 Federica Puglisi1Rachele Soma2Marta Podda3
Federica Puglisi1Rachele Soma2Marta Podda3 Simona Vetrella4Marco Rabusin5Serena Tropia6
Simona Vetrella4Marco Rabusin5Serena Tropia6 Mariaclaudia Meli2*
Mariaclaudia Meli2* Giovanna Russo2Stefania Sorrentino7Giovanni Erminio8
Giovanna Russo2Stefania Sorrentino7Giovanni Erminio8 Alfredo Pulvirenti9Martino Ruggieri10
Alfredo Pulvirenti9Martino Ruggieri10 Andrea Di Cataldo2
Andrea Di Cataldo2
- 1Unit of Neonatology and Neonatal Intensive Care Unit, AOU “Policlinico”, PO “San Marco”, University of Catania, Catania, Italy
- 2Unit of Pediatric Onco-Haematology, Department of Clinical and Experimental Medicine, Section of Pediatrics and Child Neuropsychiatry, University of Catania, Catania, Italy
- 3Pediatric Oncology Unit, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy
- 4Pediatric Oncology Unit, Santobono-Pausilipon Hospitals, Naples, Italy
- 5Institute for Maternal & Child Health (I.R.C.C.S) Burlo Garofolo, Trieste, Italy
- 6Pediatric Hematology and Oncology Unit, ARNAS “Civico, Di Cristina and Benfratelli” Hospitals, Palermo, Italy
- 7Oncology Unit, IRCCS Istituto Giannina Gaslini, Genova, Italy
- 8Epidemiology Scientific Directorate, IRCCS Istituto Giannina Gaslini, Genova, Italy
- 9Bioinformatics Unit, Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
- 10Unit of Rare Diseases of the Nervous System in Childhood, Department of Clinical and Experimental Medicine, Section of Pediatrics and Child Neuropsychiatry, University of Catania, Catania, Italy
Background: Neuroblastic tumors (NBTs) are the most common extra-cranial solid tumors of childhood. Neurofibromatosis type 1 (NF1) is the most common neurocutaneous disorder with a predisposition to tumors. The co-occurrence of NBTs in the setting of NF1 has been occasionally reported, suggesting a non-casual association and likely configuring a spectrum of neural crest–derived disorders.
Aim of the study: To explore the occurrence of NBTs within NF1 and to report on its natural history, therapeutic strategies, and outcomes in an Italian cohort of children with NF1 and in the literature.
Subjects and Methods: Study (a): a retrospective analysis of questionnaire-based data [years 1979–2017] derived from the databases of the Italian Registry for Neuroblastoma (RINB) of the Italian Society of Pediatric Onco-Haematology (AIEOP); and Study (b): a systematic review search on NF1/NB co-occurrence.
Results: Study (a) identified eight children with NBTs, 0.2% of patients registered in the RINB, fulfilling the diagnostic criteria for NF1. The primary site of NBTs was abdominal in six patients. The NBTs were neuroblastoma (NB) in five patients, ganglioneuroblastoma (GNB) in one, patient, and ganglioneuroma (GN) in two. Metastatic diffusion occurred in three out of eight children. MYCN gene testing, performed in the tumors of five patients, resulted not-amplified. The major features of NF1 included the following: NF1 family history in four patients, café-au-lait spots in all, freckling in six, Lisch nodules in three, and neurofibromas in three. With regard to the outcome, four children survived three of these for the progression of NB and one for a second tumor. Study (b) identified 12 patients with NF1/NB from the years 1966–2017, and the median age at diagnosis was 27 months (range = 0–168 months). The primary site of NB was thoracic. The prevalent histotype was NB in nine patients, GNB in two, and GN in one. Eight/nine NBs were metastatic. The MYCN gene was amplified in the only studied case. The NF1 features included NF1 family history in seven patients; the major NF1 features were café-au-lait spots in nine patients, freckling in one, Lisch nodules in none, and neurofibromas in six. The outcome was good for only two children, while eight children died of neuroblastoma, at a median age of 49.5 months (range = 2.4–174 months), with a median survival time of 21.75 months after diagnosis.
Conclusions: To our knowledge, this represents the first systematic study on the occurrence of NBTs in NF1. This confirms that NBs are rare per se in the setting of NF1 (0.2% of all NBs) and even if compared to the overall frequency of malignancies in NF1 (i.e., 14.7%). The male:female ratio in study (a) (0.6) was different from what was recorded in study (b) (1.5) and in line with the overall increased frequency of malignancies in females with NF1. The median ages at diagnosis of NB in either study (a) or (b) were concordant with what occurred in the NB population. In study (a) versus study (b), the frequency of metastatic diffusion was lower, likely indicating less awareness on work-ups for malignancies in old NF1 series in the literature. The outcome was much better in study (a) than in study (b), indicating that multidisciplinary treatment for NB is highly recommended.
Introduction
Neuroblastic tumors (NBTs) are very rare tumors, characterized by various clinical presentations and diverse prognosis of its subsets. While in some patients the tumor is successfully treated with surgery alone, or may regress spontaneously, the chances of cure in children older than 1 year with metastatic disease remain poor. Although the probability of survival for children with NBTs has improved over time, even the best published results do not parallel those obtained for most other childhood malignancies (1). A few studies have documented this finding by reporting a large series of patients diagnosed over a long period (2–6).
Most NBTs are sporadic and not correlated with any specific constitutional germline chromosomal abnormality, inherited predisposition, or associated congenital anomalies. Nevertheless, there are some exceptions. A higher incidence of NBTs has been suggested in girls with Turner syndrome (7). Patients with Kabuki syndrome and NBTs have been reported (8, 9). Hirschsprung’s disease, congenital central hypoventilation (Ondine's curse), and neurofibromatosis type 1 (NF1) have all been described in association with NBTs, suggesting the existence of a global disorder of neural crest–derived cells (i.e., neurocristopathy) (10–13).
NF1 is the most common form of neurofibromatosis and one of the most common autosomal dominant disorders in humans, with an incidence of 1 in 2,600–3,000 individuals (14, 15). Approximately one half of the cases are familial (inherited). The remainder are the result of de novo (sporadic) mutations (16). These mutations occur primarily in paternally derived chromosomes, and the likelihood of de novo NF1 increases with advanced paternal age (17). The incidence of segmental NF1 is estimated at 1 in 36,000–40,000 (18).
Patients with NF1 are predisposed to both benign and malignant tumors of neurogenic and non-neurogenic origin. Most studies that have addressed the risk of malignancy and early death have shown an approximately 8- to 15-year decrease in life expectancy in patients with NF1, mainly as a result of malignancy (19, 20). Mortality in NF1 has previously been studied in cohorts from France, Wales, United States, and Denmark (21–23). These studies have shown excess mortality rates of NF1 patients compared with those of the general population and a high proportion of deaths caused by malignancies.
Multiple studies have shown a substantial risk of nervous system malignancy, with an indisputable excess risk of gliomas and malignant peripheral nerve sheath tumor (MPNST) (24), both of which result in an excess risk of mortality (25). The onset of NBTs in patients with NF1 has also been described, but the actual incidence of this association is not known (26).
The purpose of this study is to analyze the clinical–epidemiological characteristics of this rare clinical association, as well as the therapeutic approach and prognosis, in order to allow a descriptive analysis of the development of clinical and research experience.
