the corpus callosum, respectively. Therefore, although every patient had apparent developmental delay or intellectual disability, seizure or structural brain abnormality would be an uncommon phenotype of CTNNB1-related neurodevelopmental disorder or NEDSDV.
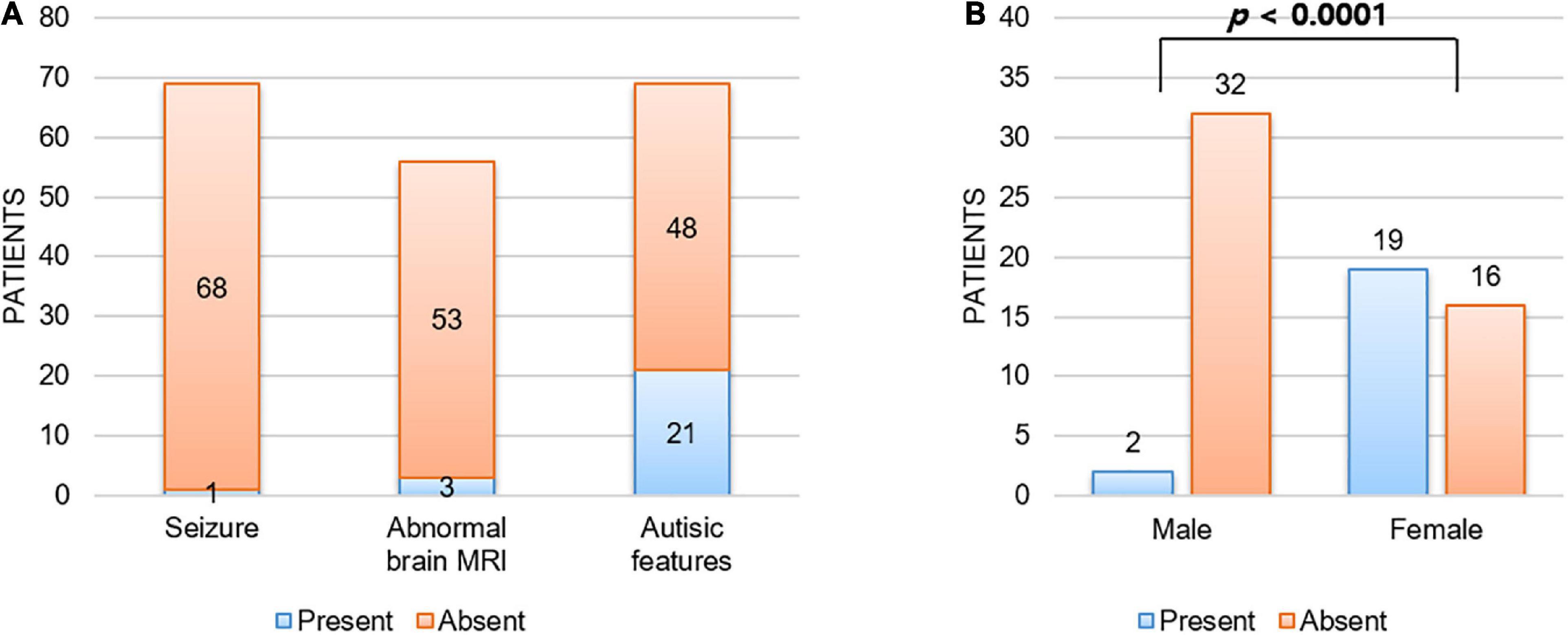
Figure 2. Unique clinical manifestations of CTNNB1-related neurodevelopmental disorder. (A) Patients with CTNNB1-related neurodevelopmental disorder were less likely to have seizure (1.4%) or structural brain abnormality (5.4%), and autistic features were shown in 30.4% of patients. Here we considered three patients with borderline brain MRI findings (delayed bilateral frontal lobe myelination, mild corpus callosum thinning, and mild dilation of ventricles) as normal. In addition, abnormal brain MRI findings noted in two patients out of three also did not suggest obvious structural abnormality (left temporal lobe atrophy and hypoplasia of corpus callosum). (B) Autistic features were significantly more frequent in female patients than in male patients. Fisher’s exact test was used to compare frequency of autistic features by gender.
