 Qiyu Li
Qiyu Li Jiaping Yu
Jiaping Yu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 29 June 2022
Sec. Pediatric Pulmonology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.937250
Background and Aim: Cystic fibrosis (CF) is a genetic disease that is difficult to treat and caused by dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Small molecules have been used to treat the symptom caused by CFTR mutations by restoring CFTR protein function. However, the data on children with CF are scarce. This meta-analysis aimed to evaluate the effectiveness and safety of this therapy in children diagnosed with CF.
Materials and Methods: Relevant studies were identified through searching medical databases before April 1, 2022. The primary outcomes of ppFEV1, lung clearance index2.5 (LCI2.5), sweat chloride concentration (SwCI), and Cystic Fibrosis Questionnaire-Revised (CFQ-R) score were pooled and analyzed. The secondary outcomes were nutritional status (weight, BMI, stature, and their z-score) and adverse events under therapy.
Results: A total of twelve studies were included. Compared with the placebo group, the pooled outcome of the ppFEV1, LCI2.5, SwCI, and CFQ-R score were improved by 7.91 {[95% confidence interval (CI), 3.71–12.12], –1.00 (95% CI, –1.38 to –0.63), –35.22 (95% CI, –55.51 to –14.92), and 4.45 (95% CI, 2.31–6.59), respectively}. Compared with the placebo group, the pooled result of the change in weight was improved by 1.53 (95% CI, 0.42–2.63). All the aforementioned results were also improved in single-arm studies. No clear differences in adverse events were found between CFTR modulator therapy and the placebo group.
Conclusion: CFTR modulators could improve multiaspect function in children with CF and result in comparable adverse events.
Cystic fibrosis (CF) is a life-shortening genetic disease characterized by a progressive decrease in lung function, pulmonary exacerbations, poor nutritional status, and eventually premature death (1, 2). The fundamental reason for CF is mutations in cystic fibrosis transmembrane conductance regulator (CFTR) genes coding the CFTR protein, which is located at the apical membrane of epithelial cells (3). The defective CFTR protein causes impairment in transporting chloride anions at epithelial surfaces, which induces the corresponding loss of function in multiple organs, including lungs, pancreas, and gastrointestinal tracts (1).
More than 2000 genetic variants have been found, which are classified into six classes (Class I to Class VI) according to their molecular mechanisms of dysfunction (4). The p.Phe508del CFTR mutation, which is found in 90% of the population, causes trafficking defects (Class II) (4). The Gly551Asp mutation (also known as G551D), which is the most common Class III, or gating mutation, is present in 4% of people with CF (PwCF) in the United States, diminishing the opening probability of the channel (4, 5). Nearly 50% of patients have homozygous p.Phe508del CFTR mutations (F/F), and almost 33% have heterozygous minimal-function CFTR mutations, which is with one mutation that is minimal function and one F508del. (F/MF) (6). Classes IV–VI cause limited CFTR dysfunction, which is called residual function (RF) mutation. Most patients with these RF (F/RF) or gating (F-gating) CFTR mutations are heterozygous for the p.Phe508del mutation (6).
According to the CFTR mutation, small molecules have been developed to restore CFTR protein function (4). Generally, the modulators can be classified as CFTR potentiators (e.g., ivacaftor, IVA) and correctors. The correctors are divided into first-generation CFTR correctors (e.g., lumacaftor, LUM and tezacaftor, TEZ) and next-generation correctors (elexacaftor, ELX). IVA was the first of these molecules that proved effective in phase III clinical trials (7), targeting F-gating CFTR mutations, with significant improvement in lung function and nutrition status (4). The following clinical trials, including dual combination therapy (LUM/IVA and TEZ/IVA) (8, 9) and triple combination therapy (ELX/TEZ/IVA) (10, 11), manifested that these molecules improved pulmonary function, CFTR function, and quality of life significantly.
However, the patients in most of these studies were adults or adolescents (older than 12 years). The safety and effectiveness of small molecules in children with CF are relatively less known. The potential reason may be associated with the risk of adverse events and uncertain efficacy compared with the favorable results in adults. Nevertheless, early treatment before any severe functional deficiency may be an optimal strategy for patients with CF. Hence, this meta-analysis was conducted to examine current studies on these small molecules in children with CF in terms of efficacy and safety, according to different mutation genotypes.
This meta-analysis was performed according to the Preferred Reporting Items for Systematic Reviews and Meta-analyses guidelines (12). The checklist is presented in Supplementary Table 1. The literature search was performed through PubMed, Web of Science, and Cochrane Library on April 1, 2022. The search terms and queries are presented in Supplementary Table 2.
Relevant studies were collected, and duplicates were removed (identification). The studies relevant to our analysis were selected for full-text review based on the titles and abstracts (screening). Studies were screened according to the inclusion and exclusion criteria. An additional search was performed on the references of the included studies to further identify potentially eligible studies.
The inclusion criteria were as follows: (1) population: children (≤ 11 years old) diagnosed with CF having at least one CFTR mutation; (2) intervention: patients who received monotherapy, dual combination therapy or triple combination therapy for CF; (3) comparison: patients who received placebo treatment; (4) outcomes: primary outcomes included the absolute change from baseline in predicted forced expiratory volume in 1 s (ppFEV1), absolute change from baseline in lung clearance index2.5 (LCI2.5), absolute change from baseline in sweat chloride concentration (SwCI), and absolute change from baseline in Cystic Fibrosis Questionnaire-Revised (CFQ-R) respiratory domain score. The secondary outcomes included adverse events, nutrition parameters (weight, BMI, and stature), pancreatic exocrine function (fecal elastase 1, FE1, and immunoreactive trypsinogen concentration, IRT); and (5) study design: single-arm study or randomized controlled trials (RCTs).
The exclusion criteria were as follows: (1) case reports, abstracts, or reviews, (2) studies with mixed data, including adolescents or adults, (3) no reporting of outcomes of interest, (4) studies published in languages other than English, and (5) preclinical studies or experiments in vitro.
A formalized table was independently used by Q.Y.L and J.P.Y to extract data from each study. The following information was included: (1) authors, (2) publication year, (3) study design, (4) setting (single center/multicenter); (5) enrollment period, (6) number of patients, (7) components of therapy, (8) components of active control therapy, (9) absolute change in ppFEV1 and LCI2.5, (10) absolute change in SwCI, (11) absolute change in CFQ-R score, (12) any adverse events, (13) mutation type, (14) absolute change in weight, weight-for-age z-score, BMI, BMI-for-age z-score, stature, and stature-for-age z-score, and (15) absolute change in FE1 and IRT.
Our meta-analysis included two types of data. One included continuous variables from single-arm studies, and the pooled single proportion rates were analyzed using Stata software, version 12.0 (2011; Stata Corp., TX, United States). The other included continuous variables from RCTs. RevMan 5.3 (Cochrane) was used for statistical analysis. The inverse variance method and mean difference (MD) were used. All pooled results were calculated with a random-effects model because it provided more conservative estimates. All statistical values were reported with 95% confidence intervals (CI). The subgroup analysis was conducted by drug and genotype to diminish the heterogeneity as much as possible.
The study flowchart is shown in Figure 1. Forty records were eligible for full-text review. Five case series or report studies were excluded. Sixteen studies were excluded for being reviews or meeting abstracts. Three studies were excluded for containing mixed data, including adolescents and adults, and four studies were excluded for being preclinical studies or experiments. Finally, 12 studies were included in the final analysis (13–24).