Materials and methods
Setting and sample
All patients with a diagnosis of a NBT and NF1 were included in this analysis. The cases were identified by searching the databases of the Italian clinical units of pediatric onco-hematology and the Italian Neuroblastoma Registry (RINB) database (from 1979 onward). The RINB was activated in 1979 and includes all subjects with any peripheral NBT [i.e., neuroblastoma (NB), ganglioneuroblastoma (GNB), and the benign ganglioneuroma (GN)], diagnosed at the institutions included in the Italian Neuroblastoma Group (ING) of the Italian Association of Pediatric Hematology and Oncology (AIEOP) (27). More than the expected number of NBT patients from Italy have been recruited through this network (28).
The data on the included patients were collected retrospectively: patients diagnosed from the years 1979 to May 2017 were considered. The patients were treated according to the AIEOP protocol till the 1990s and European SIOPEN protocols thereafter. Informed consent to the treatment and to data collection and analysis was obtained from all patients according to institutional guidelines at the time of enrolling patients in the protocols.
A standardized questionnaire was sent to all the 54 hematology-oncology units belonging to the AIEOP with the aim of collecting information about the two conditions. The survey was designed to assess data regarding epidemiological aspects, clinical features, and genotype/phenotype correlation and the management and course of the patients individuated. The questionnaire included two parts, one relating to a description of the NBT and the other relating to NF1, as given in Tables 1, 2. Tumor diagnosis, disease extension, and response to treatment were defined according to the International Neuroblastoma Staging System (INSS) and the International Neuroblastoma Risk Group (INRG) staging system as described previously (29, 30). The long-term sequelae were ascertained by contacting the clinical investigators at the various centers; no additional investigations were conducted on possible late effects for the purposes of this study.
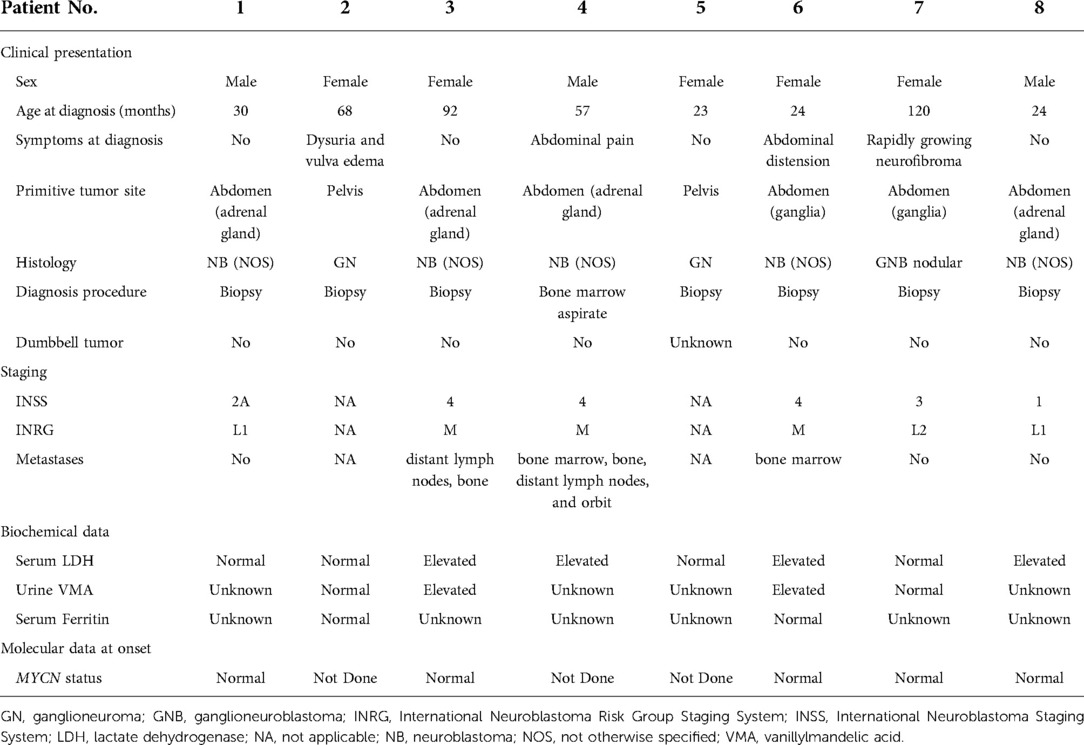
Table 1. Characteristics of the tumor in eight patients.
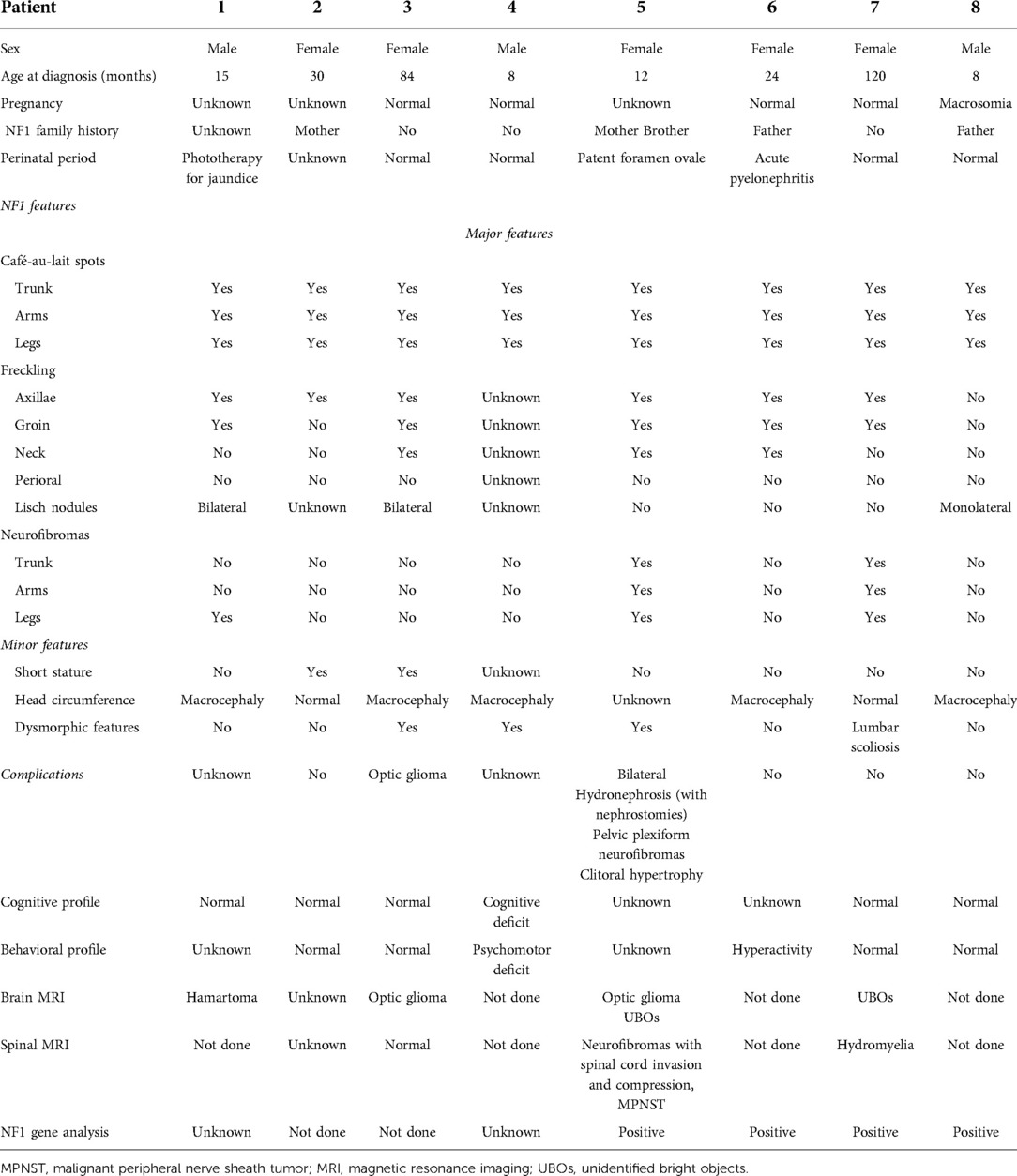
Table 2. Characteristics of NF1 in eight patients.