In addition to the above clinical presentations, there were 6 patients (46.2%, 4 males and 2 females) who showed behavioral problems; aggressive behavior in 4 patients, hyperactivity in 2 patients, and impulsivity in 2 patients (Table 1). Rett-like or autistic features were also shown in 3 patients, presenting with symptoms such as hand stereotypy, bruxism, and irregular breathing.
Interestingly, all three patients with autistic features were female in this study. Autistic features have been reported frequently (1, 3–11, 13–15), shown in around 30% of patients with CTNNB1-related neurodevelopmental disorder (Figure 2A). Consistent with our study, autistic features were significantly more frequent in female patients (54.3%, 19 out of 35) than in male patients (5.9%, 2 out of 34) with a P-value = 1.3 × 10–5 by Fisher’s exact test (Figure 2B).
Although we searched for other modifying factors determining clinical manifestations of CTNNB1-related neurodevelopmental disorder, such as mutation type and location, gender was the only significant factor associated with autistic features in our analysis. In particular, there were two pairs of unrelated patients sharing the same mutation (p.Gln623Ter and p.Arg474Ter), but their clinical presentations were different from each other despite the shared genotype (Table 1).
In this study we summarized the clinical presentations of 13 NEDSDV or CTNNB1-related neurodevelopmental disorder patients in our center. Eleven kinds of de novo mutations were identified, 5 of them being novel mutations not listed in ClinVar or HGMD. This is one of the largest single-center cohorts of NEDSDV, expanding the clinical and genetic spectrum of the disease.
As already linked with NEDSDV (MIM# 615075) and exudative vitreoretinopathy 7 (MIM# 617572), clinical features such as developmental delay, intellectual disability, spastic diplegia, and ocular problems have been well established in CTNNB1-related neurodevelopmental disorder (1, 3–8). Although most of our patients shared such key phenotypes, the severity of each symptom was different patient by patient. While half of the patients could speak sentences and read some words or sentences, two patients (Cases 2 and 12) could not speak any meaningful words at all. While most patients could walk with or without support, two patients (Cases 9 and 13) never achieved sitting or standing without support during follow-up. Likewise, ophthalmologic features also varied from strabismus to PHPV in our patients. Although we searched for associations between mutation type or location and disease phenotype, we could not find any significant genotype-phenotype correlations in our cohort. A recent study also reported that no genotype-phenotype correlations have been identified in CTNNB1-related neurodevelopmental disorder to date (15, 29).
In an animal study using a β-catenin conditional knockout mouse, the authors found an alteration in the β-catenin pathway involving cadherin-based synaptic adhesion complexes, which are essential for normal brain function (30). Although there were patients with non-syndromic familial exudative vitreoretinopathy inherited in an autosomal dominant manner, these mutations were missense or truncating mutations located in the carboxy-terminal domain of β-catenin (14). Most patients with loss-of-function mutations in CTNNB1 showed varying degrees of intellectual disability. In this study, Cases 2, 7, and 12 showed milder intellectual disability compared to the other patients. However, their full-scale intelligence quotient scores were as low as 40, 46, and 58, respectively. This finding suggests that it is quite difficult to expect normal or tolerable intelligence in patients with loss-of-function CTNNB1 mutations.
The association between seizure and developmental delay or intellectual disability has been well established, and one study reported the prevalence of epilepsy in global developmental delay patients as high as 56% (31–33). Among the 69 patients with CTNNB1-related neurodevelopmental disorder we reviewed, none of them had apparent seizure history or abnormal electroencephalogram results (1, 3–11, 13–15). In addition, while structural brain abnormalities have been detected in around 30% of developmental delay patients (34, 35), there were only three patients (5.4%) showing remarkable brain abnormalities among 56 patients. Therefore, we hypothesize that such a low frequency of seizure or brain abnormality might be rather a clinical characteristic of CTNNB1-related neurodevelopmental disorder.