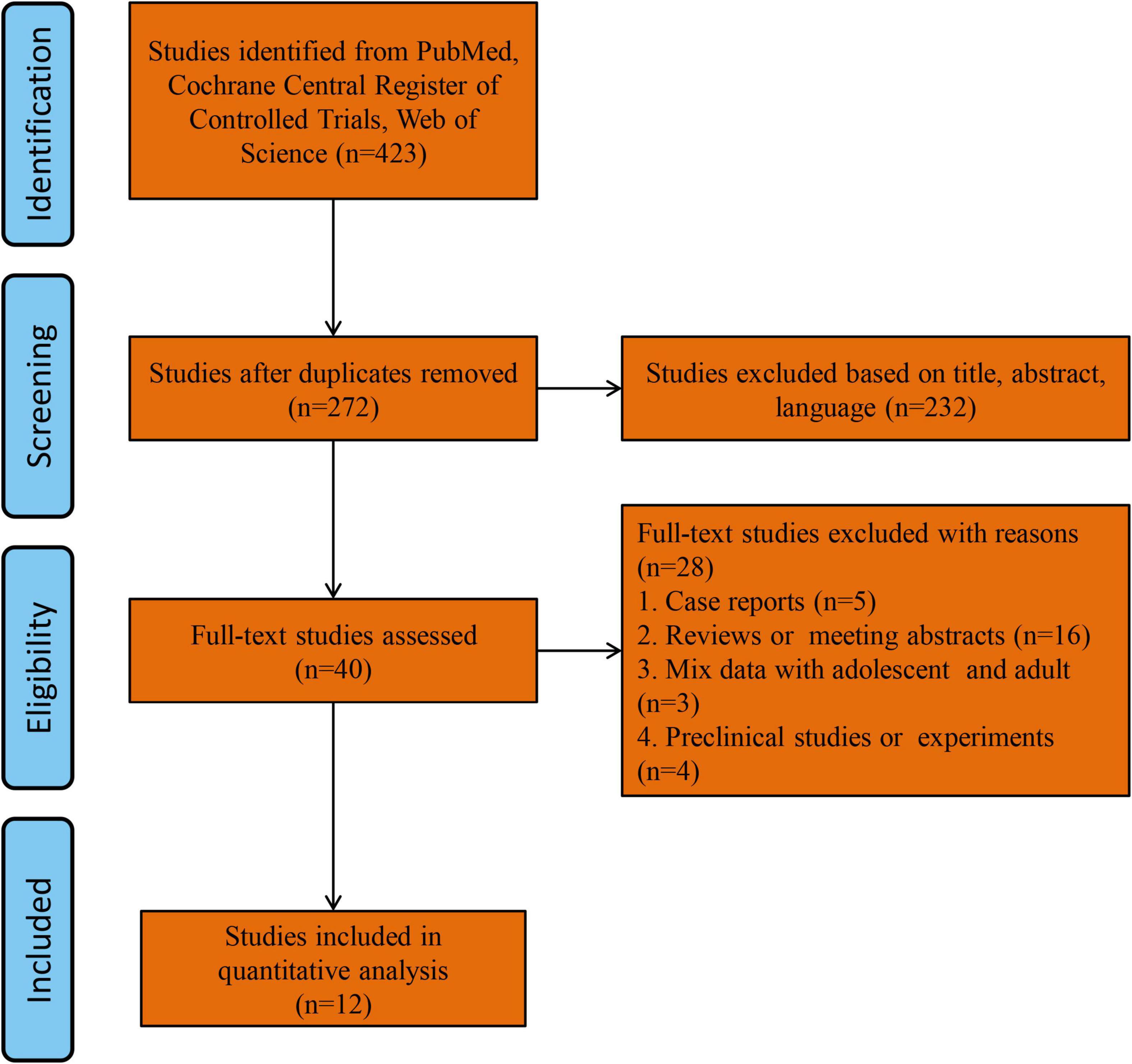
Figure 1. Flow chart of this meta-analysis.
Nine of the included studies were single-arm studies (13–21), and three were RCTs (22–24). The main characteristics of the included studies are shown in Table 1. All the included studies were multicenter phase 3 trials. Seven of 12 studies adopted a two-part study design to examine the initial safety in part A for a short period, and then conduct the rest of the trial in part B. The main information on patients and treatment are shown in Table 2. The most common length of treatment duration was 24 weeks (13–16, 18–20, 22, 23), including two long-term studies from Rosenfeld et al. (108 *weeks) (17) and Sawicki et al. (96 weeks) (21), which were the extension of three previous studies (13, 18, 24). The ages of children in the included studies were 4–12 months (19), 12–24 months (15), 2–5 years (13, 16, 17), and 6–11 years (14, 18, 20–24). The drug doses were mostly administered according to the weight of the patients and mutation types. All the CFTR gating mutations were given by IVA (monotherapy) (13, 15, 17, 19, 22). Patients with only F/F mutation were treated with LUM + IVA (dual combination therapy) (14, 16, 23), and patients with F/F or F/RF mutation were treated with TEZ + IVA (dual combination therapy) (18, 21, 24). Only one triple combination therapy (ELX + TEZ + IVA) was used in patients with F/F or F/MF mutation (20).
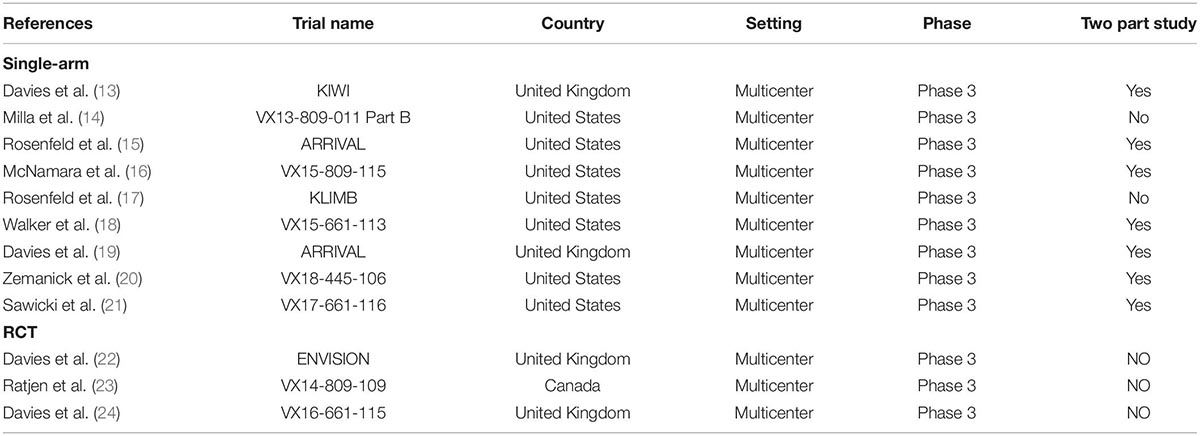
Table 1. The main characteristics of included studies.