Statistical analysis was conducted within the R statistical System. To identify possible correlations among the subgroup of patients, we performed principal component analysis using the variables characterizing both NF1 and NBTs.
Results
Demographic information
Overall, eight patients with NBTs and NF1 were registered, who included three males and five females, with a male:female ratio of 0.6. They represented 0.2% of all patients registered in the RINB (3,976 patients). The age at diagnosis of neuroblastoma was between 23 and 120 months, with an average of 54.7 months and a median age of 43.5 months. The age at diagnosis of NF1 was between 8 and 120 months, with an average of 37.6 and a median age of 19.5 months. The patients’ demographic data are given in Table 3.
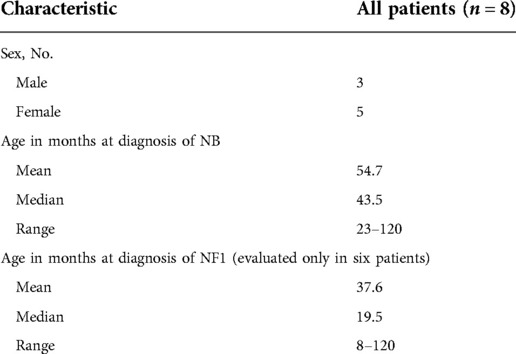
Table 3. Demographic information of the study patients.
Clinical presentation
The abdomen was the primary site for six patients, the adrenal gland for four patients, paravertebral ganglia for two, and pelvis for two. Symptoms at diagnosis were present in half of the patients and depended on the primary site: one child had dysuria and vulva edema, two had abdominal pain, and only one patient had a symptom correlated with NF1, an increase in the size of a neurofibroma in the same site. No patient had a dumbbell tumor (Tables 1, 2).
The histology most represented was NB in five patients. One patient had a GNB nodular, and two had GN. Among these five patients with neuroblastoma, metastatic disease was present at onset in three patients; the other two cases with NB,were both stage L1 for the INRG and stage 2A and 1 for the INSS. The sites of metastasis were distant lymph nodes, bone, and bone marrow in two patients and orbit in one.
With regard to biologic studies at diagnosis, serum lactate dehydrogenase (LDH) was studied in all patients and was found to be above normal value in four patients; serum ferritin was normal in the two studied cases, while urinary vanillylmandelic acid (VMA) was above normal value in two of the four studied cases. The MYCN gene was not amplified in the five studied cases.
In six patients, NF1 was diagnosed before the diagnosis of NBTs, while in two patients, it was diagnosed at the same time. Half of the patients had a family history of NF1. The perinatal period was uneventful for four patients, one was treated for jaundice at birth, one was treated for urinary tract infection, and one had a patent foramen ovale at birth. At diagnosis, café-au-lait spots were found on all patients, distributed over the trunk, arms, and groin in all of them; freckling was recorded in seven patients; bilateral ocular Lisch nodules were detected in two patients and monolateral ocular Lish nodules in one. Three children developed neurofibromas. For what was regarded as minor features, macrocephaly was reported in five patients, short stature in two, and dysmorphic features in four. With regard to the cognitive and behavioral profile, one case of hyperactivity was reported and there was one case of cognitive and psychomotor deficit. On brain magnetic resonance imaging (MRI), optic glioma was detected in two patients and hamartoma in one; unidentified bright objects (UBOs) were observed in two. Furthermore, spinal MRI showed the presence of neurofibromas with a compression of the spinal cord in one patient and hydromyelia in another. Genetic testing was performed in four patients to confirm the diagnosis. Patient No. 5 was the one who presented the most significant number of complications related to NF1, with the presence of bilateral hydronephrosis (with nephrostomies), clitoral hypertrophy, pelvic plexiform neurofibromas, neurofibromas with invasion of the spinal canal, and a compression of the spinal cord and MPNST.
Because of the availability of only a few samples, we were not able to establish any statistically significant relationship among these patients.
Treatment
Treatment and outcome are summarized in Table 4. As first-line treatment, surgery was performed on two patients, and it involved a complete resection. No postoperative complications were reported. Four patients received multidrug chemotherapy according to their time protocols. Only one patient received, after induction chemotherapy, a consolidation treatment with megatherapy and stem cell rescue, radiotherapy, immunotherapy, and cis-retinoic acid. Only one patient underwent 131I-metaiodobenzylguanidine (MIBG) therapy, given the intense capture of the documented mass with MIBG scintigraphy.

Table 4. Treatment details and outcome for eight patients.
Outcome
Three patients reached the stage of complete remission (CR) after first-line treatment, two reached the surgery alone stage, and one the chemotherapy stage, and these patients are in CR at 44.5, 53, and 7 months, respectively, after diagnosis; only one patient was observed and is in a condition of stable disease (SD) 8 years after diagnosis. Disease progressed in four patients after first-line therapy in two, during first-line therapy in one, and after MIBG therapy in one. All of them received rescue chemotherapy, but they all died, three of NB at 6, 19, and 20 months from diagnosis and one from a second tumor (mediastinal MPNST), but their time of death was not reported.
Literature review and discussion
A literature study was performed using MEDLINE/PubMed up to May 2017. The following MeSH headings and text words were used: “neuroblastoma AND neurofibromatosis type 1” and “neuroblastoma AND von Recklinghausen disease”. We did not impose any language restrictions. Two additional articles from the reference lists were obtained. Finally, all papers identified in the literature scan were examined and relevant papers were reviewed and summarized for inclusion in this report (31–38) (Figure 1).