Autistic or Rett-like phenotypes have been commonly reported in patients with CTNNB1-related neurodevelopmental disorder (1, 3–11, 13–15), and recent studies suggest that the Wnt signaling pathway is one of the major developmental pathways affecting autistic behaviors and could be a therapeutic target of the disease (36, 37). CTNNB1 is a key regulator of the canonical Wnt pathway and plays a key role in neurodevelopment. Interestingly, by analyzing patients of previous studies including those of our cohort, we revealed that autistic features were predominantly found in female NEDSDV patients. There might be some gender differences in molecular pathways that result in female predominance. Further studies are required to confirm this tendency and reveal its underlying mechanisms. On the other hand, six of our patients and several patients in other studies had various kinds of behavior problems. Such clinical features need to be investigated in more patients with CTNNB1-related neurodevelopmental disorder.
Mutations causing disease by a loss-of-function mechanism were generally located throughout the gene rather than concentrated in a hot spot. However, two mutations identified in our cohort, c.1867C > T (p.Gln623Ter) and c.1420C > T (p.Arg474Ter), were found in two unrelated patients, respectively (Figure 1). These amino-acid positions might be fragile sites of CTNNB1, and further studies are required to investigate such mutation-prone sequences in the human genome. On the other hand, as shown in Table 1, their clinical features were different from each other despite having identical mutations. We suggest that there are other genetic and/or environmental factors that modify the clinical presentation, such as gender or other genes involved in the Wnt/β-catenin signaling pathway.
This study presented the genetic and clinical spectrum of CTNNB1-related neurodevelopmental disorder and identified some key clinical features of the disease. More clinical studies are required to validate our findings, and molecular studies are also needed to discover novel therapeutic targets for treatment of neurodevelopmental disorders.
The data presented in this study are deposited in the National Center for Biotechenology Information (NCBI) BioProject repostitory, accession number: PRJNA855946.
The studies involving human participants were reviewed and approved by Institutional Review Board of Seoul National University Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
BL and JC: study conception and design. SK, BL, and JC: data collection. SL, SJ, JY, and SP: data analysis. SL and JC: results interpretation. SL: drafting the manuscript. All authors contributed to the manuscript revision and approval.
This research was supported by the Research of Korea Disease Control and Prevention Agency (2021-ER0701-01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.960450/full#supplementary-material
1. de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. (2012) 367:1921–9. doi: 10.1056/NEJMoa1206524
2. Hamosh A, Scott AF, Amberger J, Valle D, McKusick VA. Online Mendelian Inheritance in Man. Hum Mutat. (2000) 15:57–61. doi: 10.1002/(SICI)1098-1004(200001)15:1<57::AID-HUMU12>3.0.CO;2-G
3. Dubruc E, Putoux A, Labalme A, Rougeot C, Sanlaville D, Edery P. A new intellectual disability syndrome caused by CTNNB1 haploinsufficiency. Am J Med Genet A. (2014) 164A:1571–5. doi: 10.1002/ajmg.a.36484
4. Tucci V, Kleefstra T, Hardy A, Heise I, Maggi S, Willemsen MH, et al. Dominant beta-catenin mutations cause intellectual disability with recognizable syndromic features. J Clin Invest. (2014) 124:1468–82. doi: 10.1172/JCI70372