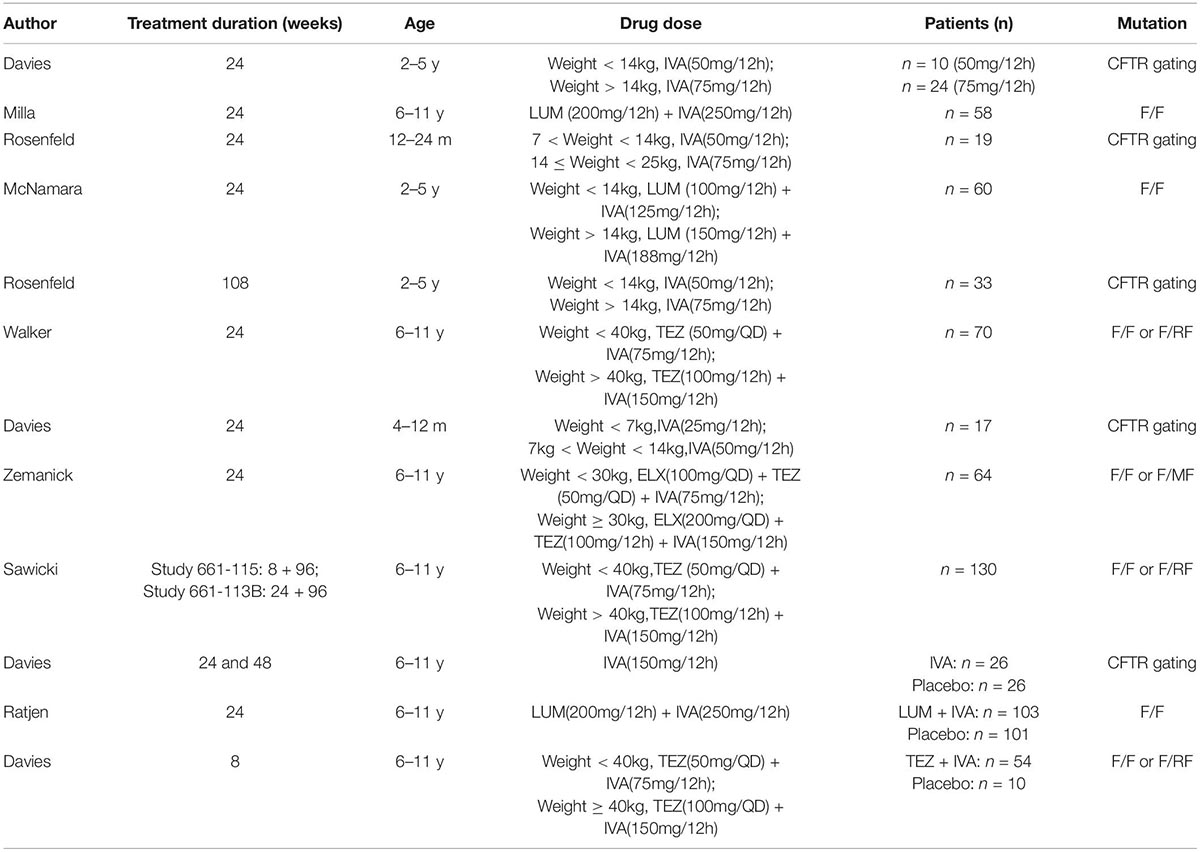
Table 2. The main information of patients and treatment.
In RCTs, the pooled result of ppFEV1 in the treatment group was much more improved than that in the placebo group (MD, 7.91; 95% CI, 3.71 to 12.12) with significant heterogeneity (I2 = 96%) (Figure 2A). Similarly, the pooled effect of LCI2.5 was also improved in the treatment group compared with that in the placebo group (MD, –1.00; 95% CI, –1.38 to –0.63) with small heterogeneity (I2 = 11%) (Figure 2B).
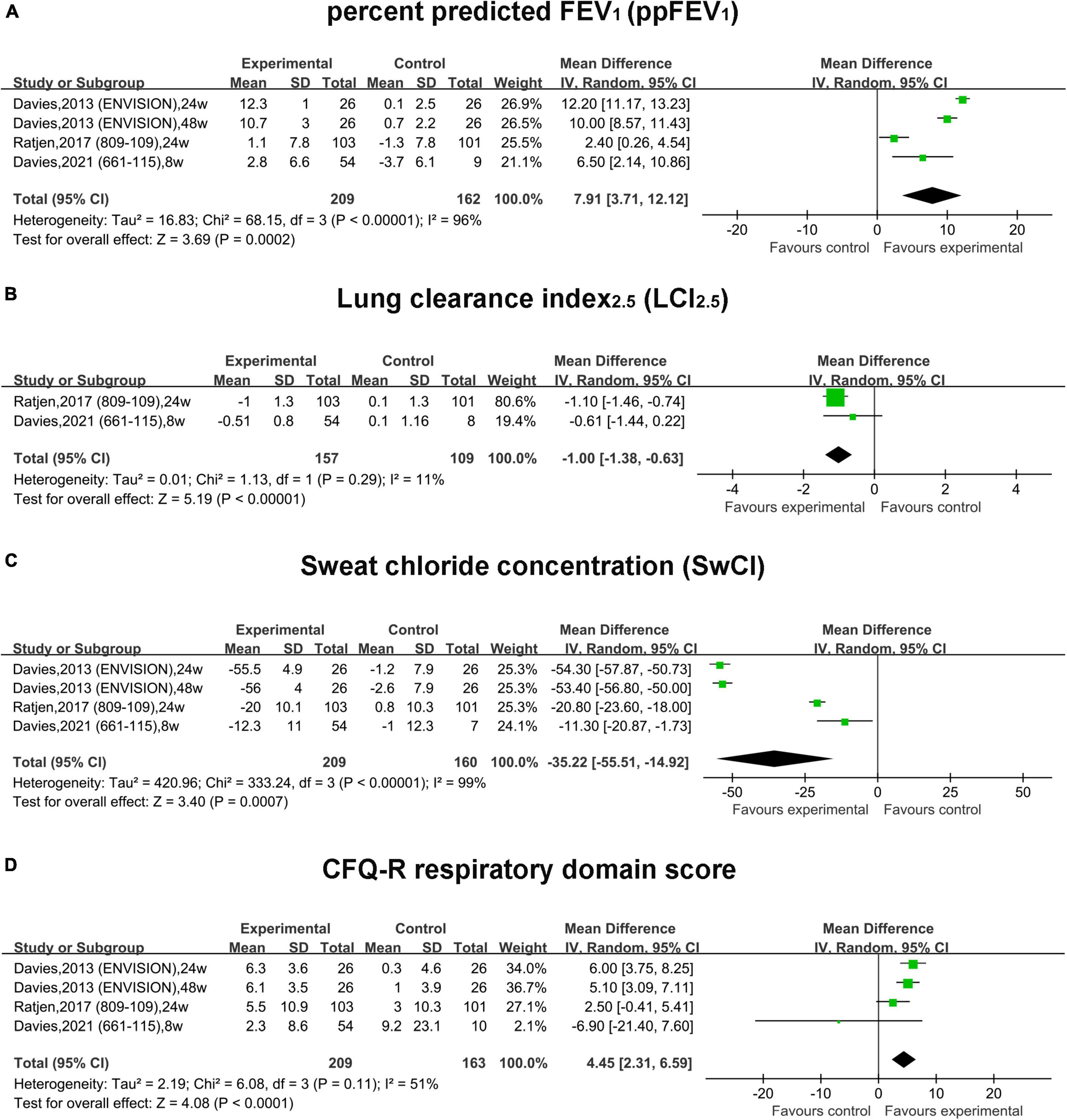
Figure 2. Forest plots of the RCT studies evaluating the efficacy of therapy vs. placebo. (A) ppFEV1. (B) LCI2.5. (C) Sweat chloride concentration. (D) CFQ-R respiratory domain score.