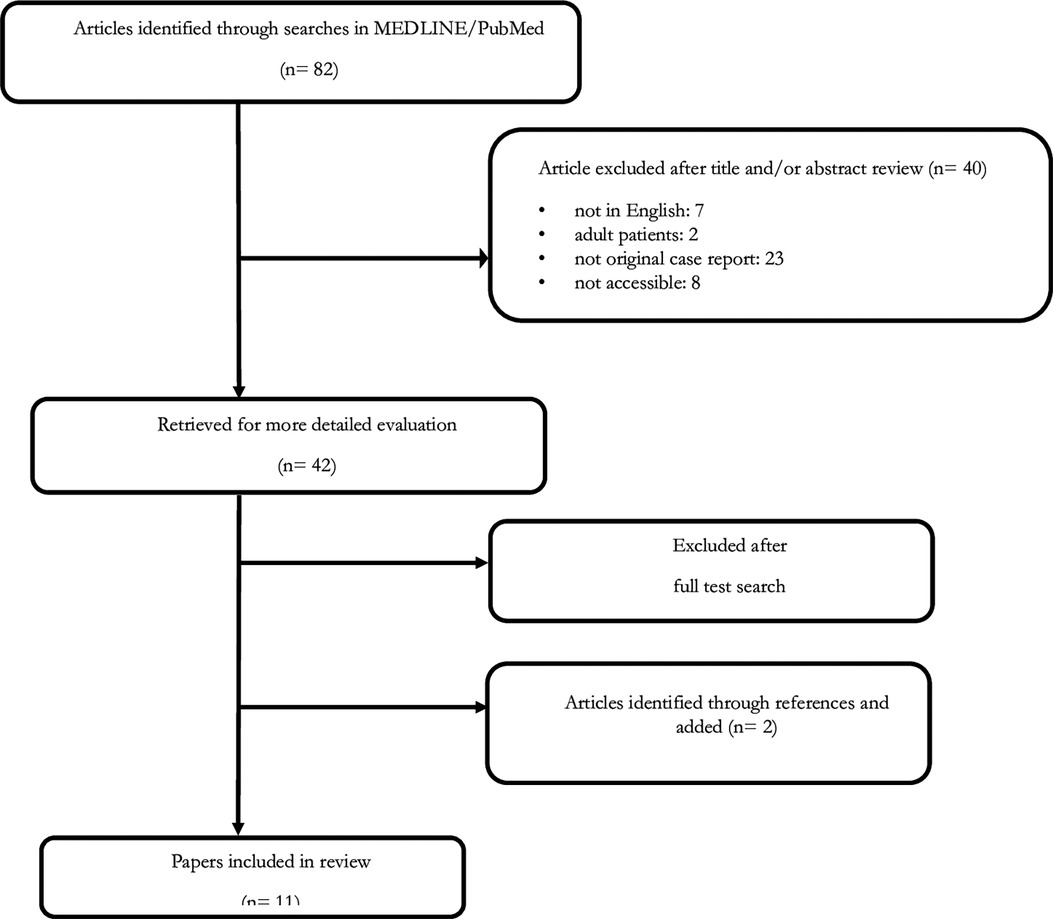
Figure 1. Selection process for papers included in the review.
Overall, 12 cases of patients with NBTs and NF1 were reported between 1966 and 2013: 7 males and 3 females (the sex of the other two patients was not available). The age at diagnosis of NBTs was between 0 and 168 months, with an average of 39 months and a median age of 27 months. The age at diagnosis of NF1 was specified only in five patients, while in three patients, it was not reported, and in four patients, NF1 was diagnosed at the same time of NB diagnosis. The patients’ demographic data are presented in Table 5.

Table 5. Demographic information of the case reports.
A detailed description of the case reports is given in Tables 6A,B. The primary site of NBTs was known in 11 out of 12 patients. It was in the mediastinum in five patients, in the abdomen in four patients, and in the neck in one, and in one patient, there were two masses, both in the thorax and in the abdomen. Diagnostic symptoms were present in all patients and correlated with the primary site. Dumbbell tumor was found in two patients, one with NB and one with GNB.
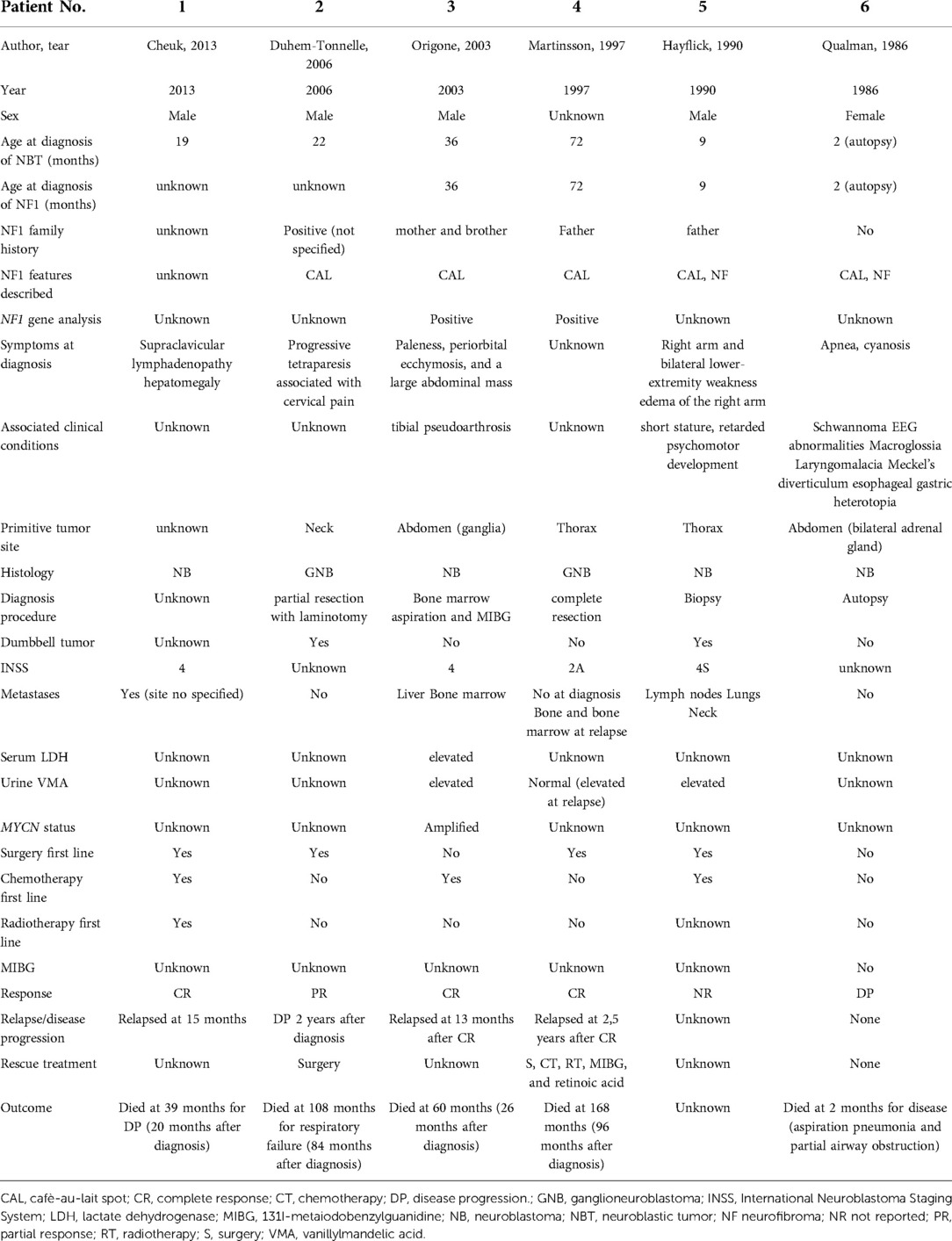
Table 6A. Description of the cases reports analyzed by the literature (part 1).