5. Kuechler A, Willemsen MH, Albrecht B, Bacino CA, Bartholomew DW, van Bokhoven H, et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum Genet. (2015) 134:97–109. doi: 10.1007/s00439-014-1498-1
6. Kharbanda M, Pilz DT, Tomkins S, Chandler K, Saggar A, Fryer A, et al. Clinical features associated with CTNNB1 de novo loss of function mutations in ten individuals. Eur J Med Genet. (2017) 60:130–5. doi: 10.1016/j.ejmg.2016.11.008
7. Percy AK, Lane J, Annese F, Warren H, Skinner SA, Neul JL. When Rett syndrome is due to genes other than MECP2. Transl Sci Rare Dis. (2018) 3:49–53. doi: 10.3233/TRD-180021
8. Ho S, Tsang MH, Fung JL, Huang H, Chow CB, Cheng SS, et al. CTNNB1-related neurodevelopmental disorder in a Chinese population: a case series. Am J Med Genet A. (2022) 188:130–7. doi: 10.1002/ajmg.a.62504
9. Verhoeven WMA, Egger JIM, Jongbloed RE, van Putten MM, de Bruin-van Zandwijk M, Zwemer AS, et al. A de novo CTNNB1 novel splice variant in an adult female with severe intellectual disability. Int Med Case Rep J. (2020) 13:487–92. doi: 10.2147/IMCRJ.S270487
10. Ke Z, Chen Y. Case report: a de novo CTNNB1 nonsense mutation associated with neurodevelopmental disorder, retinal detachment, polydactyly. Front Pediatr. (2020) 8:575673. doi: 10.3389/fped.2020.575673
11. Dashti S, Salehpour S, Ghasemi MR, Sadeghi H, Rostami M, Hashemi-Gorji F, et al. Identification of a novel de novo mutation in the CTNNB1 gene in an Iranian patient with intellectual disability. Neurol Sci. (2022) 43:2859–63. doi: 10.1007/s10072-022-05904-4
12. Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. (2017) 2017:1–16.
13. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. (2018) 46:D1062–7. doi: 10.1093/nar/gkx1153
14. Panagiotou ES, Sanjurjo Soriano C, Poulter JA, Lord EC, Dzulova D, Kondo H, et al. Defects in the cell signaling mediator beta-catenin cause the retinal vascular condition FEVR. Am J Hum Genet. (2017) 100:960–8. doi: 10.1016/j.ajhg.2017.05.001
15. Rossetti LZ, Bekheirnia MR, Lewis AM, Mefford HC, Golden-Grant K, Tarczy-Hornoch K, et al. Missense variants in CTNNB1 can be associated with vitreoretinopathy-Seven new cases of CTNNB1-associated neurodevelopmental disorder including a previously unreported retinal phenotype. Mol Genet Genomic Med. (2021) 9:e1542. doi: 10.1002/mgg3.1542
16. Sun W, Xiao X, Li S, Jia X, Wang P, Zhang Q. Germline Mutations in CTNNB1 associated with syndromic FEVR or norrie disease. Invest Ophthalmol Vis Sci. (2019) 60:93–7. doi: 10.1167/iovs.18-25142
17. Shashi V, McConkie-Rosell A, Rosell B, Schoch K, Vellore K, McDonald M, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med. (2014) 16:176–82. doi: 10.1038/gim.2013.99
18. Adams DR, Eng CM. Next-Generation sequencing to diagnose suspected genetic disorders. N Engl J Med. (2018) 379:1353–62. doi: 10.1056/NEJMra1711801
19. Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. (2019) 21:2413–21. doi: 10.1038/s41436-019-0554-6
20. Tran Mau-Them F, Moutton S, Racine C, Vitobello A, Bruel AL, Nambot S, et al. Second-tier trio exome sequencing after negative solo clinical exome sequencing: an efficient strategy to increase diagnostic yield and decipher molecular bases in undiagnosed developmental disorders. Hum Genet. (2020) 139:1381–90. doi: 10.1007/s00439-020-02178-8
21. Lee S, Eum J, Park S, Ki S, Hwang BJ, Kee Y, et al. TNNT1 myopathy with novel compound heterozygous mutations. Neuromuscul Disord. (2022) 32:176–84. doi: 10.1016/j.nmd.2021.12.003
22. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
23. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
24. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43.
25. Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. (2017) 136:665–77. doi: 10.1007/s00439-017-17796
26. Lefter M, Vis JK, Vermaat M, den Dunnen JT, Taschner PEM, Laros JFJ. Next Generation HGVS Nomenclature Checker. Bioinformatics. (2021) 37:2811–7. doi: 10.1093/bioinformatics/btab051
27. Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet. (2016) 48:4–6. doi: 10.1038/ng.3466
28. den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. (2016) 37:564–9. doi: 10.1002/humu.22981
29. Ho SKL, Tsang MHY, Lee M, Cheng SSW, Luk H, Lo IFM, et al. In: MP Adam, GM Mirzaa, RA Pagon, SE Wallace, LJH Bean, KW Gripp, et al. editors. CTNNB1 Neurodevelopmental Disorder. Seattle (WA): GeneReviews((R)) (1993).
30. Wickham RJ, Alexander JM, Eden LW, Valencia-Yang M, Llamas J, Aubrey JR, et al. Learning impairments and molecular changes in the brain caused by beta-catenin loss. Hum Mol Genet. (2019) 28:2965–75. doi: 10.1093/hmg/ddz115
31. Oostrom KJ, Smeets-Schouten A, Kruitwagen CL, Peters AC, Jennekens-Schinkel A. Dutch Study Group of Epilepsy in C. Not only a matter of epilepsy: early problems of cognition and behavior in children with “epilepsy only”–a prospective, longitudinal, controlled study starting at diagnosis. Pediatrics. (2003) 112:1338–44. doi: 10.1542/peds.112.6.1338
32. Albaradie R, Habibullah H, Mir A, Alshammari AK, Alajmi MS, Alsubaie FA, et al. The prevalence of seizures in children with developmental delay. Neurosciences (Riyadh). (2021) 26:186–91.
33. Berg AT, Langfitt JT, Testa FM, Levy SR, DiMario F, Westerveld M, et al. Global cognitive function in children with epilepsy: a community-based study. Epilepsia. (2008) 49:608–14. doi: 10.1111/j.1528-1167.2007.01461.x
34. Kjos BO, Umansky R, Barkovich AJ. Brain MR imaging in children with developmental retardation of unknown cause: results in 76 cases. AJNR Am J Neuroradiol. (1990) 11:1035–40.
35. Alamri A, Aljadhai YI, Alrashed A, Alfheed B, Abdelmoaty R, Alenazi S, et al. Identifying clinical clues in children with global developmental delay / intellectual disability with abnormal brain magnetic resonance imaging (MRI). J Child Neurol. (2021) 36:432–9. doi: 10.1177/0883073820977330
36. Baranova J, Dragunas G, Botellho MCS, Ayub ALP, Bueno-Alves R, Alencar RR, et al. Autism spectrum disorder: signaling pathways and prospective therapeutic targets. Cell Mol Neurobiol. (2021) 41:619–49. doi: 10.1007/s10571-020-00882-7
Keywords: CTNNB1, β-catenin, neurodevelopmental disorder with spastic diplegia and visual defects, exudative vitreoretinopathy 7, Rett-like phenotype, autism spectrum disorder
Citation: Lee S, Jang SS, Park S, Yoon JG, Kim SY, Lim BC and Chae JH (2022) The extended clinical and genetic spectrum of CTNNB1-related neurodevelopmental disorder. Front. Pediatr. 10:960450. doi: 10.3389/fped.2022.960450
Received: 03 June 2022; Accepted: 28 June 2022;
Published: 22 July 2022.
Edited by:
Wang-Tso Lee, National Taiwan University Hospital, TaiwanReviewed by:
Ihtisham Bukhari, Henan Provincial People’s Hospital, ChinaCopyright © 2022 Lee, Jang, Park, Yoon, Kim, Lim and Chae. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jong Hee Chae, Y2hhZXBlZDFAc251LmFjLmty
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.