In single-arm studies, the pooled estimate of the absolute change in ppFEV1 of the IVA subgroup was improved by 11.73 (95% CI, 9.88–13.59) with significant heterogeneity (I2 = 89.3%) (Figure 3A). In the LUM + IVA subgroup and TEZ + IVA subgroup, the pooled estimate of ppFEV1 was elevated by 1.4 (95% CI, 0.11–2.69) and 2.96 (95% CI, 1.15–4.77), respectively (Figure 3A). The heterogeneity of these two subgroups was relatively non-significant (Figure 3A). The triple combination could increase ppFEV1 by 10.2 (95% CI, 7.88–12.52) (Figure 3A).
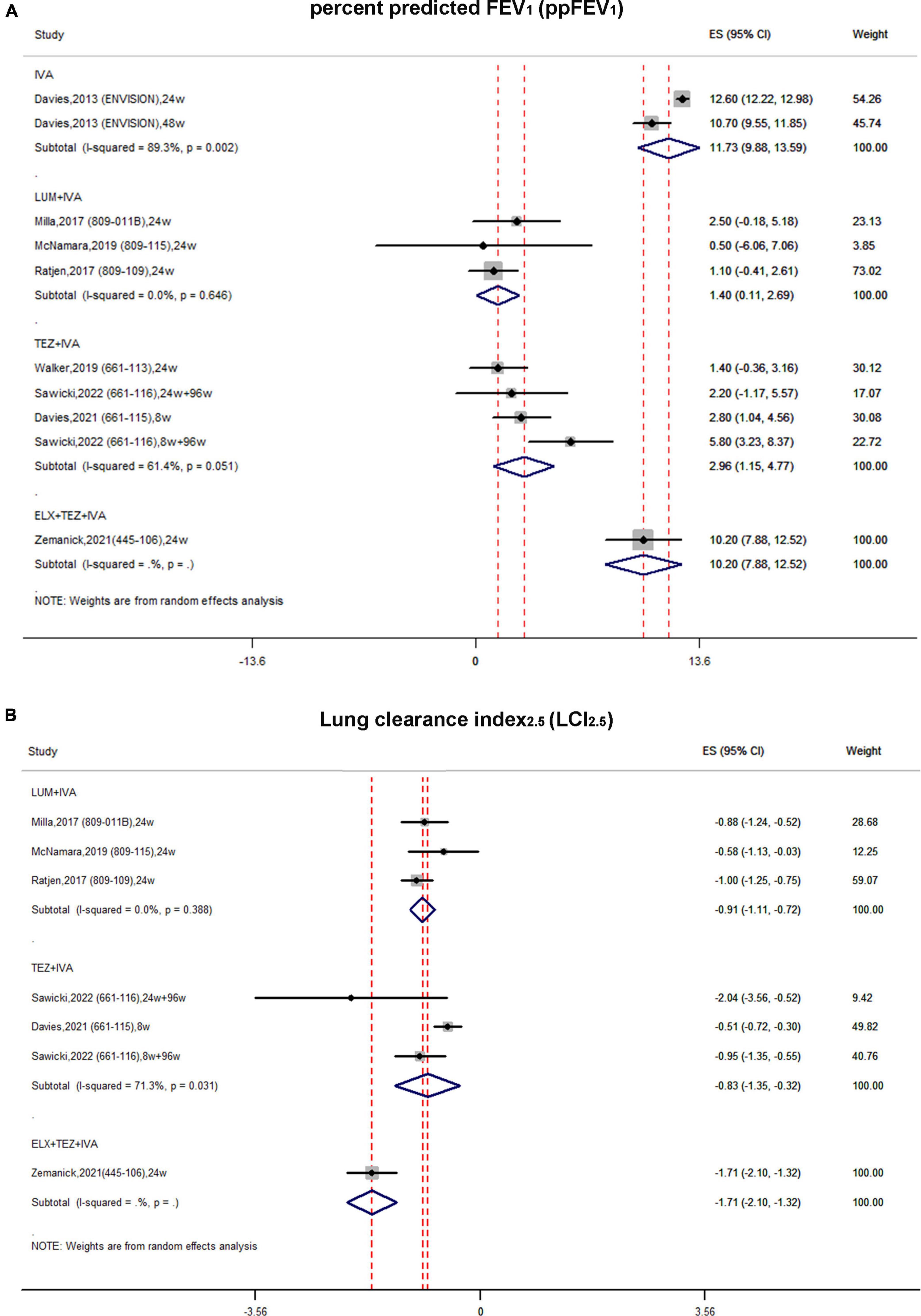
Figure 3. Forest plots of the single-arm studies evaluating the effectiveness of (A) ppFEV1 and (B) LCI2.5.
In single-arm studies, the pooled estimate of the absolute change in LCI2.5 in the LUM + IVA subgroup was improved by –0.91 (95% CI, –1.11 to –0.72) with no obvious heterogeneity (I2 = 0%) (Figure 3B). Similarly, the pooled estimate of LCI2.5 in the TEZ + IVA group was improved up to –0.83 (95% CI, –1.35 to –0.32) with significant heterogeneity (Figure 3B). The triple combination improved LCI2.5 by –1.71 (95% CI, –2.1 to –1.32) (Figure 3B).
In RCTs, the pooled absolute change of SwCI in the treatment group was improved by –35.22 mmol/L (95% CI, –55.51 to –14.92) compared with the placebo group with significant heterogeneity (I2 = 96%) (Figure 2C). The pooled estimate of the CFQ-R score was increased by 4.45 (95% CI, 2.31–6.59) in the treatment group compared with the placebo group with non-significant heterogeneity (I2 = 51%, P = 0.11) (Figure 2D).
In single-arm studies, the pooled estimate of the absolute change in SwCI in the IVA subgroup was improved by –55.94 mmol/L (95% CI, –58.86 to –53.03) with moderate heterogeneity (I2 = 57.9%) (Figure 4A). In the LUM + IVA subgroup, the pooled result of SwCI was improved by –25.36 mmol/L (95% CI, –32.58 to –18.14) with significant heterogeneity (I2 = 92.9%) (Figure 4A). In the TEZ + IVA group, the pooled estimate of SwCI was improved by –13.77 mmol/L (95% CI, –15.48 to –12.06) with non-significant heterogeneity (I2 = 0%) (Figure 4A). In the triple combination subgroup, the pooled estimate of SwCI was significantly improved by –60.9 mmol/L (95% CI, –63.6 to –58.2) (Figure 4A).