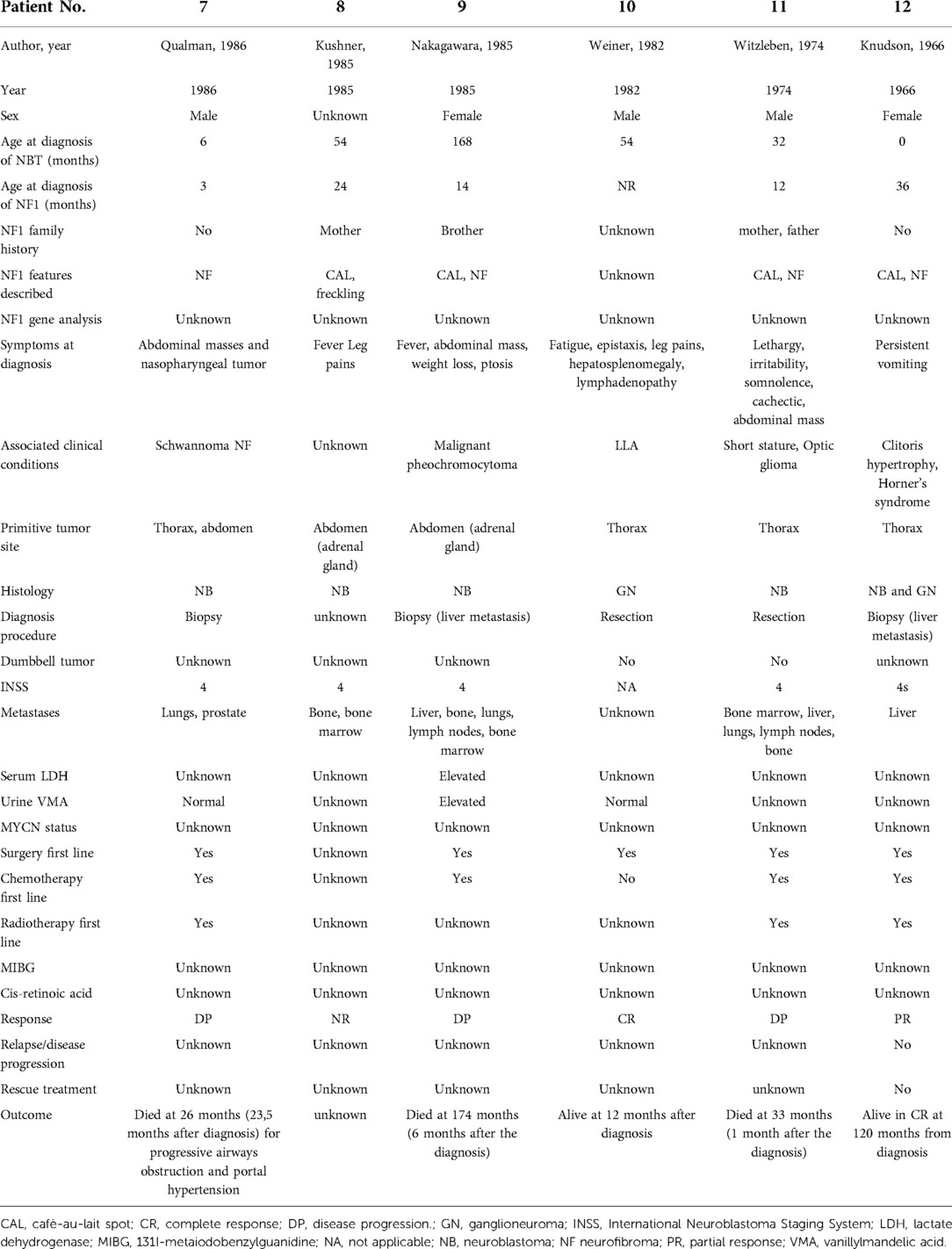
Table 6B. Description of the cases reports analyzed by the literature (part 2).
The histotype was NB in nine patients, GNB in two, and GN in one. One diagnosis of NB (patient 12) was done on liver biopsy, while the primary thoracic tumor, whose histology was analyzed some months later, was a ganglioneuroma, probably due to the maturation of a NB. Among these nine patients with NB, eight were metastatic. In one patient with GNB, metastasis appeared at relapse in the bone and bone marrow.
The results of the biologic studies were reported only for six patients. Above normal values were recorded in two patients for LDH and in three for VMA. An MYCN gene study was performed in only one patient and it was amplified.
Seven patients had an NF1 family history. At diagnosis, cafe-au-lait spots were present on nine patients, and freckling was recorded in one. Lisch nodules were not detected in any patient. Six patients developed neurofibromas. With regard to the associated clinical conditions, short stature was reported in two patients, and tibial pseudarthrosis and clitoris hypertrophy, respectively, in one. One patient presented with macroglossia, laryngomalacia, Meckel's diverticulum, and esophageal gastric heterotopia. Two cases of schwannoma and one case of optic glioma were registered. Macrocephaly was not described. With regard to the cognitive and behavioral profile, only one case of retarded psychomotor development was reported. Genetic testing was performed in two patients.
A diagnostic biopsy was performed on four patients, two for primary and two for liver metastases, while a major resection was obtained in four patients, in one, together with laminotomy; in one patient, diagnosis was made by using a bone marrow aspirate and MIBG scintigraphy and in one patient after autopsy.
Among 11 patients whose treatment was reported, three received only chemotherapy during the first-line treatment and four both chemotherapy and radiotherapy. For patients with GN and GNB, only surgery was the first-line treatment. Patient No. 4 received chemotherapy, radiotherapy, retinoic acid, and therapeutic MIBG at relapse. Patient No. 1 was the only one who received, in addition to chemotherapy during the first-line treatment, megatherapy with stem cell rescue.
The outcome was not reported for two patients. Four patients survived after first-line therapy, in CR, but only one maintained CR and was described as “alive” 12 months after diagnosis, while the other three eventually relapsed and died of the disease. Two patients obtained a PR, but only one is now alive in CR, while the other relapsed and died of NB. The remaining four patients showed rapid progress but eventually died of NB. The median age at death was 49.5 months (range, 2–174 months), with a median survival of 21.75 months after diagnosis.
To the best of our knowledge, this is the first study on the spectrum of NBT in Italian patients with NF1, which is one of the cancer-predisposing genetic disorders. To provide additional information, we reviewed the clinical and survival data of children with NBT and NF1 enrolled in the RINB over a 38-year period.
The frequencies of benign and malignant tumor development are increased in children with NF1 compared with the healthy population. In a recent study, it was found that the incidence of malignancy in children with NF1 younger than 16 years was 14.7% (39). Our report confirms that NBTs are very rare in patients with NF1 (only 0.2% of all patients registered in the RINB). In most NF1-related malignancies, including astrocytomas, MPNSTs, and neuroblastomas, a biallelic mutation of the NF1 gene function is found in the affected cells (40–43) demonstrated homozygous inactivation of the NF1 gene found in NB cells of a patient affected by familial NF1 and stage 4 NB. It is known that patients with stage 4 NB display an aggressive behavior, which is possibly connected with genetic abnormalities like MYCN gene amplification, chromosome 1p36 deletion, and chromosome 17q gain (44). Furthermore, an analysis of the NF1 gene in NB cells revealed a somatic paternal allelic deletion encompassing the introns 26 and 27b and a maternal-derived constitutional T → C transition in the donor splice site of intron 14. Martinsson et al. (43) described a homozygous deletion of the NF1 gene in a patient affected by NB. This patient showed a large biallelic deletion of NF1 in the tumor cells that also revealed chromosome 1p36 deletion but not MYCN amplification (43).
Although this particular patient had a prognostically favorable localized disease (stage 2), his tumor underwent progression, leading (like in our own case) to his death. This suggests that the mutation of the NF1 gene may be associated with a more aggressive tumor behavior. Hence, homozygous inactivation of the NF1 gene could result in a partial or total abrogation of neurofibromin activity, which would lead to increased intracellular RAS signaling; this could lead to abnormal cell proliferation. A maternal or paternal germline mutation associated with a paternal or maternal somatic deletion found in NB cells indicate that an inactivation of the NF1 gene in tumor cells occurred according to the “two-hit” model. Moreover, this mutation could be also inherited in a de novo way. The two-hit hypothesis has also been suggested by Martinsson et al. to explain the homozygous inactivation of the NF1 gene. A somatic inactivation of the still functioning NF1 allele is believed to be required for tumor formation. This “second hit” creates a reduced function of neurofibromin in the affected cells, diminishing its normal functions, including those of controlling cell growth and proliferation. With this role of the NF1 gene as a tumor suppressor, it is not surprising that somatic mutations of the NF1 gene are also commonly found in different non-NF1-associated tumors. Both patients died of tumor progression, even though one had favorable clinical characteristics, suggesting that the biallelic inactivation of NF1 might induce a more aggressive growth behavior of the disease.