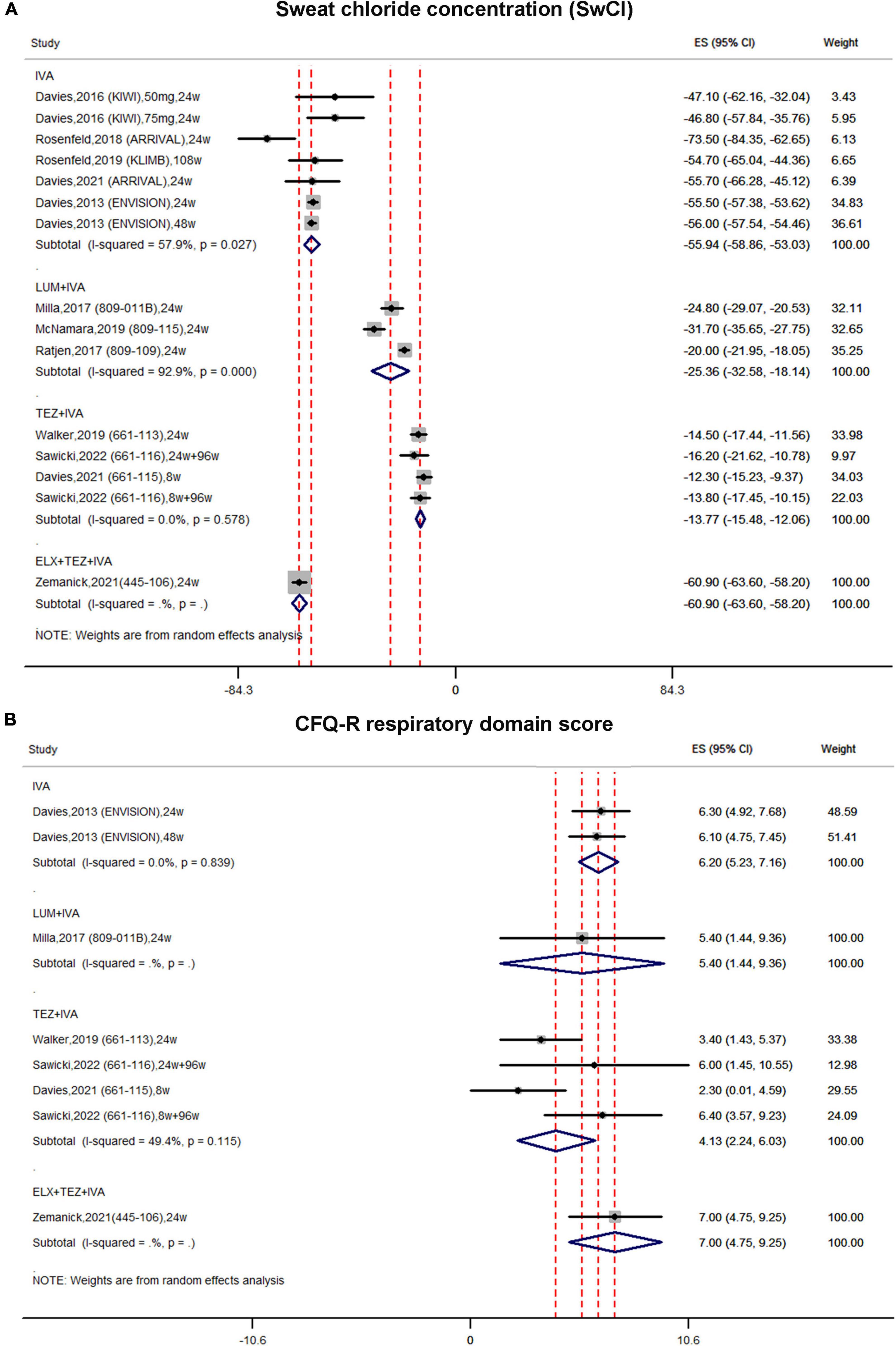
Figure 4. Forest plots of the single-arm studies evaluating the effectiveness of (A) sweat chloride concentration and (B) CFQ-R respiratory domain score.
In single-arm studies, the pooled estimate of the CFQ-R score in the IVA subgroup was increased by 6.2 (95% CI, 5.23–7.16) (Figure 4B). Similarly, the pooled estimate of the CFQ-R score was also improved in both the LUM + IVA subgroup and the TEZ + IVA subgroup by 5.4 (95% CI, 1.44–9.36) and 4.13 (95% CI, 2.24–6.03), respectively (Figure 4B). In the triple combination group, the pooled result of the CFQ-R score was also improved by 7.00 (95% CI, 4.75–9.25) (Figure 4B).
In RCTs, the pooled change in weight in the treatment group was slightly increased by 1.53 kg (95% CI, 0.42–2.63) compared with that in the placebo group, and the heterogeneity was significant (I2 = 98%) (Figure 5A). The pooled change in the BMI and BMI-for-age z-score was slightly increased compared with that in the placebo group by 0.05 (95% CI, –0.10 to 0.20) and 0.24 (95% CI, –0.07 to 0.54), respectively. However, no statistical significance was observed (Figures 5B,C).
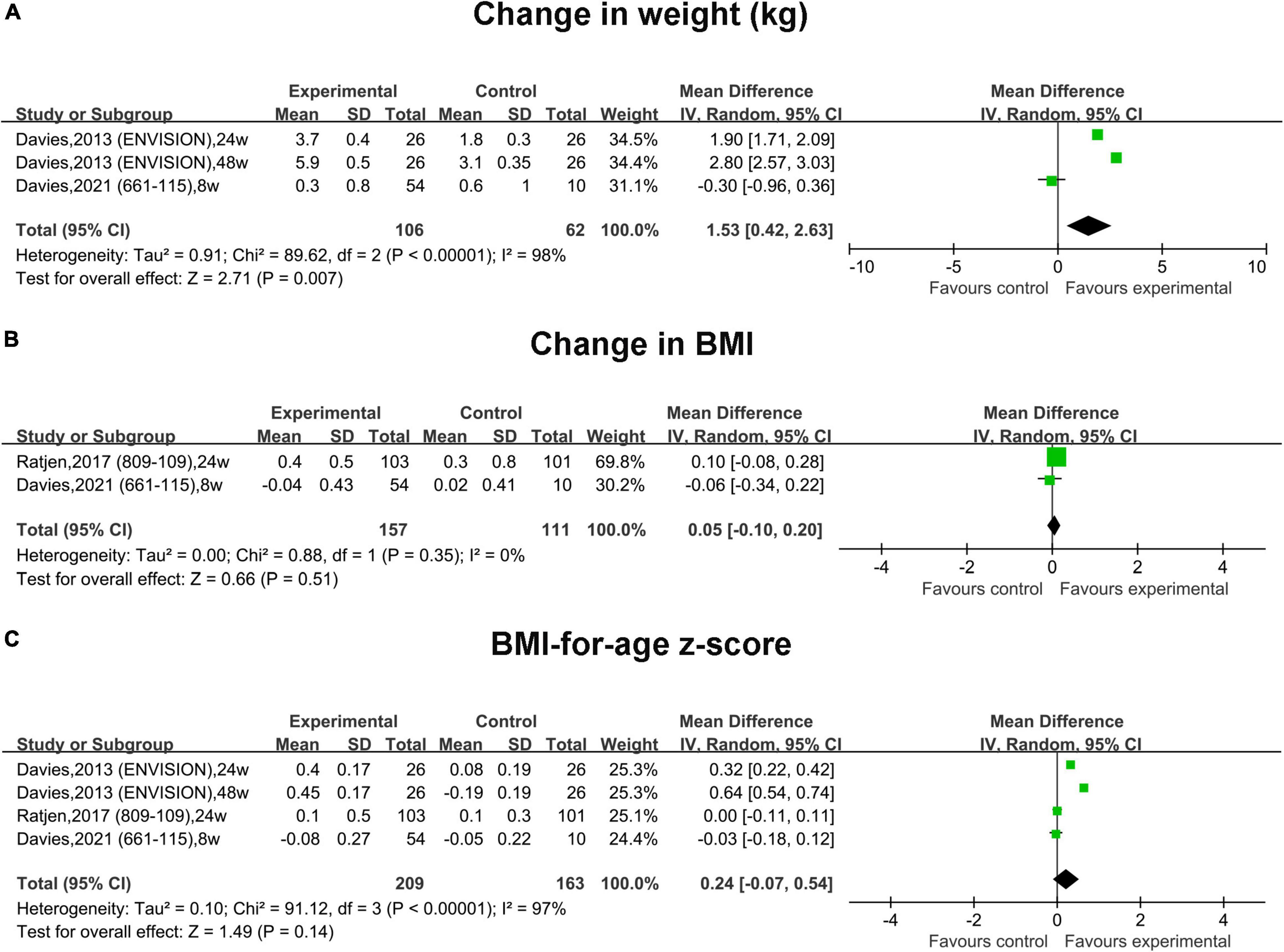
Figure 5. Forest plots of the RCT studies evaluating the efficacy of therapy vs. placebo. (A) Change in weight. (B) Change in BMI. (C) BMI-for-age z-score.
In single-arm studies, the pooled change in weight in the IVA subgroup was increased by 4.80 kg (95% CI, 2.64–6.96) (Supplementary Figure 1A). The pooled estimates in the LUM + IVA subgroup and the TEZ + IVA subgroup were also improved by 1.40 kg (95% CI, 1.17–1.63) and 4.62 kg (95% CI, 1.96–7.28), respectively (Supplementary Figure 1A). The pooled change in the weight-for-age z-score in the IVA subgroup and the LUM + IVA group was increased by 0.27 (95% CI, 0.06–0.49) and 0.26 (95% CI, 0.15–0.37), respectively (Supplementary Figure 1B). However, the pooled change in the weight-for-age z-score in the TEZ + IVA group was basically not changed (95% CI, –0.06 to 0.05) (Supplementary Figure 1B). The pooled change in the BMI in the LUM + IVA subgroup was improved by 0.44 (95% CI, 0.26–0.62) (Supplementary Figure 2A). Although the pooled change in the BMI in the TEZ + IVA subgroup was also improved by 0.83, no statistically significant difference was noted (95% CI, –0.09 to 1.75) (Supplementary Figure 2A). As for the BMI-for-age z-score, the pooled results in the IVA subgroup and the LUM + IVA group was improved by 0.37 (95% CI, 0.29–0.45) and 0.16 (95% CI, 0.08–0.25), respectively (Supplementary Figure 2B). The pooled estimates in the TEZ + IVA subgroup were slightly decreased by –0.04 (95% CI, –0.09 to 0.00); however, no statistically significant difference was found (Supplementary Figure 2B).
As for the change in stature, the pooled result in the IVA subgroup and the TEZ + IVA subgroup was improved by 3.6 cm (95% CI, 3.29–3.91) and 7.18 cm (95% CI, 3.91–10.45), respectively (Supplementary Figure 3A). The pooled outcome in the stature-for-age z-score in the IVA subgroup was increased by 0.13 (95% CI, 0.00–0.27) (Supplementary Figure 3B); however, the statistically significant difference was not significant. In the LUM + IVA subgroup and the TEZ + IVA subgroup, the pooled estimate of the stature-for-age z-score was slightly improved by 0.09 (95% CI, 0.03–0.15) and 0.02 (95% CI, –0.02 to 0.06), respectively (Supplementary Figure 3B).
The pooled estimate of FE1 in the IVA subgroup was 129.09 μg/g (95% CI, 95.06–163.11) with non-significant heterogeneity (I2 = 7.3%) (Supplementary Figure 4A). In the LUM + IVA subgroup and the TEZ + IVA subgroup, the pooled result of FE1 was 52.6 μg/g (95% CI, 23.51–81.69) and 2.50 (95% CI, –1.60 to 6.59), respectively (Supplementary Figure 4A). As for the IRT concentration, the pooled result in the IVA group and the LUM + IVA group was –130.86 ng/g (95% CI, –196.01 to –65.71) and –130.20 ng/g (95% CI, –190.91 to –69.49), respectively (Supplementary Figure 4B), which were almost the same. In the TEZ + IVA group, the pooled result was –61.45 ng/g (95% CI, –89.26 to –33.64) (Supplementary Figure 4B).
In single-arm studies, the rate of any adverse event was almost the same across the studies, ranging from 92.9 to 100% (Table 3). The rate of serious adverse events was different among the studies, ranging from 1.5 to 33.3% (Table 3). The rate of interruption of treatment due to an adverse event varied obviously (from 0 to 11.8%); however, the rate of discontinuation of treatment due to an adverse event was similar among most studies (Table 3). The most common adverse events were cough (from 35.7 to 73.7%), vomiting (from 10.0 to 39.4%), nasal congestion (from 14.3 to 26.5%), rhinorrhea (from 10.0 to 31.6%), and pyrexia (from 10.3 to 39.4%%) (Table 3). The rates of ALT or AST > 5 × upper limit of normal in most of the studies were more than 5% (Table 3).
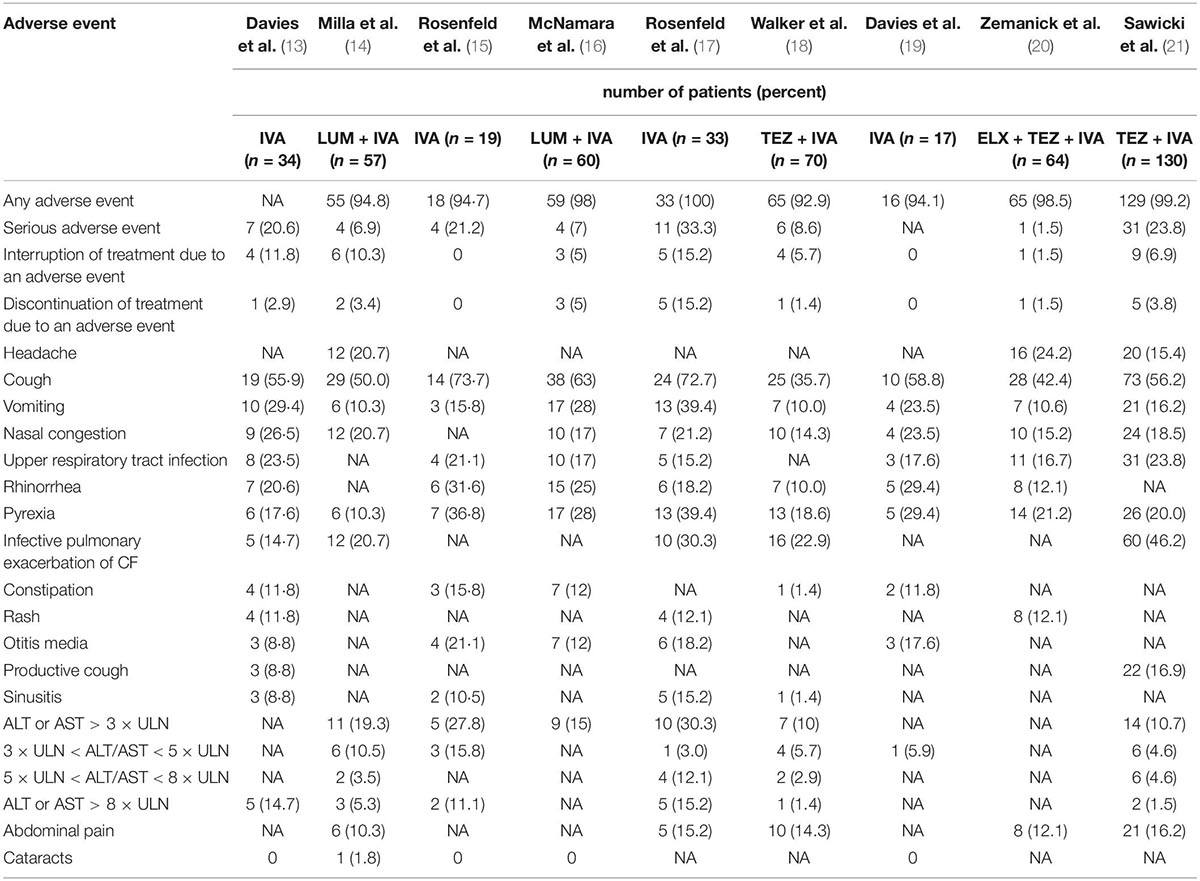
Table 3. Adverse events for single-arm studies.