Both in our study and in our review, the media and median age at diagnosis of neuroblastoma are similar, which is in agreement with the data that 95% of all neuroblastomas occur in children under 5 years of age. However, there is a difference in terms of the male-to-female ratio. In the review study, there is a prevalence of males, which is in concordance with the majority of reviewed studies in which neuroblastoma is found to be slightly more common among boys than among girls. However, the cases analyzed in the Italian study show a slightly greater frequency in females. This is in agreement with some studies that reported that females with NF1 had a higher risk of malignancy than males (26), but we did not replicate this observation, which may be attributed to our relatively small sample size limiting the statistical power to detect a difference.
Neuroblastomas have a very broad spectrum of clinical behavior, which can include spontaneous regression, maturation to a benign GN, or aggressive disease, with metastatic dissemination leading to death. This pattern of malignancies was similar to what we found in our NF1 patients and included a variety of different histotypes with a general prevalence of neuroblastoma on ganglioneuroblastoma and ganglioneuroma. Diagnostic dilemmas may arise in patients with NF1 and malignancy because neurofibroma and neuroblastoma share a common origin in the neural crest. Both show rapid growth and extensive invasion. Diagnosis is further complicated by the potential for different histologic components existing within one tumor mass. The biopsy of a portion of a heterogeneous tumor may not be representative of the spectrum of pathology.
Adrenal glands were the most common primary tumor site in our series and presented as an abdominal mass with symptoms of compression of the abdominal viscera. This finding was consistent with the findings of previous studies, followed by retroperitoneal and pelvic sympathetic ganglia (Figure 2). Thoracic localization was reported in cases studied in the literature, but not in ours. The SEER monograph reported that, regardless of age, neuroblastomas most frequently occurred in the adrenal gland (45).
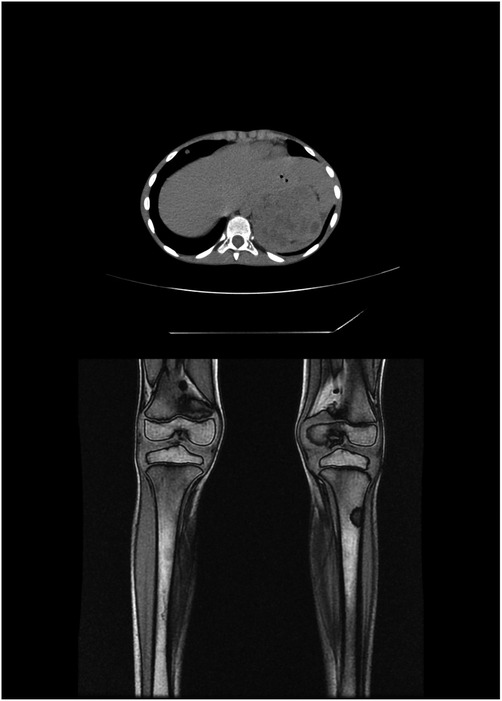
Figure 2. Patient No. 3’s MRI images. MRI, magnetic resonance imaging.
The benefits of early diagnosis are well documented and include monitoring for malignancy. A possible pathogenic relationship between NF1 and NBT is based on several observations. Neurofibromatosis is a neurocristopathy. The clinical features are pathogenetically united in their origin in neural crest dysgenesis. NBTs arise from cells of the neural crest as well. INSS stage 4s neuroblastoma is characterized by a small primary tumor in infants less than 12 months old in whom remote involvement is confined to the liver, skin, or bone marrow. The tumor can regress to lesions that are pathogenetically indistinguishable from neurofibroma. Early in its development, neurofibroma may cytologically resemble a differentiating neuroblastoma (46). In children, an extremely rapid growth of a neurofibroma is uncharacteristic and warrants investigation for malignancy. In addition to physical examination, imaging studies and urinary catecholamine measurements are indicated. Because management decisions depend on distinguishing malignant from non-malignant tumor, direct tissue diagnosis of both neuroblastoma and neurofibroma is essential.
In the present study, the staging distribution is fairly heterogeneous, with the majority of patients with an INSS stage 4 neuroblastoma. However, it is interesting to note how, compared with the review, the percentage of metastatic tumors has decreased over time. The high percentage of stage 4 in the review study may be attributed to a lack of awareness shown by general practitioners of the probability of cancer, especially in infancy when localized stages are more common, and to the lack of ultrasound use, such that tumors were not initially identified and subsequently regressed or later diagnosed at a more advanced stage in a tertiary hospital. This explanation may also account for similar findings in the postulation by Spix et al., in a study of neuroblastoma in Europe between 1978 and 1992, that the variation in stage distribution between countries may be explained by differences in the frequency of diagnosis of localized cases (5).
In our study, as described by the INSS, the rate of patients with elevated VMA and LDH at diagnosis varies according to the stage, with high-stage tumors being more likely to have pathological values. MYCN amplification, performed in five tested tumors, was absent, while the literature refers to only one case in 2003 with amplified MYCN (42).
In general, the treatment of neuroblastoma in NF1 patients is similar to that in patients without NF1. When dealing with biopsy specimens, the main concern is related to arriving at a diagnosis. The material should, therefore, be subdivided into at least two parts: one for diagnosis and the definition of cellular composition and the other for touch preparations and molecular biology investigations. In our series, only one patient (No. 4) did not undergo surgery, and diagnosis was made by using bone marrow aspiration; on the remaining patients, biopsy was performed. In our series, multiple-agent chemotherapy was adopted systematically and judged, and it was preferable to invasive surgery as an initial approach. This had numerous advantages for the patients, because no major surgical complications were reported and tumor shrinkage after chemotherapy allowed delayed surgery to be less aggressive. Unfortunately, complete tumor resection could be still difficult to achieve, but our chemotherapy and radiotherapy may be enough to control postoperative residuals, and some patients were cured without having to undergo any major surgery.
An interesting presentation is that of one of our patients (patient No. 5), who showed the most significant number of complications related to NF1, with bilateral hydronephrosis (with nephrostomies), clitoral hypertrophy, pelvic plexiform neurofibromas, neurofibromas with invasion of the spinal canal and compression of the spinal cord, and MPNST. This patient developed a ganglioneuroma, which, as already mentioned, is considered benign (47) and the prognosis is excellent, even when complete tumor removal is not possible. Nevertheless, the patient had a pelvic mass infiltrating the uterus, urethra, and sciatic nerve, with the nodules reaching the right gluteus. The histological examination carried out following biopsy revealed a diagnosis of plexiform neurofibromas with ganglioneuroma areas. Given the extent of the disease and the infiltration of the pelvic organs, surgical treatment was excluded, and it was decided to perform a strict radiological clinical follow-up. Because of the dimensional increase in the mass, the appearance of pain, and the accentuation of itching, the patient was subjected to metabolic radiotherapy with MIBG, given the intense capture of the documented mass with MIBG scintigraphy. After two cycles with no response, attempts were made with different chemotherapy cycles, first with cisplatin and etoposide, then with cyclophosphamide, and finally with methotrexate and vinorelbine. Finally, the patient showed a steady state of the mass with the administration of lenalidomide. Unfortunately, however, the patient died subsequently following the development of a mediastinal MPNST. This presentation should be regarded as an exception.