In RCTs, the rates of any adverse event and serious adverse events were almost the same between the LUM + IVA group and the placebo group (Table 4). The most common adverse events were cough, pulmonary exacerbation, oropharyngeal pain, headache, pyrexia, upper abdominal pain, upper respiratory tract infection, nasal congestion, and abdominal pain, which were almost even between the treatment group and the placebo group (Table 4).
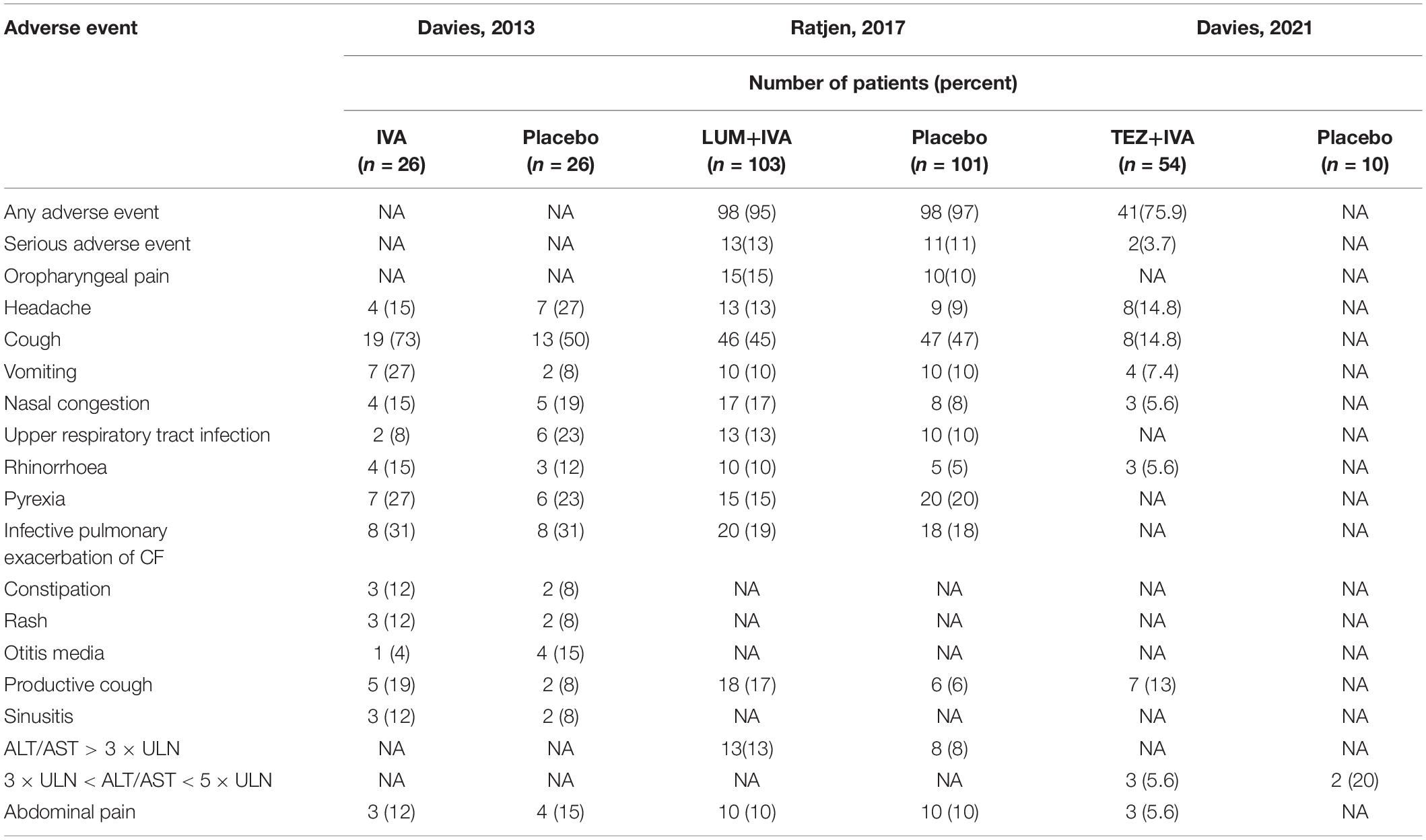
Table 4. Adverse events for RCT studies.
Cystic fibrosis (CF) begins early in life, with injury to the pancreas beginning in utero (25), and lung disease can appear at birth and progress throughout childhood (26). Hence, early treatment using a CFTR modulator in life is expected to prevent the development of end-organ damage. The physiological status of children is different from that of adults, and hence the effectiveness and safety of the CFTR modulator need to be explored.
Ivacaftor (IVA) was first conducted in double-blind RCTs in patients older than 12 years (STRIVE) (7) and children aged 6–11 years (ENVISION) (22), which demonstrated a significant improvement in ppFEV1, reduction in SwCI, and weight gain. Hence, IVA was approved for treating patients with CF having CFTR gating mutation. The first-generation CFTR correctors LUM and TEZ (which were designed on the basis of LUM) were used to repair aberrant assembly of the full-length proteins. However, p.Phe508del mutation led to not only a processing and trafficking defect but also premature degradation and defects in the gating and stability for CFTR. This was the reason why LUM or TEZ monotherapy was not effective in patients with F/F mutation (27). Hence, dual combination treatment, which includes IVA (LUM + IVA or TEZ + IVA), should be used for patients with F/F or F/RF mutation.
In the meta-analysis of the included RCTs, the ppFEV1 in the treatment group was significantly increased compared with that in the placebo group. Simultaneously, the heterogeneity was significant among the studies due to different drugs and genotype mutations. The ppFEV1 in the studies by Ratjen et al. (23) and Davies et al. (24) remained stable in the treatment group but declined in the placebo group. This finding proved that lung function progressively declined in patients with CF; hence, early preservation of lung function is important for pediatric patients (23).
The subgroup analysis could not be conducted because of the limited number of RCTs. However, in single-arm studies, the heterogeneity among the included studies moderately decreased, especially in the LUM + IVA subgroup. In the TEZ + IVA group, the children with F/F and F/RF mutations were simultaneously included in the studies, but these two mutations reacted to the drug combination differently, causing heterogeneity. Hence, the CFTR modulator manifested an improvement in ppFEV1 for children with gating mutation or at least one p.Phe508del mutation. Evidence suggested that LCI2.5 was more sensitive to detect early structural lung abnormalities, particularly in younger children, compared with ppFEV1 (28, 29). In this meta-analysis, irrespective of RCTs or single-arm studies, LCI2.5 improved. Therefore, intervention with CFTR modulators in children with CF improved lung function and offered the opportunity to slow lung function decline that occurred over time.
Sweat chloride concentration (SwCI) is an established measurement of CFTR function (21). The concentration of SwCI reduced significantly compared with that in the placebo group. The concentration of SwCI was more reduced in the LUM + IVA and TEZ + IVA groups than in the IVA monotherapy group. The reason was perhaps related to different genotype mutations. CFTR gating mutation was more reactive than F/F or F/RF mutation to IVA (27).