The analysis of our series of cases shows a death outcome in half of the patients. In the literature review, only two of ten patients reported survived after treatment.
The results indicate that the survival rates reported in our series are higher than those described in previously published reports. For this, many explanations should be considered: the small number of patients in both series, that is, in ours and in the literature; most of our patients have been diagnosed later than those in the literature. Moreover, this may be due to the systematic use of a multidisciplinary approach in all the patients concerned and to the ongoing trials that are exploring the efficacy of new drugs and novel immunological approaches in order to save a greater number of these patients. Unfortunately, the need for an aggressive treatment approach can also mean severe side effects, and this issue should be addressed when planning future treatments.
The current study has several limitations. It is a retrospective study. Compared with the reported national studies in other countries, the relatively small sample size in ours was an important limitation affecting statistical power. However, this is already the largest study on neuroblastoma in patients with NF1, and considering the fact that we do not have a registry on malignancies in NF1 patients, these are the best currently available data.
Undoubtedly, case reports will not have as much potential impact on the science or practice of healthcare as a randomized controlled trial or other research projects on cancerous diseases. However, case reports have a role to play only in evidence-based medicine and constitute the first line of evidence, also considering that in cancer therapies, the number of patients required to conduct such studies cannot always be recruited. It may be the only way to make others in the field aware of unusual presentations or complications, and it is a time-honored vehicle for teaching others.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical review and approval were not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the 2016/2018 Research Plan of the University of Catania, Department of Clinical and Experimental Medicine (project #B: “Evaluation of long-term harmful effects of chemotherapy and radiotherapy on various organs in childhood cancer survivors”).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer ADP declared a shared affiliation with the author(s) FP, RS, MM, GR, AP, MR and ADC to the handling editor at the time of review.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gatta G, van der Zwan JM, Casali PG, Siesling S, Dei Tos AP, Kunkler I, et al. Rare cancers are not so rare: the rare cancer burden in Europe. Eur J Cancer. (2011) 47(17):2493–511. doi: 10.1016/j.ejca.2011.08.008
2. Bernstein ML, Leclerc JM, Bunin G, Brisson L, Robison L, Shuster J, et al. A population-based study of neuroblastoma incidence, survival, and mortality in North America. J Clin Oncol. (1992) 10(2):323–9. doi: 10.1200/JCO.1992.10.2.323
3. De Bernardi B, Nicolas B, Boni L, Indolfi P, Carli M, Cordero Di Montezemolo L, et al. Disseminated neuroblastoma in children older than one year at diagnosis: comparable results with three consecutive high-dose protocols adopted by the Italian co-operative group for neuroblastoma. J Clin Oncol. (2003) 21:1592–601. doi: 10.1200/JCO.2003.05.191
4. Cotterill SJ, Pearson AD, Pritchard J, Foot AB, Roald B, Kohler JA, et al. Clinical prognostic factors in 1277 patients with neuroblastoma: results of The European Neuroblastoma Study Group “Survey” 1982–1992. Eur J Cancer. (2000) 36(7):901–8. doi: 10.1016/s0959-8049(00)00058-7
5. Spix C, Aareleid T, Stiller C, Magnani C, Kaatsch P, Michaelis J. Survival of children with neuroblastoma. Time trends and regional differences in Europe, 1978–1992. Eur J Cancer. (2001) 37(6):722–9. doi: 10.1016/s0959-8049(01)00003-x
6. Schroeder H, Wacher J, Larsson H, Rosthoej S, Rechnitzer C, Petersen BL, et al. Unchanged incidence and increased survival in children with neuroblastoma in Denmark 1981–2000: a population-based study. Br J Cancer. (2009) 100(5):853–7. doi: 10.1038/sj.bjc.6604922
7. Blatt J, Olshan AF, Lee PA, Ross JL. Neuroblastoma and related tumors in Turner's syndrome. J Pediatr. (1997) 131(5):666–70. doi: 10.1016/s0022-3476(97)70090-8
8. Merks JH, Caron HN, Hennekam RC. High incidence of malformation syndromes in a series of 1,073 children with cancer. Am J Med Genet A. (2005) 134A(2):132–43. doi: 10.1002/ajmg.a.30603
9. Tumino M, Licciardello M, Sorge G, Cutrupi MC, Di Benedetto F, Amoroso L, et al. Kabuki syndrome and cancer in two patients. Am J Med Genet A. (2010) 152A(6):1536–9. doi: 10.1002/ajmg.a.33405
10. Shahar E, Shinawi M. Neurocristopathies presenting with neurologic abnormalities associated with Hirschsprung’s disease. Pediatr Neurol. (2003) 28(5):385–91. doi: 10.1016/s0887-8994(03)00010-9
11. Nemecek ER, Sawin RW, Park J. Treatment of neuroblastoma in patients with neurocristopathy syndromes. J Pediatr Hematol Oncol. (2003) 25(2):159–62. doi: 10.1097/00043426-200302000-00015
12. Stovroff M, Dykes F, Teague WG. The complete spectrum of neurocristopathy in an infant with congenital hypoventilation, Hirschsprung’s disease, and neuroblastoma. J Pediatr Surg. (1995) 30(8):1218–21. doi: 10.1016/0022-3468(95)90027-6
13. Kushner BH, Hajdu SI, Helson L. Synchronous neuroblastoma and von Recklinghausen’s disease: a review of the literature. J Clin Oncol. (1985) 3(1):117–20. doi: 10.1200/JCO.1985.3.1.117
14. Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. (2005) 141(1):71–4. doi: 10.1001/archderm.141.1.71
15. Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Berman Y, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. (2021) 23(8):1506–13. doi: 10.1038/s41436-021-01170-5
16. Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. (2010) 152A(2):327–32. doi: 10.1002/ajmg.a.33139
17. Stephens K, Kayes L, Riccardi VM, Rising M, Sybert VP, Pagon RA. Preferential mutation of the neurofibromatosis type 1 gene in paternally derived chromosomes. Hum Genet. (1992) 88(3):279–82. doi: 10.1007/bf00197259
18. Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology. (2001) 56(11):1433–43. doi: 10.1212/wnl.56.11.1433
19. Seminog OO, Goldacre MJ. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: population-based record-linkage study. Br J Cancer. (2013) 108(1):193–8. doi: 10.1038/bjc.2012.535
20. Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. (2001) 68(5):1110–8. doi: 10.1086/320121
21. Sorensen SA, Mulvihill JJ, Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis. Survival and malignant neoplasms. N Engl J Med. (1986) 314(16):1010–5. doi: 10.1056/NEJM198604173141603
22. Duong TA, Sbidian E, Valeyrie-Allanore L, Vialette C, Ferkal S, Hadj-Rabia S, et al. Mortality associated with neurofibromatosis 1: a cohort study of 1895 patients in 1980-2006 in France. Orphanet J Rare Dis. (2011) 6:18. doi: 10.1186/1750-1172-6-18
23. Evans DG, O’Hara C, Wilding A, Ingham SL, Howard E, Dawson J, et al. Mortality in neurofibromatosis 1: in North West England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet. (2011) 19(11):1187–91. doi: 10.1038/ejhg.2011.113
24. Airewele GE, Sigurdson AJ, Wiley KJ, Frieden BE, Caldarera LW, Riccardi VM, et al. Neoplasms in neurofibromatosis 1 are related to gender but not to family history of cancer. Genet Epidemiol. (2001) 20(1):75–86. doi: 10.1002/1098-2272(200101)20:1%3C75::AID-GEPI7%3E3.0.CO;2-Z
25. Uusitalo E, Leppavirta J, Koffert A, Suominen S, Vahtera J, Vahlberg T, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. (2015) 135(3):904–6. doi: 10.1038/jid.2014.465
26. Walker L, Thompson D, Easton D, Ponder B, Ponder M, Frayling I, et al. A prospective study of neurofibromatosis type 1 cancer incidence in the UK. Br J Cancer. (2006) 95(2):233–8. doi: 10.1038/sj.bjc.6603227
27. Conte M, Parodi S, De Bernardi B, Milanaccio C, Mazzocco K, Angelini P, et al. Neuroblastoma in adolescents: the Italian experience. Cancer. (2006) 106(6):1409–17. doi: 10.1002/cncr.21751
28. AIRTUM Working Group; CCM; AIEOP Working Group. Italian Cancer figures, report 2012: cancer in children and adolescents. Epidemiol Prev. (2013) 37(1 Suppl 1):211–6.
29. Brodeur GM, Prichard Jones K, Berthold F, Carlsen NL, Castel V, Castelberry RP, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging and response to treatment. J Clin Oncol. (1993) 11(8):1466–77. doi: 10.1200/JCO.1993.11.8.1466
30. Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmeset K, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol. (2009) 27(2):298–303. doi: 10.1200/JCO.2008.16.6876
31. Cheuk DK, Chiang AK, Ha SY, Chan GC. Malignancies in Chinese patients with neurofibromatosis type 1. Hong Kong Med J. (2013) 19(1):42–9.23378354
32. Duhem-Tonnelle V, Vinchon M, Defachelles AS, Cotten A, Dhellemmes P. Mature neuroblastic tumors with spinal cord compression: report of five pediatric cases. Childs Nerv Syst. (2006) 22(5):500–5. doi: 10.1007/s00381-005-0009-z
33. Hayflick SJ, Hofman KJ, Tunnessen WW Jr., Leventhal BG, Dudgeon DL. Neurofibromatosis 1: recognition and management of associated neuroblastoma. Pediatr Dermatol. (1990) 7(4):293–5. doi: 10.1111/j.1525-1470.1990.tb01028.x
34. Knudson AG Jr., Amromin GD. Neuroblastoma and ganglioneuroma in a child with multiple neurofibromatosis. Implications for the mutational origin of neuroblastoma. Cancer. (1966) 19(7):1032–7. doi: 10.1002/1097-0142(196607)19:7%3C1032::aid-cncr2820190719%3E3.0.co;2-7
35. Nakagawara A, Ikeda K, Tsuneyoshi M, Daimaru Y, Enjoji M. Malignant pheochromocytoma with ganglioneuroblastoma elements in a patient with von Recklinghausen’s disease. Cancer. (1985) 55(12):2794–8. doi: 10.1002/1097-0142(19850615)55:12%3C2794::aid-cncr2820551213%3E3.0.co;2-l
36. Qualman SJ, Green WR, Brovall C, Leventhal BG. Neurofibromatosis and associated neuroectodermal tumors: a congenital neurocristopathy. Pediatr Pathol. (1986) 5(1):65–78. doi: 10.3109/15513818609068849
37. Weiner MA, Harris MB, Siegel RB, Klein G. Ganglioneuroma and acute lymphoblastic leukemia in association with neurofibromatosis. Am J Dis Child. (1982) 136(12):1090–1. doi: 10.1001/archpedi.1982.03970480056012
38. Witzleben CL, Landy RA. Disseminated neuroblastoma in a child with von Recklinghausen’s disease. Cancer. (1974) 34(3):786–90. doi: 10.1002/1097-0142(197409)34:3%3C786::aid-cncr2820340339%3E3.0.co;2-9
39. McKeever K, Shepherd CW, Crawford H, Morrison PJ. An epidemiological, clinical and genetic survey of neurofibromatosis type 1 in children under sixteen years of age. Ulster Med J. (2008) 77(3):160–3.18956796
40. Upadhyaya M, Kluwe L, Spurlock G, Monem B, Majounie E, Mantripragada K, et al. Germline and somatic NF1 gene mutation spectrum in NF1-associated malignant peripheral nerve sheath tumors (MPNSTs). Hum Mutat. (2008) 29(1):74–82. doi: 10.1002/humu.20601
41. Gutmann DH, Donahoe J, Brown T, James CD, Perry A. Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol Appl Neurobiol. (2000) 26(4):361–7. doi: 10.1046/j.1365-2990.2000.00258.x
42. Origone P, Defferrari R, Mazzocco K, Lo Cunsolo C, De Bernardi B, Tonini GP. Homozygous inactivation of NF1 gene in a patient with familial NF1 and disseminated neuroblastoma. Am J Med Genet A. (2003) 118A(4):309–13. doi: 10.1002/ajmg.a.10167
43. Martinsson T, Sjoberg RM, Hedborg F, Kogner P. Homozygous deletion of the neurofibromatosis-1 gene in the tumor of a patient with neuroblastoma. Cancer Genet Cytogenet. (1997) 95(2):183–9. doi: 10.1016/s0165-4608(96)00259-2
44. Bown N, Cotterill S, Lastowska M, O’Neill S, Pearson AD, Plantaz D, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med. (1999) 340(25):1954–61. doi: 10.1056/NEJM199906243402504
45. Goodman M, Gurney J, Smith M, Olshan A. Sympathetic nervous system tumors. In: Cancer Incidence and Survival among Children and Adolescents: United States SEER Program, 1975–1995. Retrieved from NIH Pub (1999).
46. Bolande RP, Towler WF. A possible relationship of neuroblastoma to Von Recklinghausen’s disease. Cancer. (1970) 26(1):162–75. doi: 10.1002/1097-0142(197007)26:1%3C162::aid-cncr2820260122%3E3.0.co;2-y
Keywords: neurofibromatosis type 1, NF1, neuroblastoma, child, cancer
Citation: Puglisi F, Soma R, Podda M, Vetrella S, Rabusin M, Tropia S, Meli M, Russo G, Sorrentino S, Erminio G, Pulvirenti A, Ruggieri M and Di Cataldo A (2022) Neuroblastic tumors and neurofibromatosis type 1: A retrospective multicenter study in Italy and systematic review of the literature. Front. Pediatr. 10:950911. doi: 10.3389/fped.2022.950911
Received: 23 May 2022; Accepted: 27 September 2022;
Published: 4 November 2022.
Edited by:
Chi-kong Li, The Chinese University of Hong Kong, ChinaReviewed by:
Simonetta Bellone, University of Eastern Piedmont, ItalyAndrea Domenico Praticò, University of Catania, Italy
Eric Legius, KU Leuven, Belgium
© 2022 Puglisi, Soma, Podda, Vetrella, Rabusin, Tropia, Meli, Russo, Sorrentino, Erminio, Pulvirenti, Ruggieri and Di Cataldo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariaclaudia Meli bWNsYXVkaWFtZWxpQGdtYWlsLmNvbQ==
Specialty Section: This article was submitted to Pediatric Oncology, a section of the journal Frontiers in Pediatrics