The pooled CFQ-R score demonstrated an increase in the treatment group relative to the placebo group. According to the adult version of the CFQ-R score, the minimally clinically important difference was ≥ 4 points (21, 23). Under this criterion, CFTR modulators could increase the life quality of children with CF. However, the child’s version did not have a widely accepted minimal clinically important difference (18). Moreover, the baseline of the CFQ-R score in the included studies was around 80 points, which suggested that the children had a relatively low burden of respiratory symptoms at baseline (21). Hence, the improvement in the CFQ-R score should be interpreted carefully.
The nutritional status is an important consideration in pediatric patients because improvements in growth are associated with better lung function (23, 24). A slow gain in the weight of patients with CF may be attributed to multiple factors, including pancreatic insufficiency leading to intestinal malabsorption, reduced appetite, diabetes, and higher metabolic rate caused by chronic lung inflammation (22). The weight increased in the treatment group compared with the placebo group. However, the BMI and BMI-for-age z-score were not statistically different between the treatment and placebo groups. Similarly, the weight, BMI, and stature were improved under treatment in single-arm studies; however, their associated z-scores were not obviously increased. The potential reason for inconsistent results might be the limited number of patients in the included studies.
FE1 concentration is a measurement for evaluating exocrine pancreatic function. If the FE1 level is < 200 μg/g, it means that exocrine pancreatic function is under insufficiency. The pooled estimates of FE1 increased obviously in the IVA and LUM + IVA group, but the FE1 level of most of the children was still under 200 μg/g after treatment. Another non-specific marker of pancreatic injury is serum IRT, which decreased in the IVA subgroup, LUM + IVA group, and TEZ + IVA group. Although the FE1 level was not over the clinical cutoff level, CFTR modulators might reduce pancreatic inflammation and injury to preserve pancreatic function (24). Additional studies in larger populations are needed to elucidate the precise relationship between FE1 or IRT.
Beyond the prominent benefits of the therapy, the safety was also favorable compared with that of placebo. The adverse events in the therapy groups were nearly the same as those in the placebo groups. Most of the adverse events were mild or moderate; the rate of discontinuation of treatment due to an adverse event in most studies was less than 5%, even in long-term treatment studies (21). The elevated levels of AST or ALT were reported in most of the included studies, which were generally asymptomatic. However, it reminded us that liver function should be assessed before the initiation of CFTR modulator treatment and monitored during treatment. Another particular rare adverse event was cataract, which was found in an animal experiment study (22). One patient developed a cataract of mild severity by Week 24 of the study (14). Therefore, regular optical inspection is needed for children during the treatment.
Only one triple combination therapy (ELZ + TEZ + IVA) has been published, which was approved in the United States to treat CF ≥ 6 years with at least one copy of p.Phe508del mutation (24). The overall manifestations in improving lung function, CFTR protein, CFQ-R score, and nutritional status were obviously significant. The next-generation corrector (ELZ) with a different structure and mechanism of action has been found to increase CFTR processing, trafficking, and function in vitro (30, 31). The combination of a next-generation corrector and tezacaftor increased the efficacy of CFTR function to a greater extent than either compound alone (32). However, no RCT result about triple therapy in children is available to date.
This was the first meta-analysis evaluating the effectiveness and safety of CFTR modulators in treating children with CF. The strengths of this meta-analysis were as follows. First, all included single-arm studies were analyzed by the same drug and genotype mutation, thus minimizing bias among the studies as much as possible. Second, the comparison was conducted among RCTs, enhancing the potency of CFTR modulator treatment. Third, similar adverse events were found between the therapy and placebo groups, which demonstrated acceptable safety.
Despite the advantages of CFTR modulator therapy, some limitations of this study should also be considered. First, the number of patients included in the studies was limited, leading to bias in the outcomes. Second, only three RCTs were included, and the treatment of drugs and genotypes were different from each other. Also, the heterogeneity was significant, which made the conclusion less confirmed. Third, the results from the included studies were mostly short term (24 weeks). Hence, additional long-term studies are needed to confirm the results.
In conclusion, CFTR modulator therapy significantly improved or preserved the lung function, CFTR protein function, CFQ-R score, and nutritional status of children with CF using a safe approach. More well-designed RCTs are needed to support the effectiveness and safety, and extend the indications for younger patients diagnosed with CF, to achieve radical treatment for CF before the development of the disease.
The original contributions presented in this study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
QL and JY: study conception and design and data acquisition. SL and XM: analysis and data interpretation. QL: drafting of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.937250/full#supplementary-material
Supplementary Figure 1 | Forest plots of the single-arm studies evaluating the effectiveness of (A) change in weight and (B) weight-for-age z-score.
Supplementary Figure 2 | Forest plots of the single-arm studies evaluating the effectiveness of (A) change in BMI and (B) BMI-for-age z-score.
Supplementary Figure 3 | Forest plots of the single-arm studies evaluating the effectiveness of (A) change in stature and (B) stature-for-age z-score.
Supplementary Figure 4 | Forest plots of the single-arm studies evaluating the effectiveness of (A) FE1 concentration and (B) IRT concentration.
2. Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet. (2019) 394:1940–8. doi: 10.1016/S0140-6736(19)32597-8
3. Berger HA, Anderson MP, Gregory RJ, Thompson S, Howard PW, Maurer RA, et al. Identification and regulation of the cystic fibrosis transmembrane conductance regulator-generated chloride channel. J Clin Investig. (1991) 88:1422–31. doi: 10.1172/JCI115450
4. Gramegna A, Contarini M. From ivacaftor to triple combination: a systematic review of efficacy and safety of CFTR modulators in people with cystic fibrosis. Int J Mol Sci. (2020) 21:5882. doi: 10.3390/ijms21165882
5. Aoyama BC, Mogayzel PJ. Ivacaftor for the treatment of cystic fibrosis in children under six years of age. Exp Rev Respir Med. (2020) 14:547–57. doi: 10.1080/17476348.2020.1741352
6. Barry PJ, Mall MA, Álvarez A, Colombo C, de Winter-de Groot KM, Fajac I, et al. Triple therapy for cystic fibrosis Phe508del-gating and -residual function genotypes. N Engl J Med. (2021) 385:815–25.
7. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. (2011) 365:1663–72.
8. Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. (2015) 373:220–31.
9. Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, et al. Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med. (2017) 377:2024–35. doi: 10.1056/NEJMoa1709847
10. Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. (2019) 381:1809–19. doi: 10.1056/NEJMoa1908639
11. Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, et al. VX-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. (2018) 379:1612–20. doi: 10.1056/NEJMoa1807120
12. Vrabel M. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Revista Espaola De Nutrición Humana Y Dietética. (2009) 18:e123.
13. Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. (2016) 4:107–15. doi: 10.1016/S2213-2600(15)00545-7
14. Milla CE, Ratjen F, Marigowda G, Liu F, Waltz D, Rosenfeld M. * VXPBIG: lumacaftor/ivacaftor in patients aged 6-11 years with cystic fibrosis and homozygous for F508del-CFTR. Am J Respir Crit Care Med. (2017) 195:912–20.
15. Rosenfeld M, Wainwright CE, Higgins M, Wang LT, McKee C, Campbell D, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir Med. (2018) 6:545–53. doi: 10.1016/S2213-2600(18)30202-9
16. McNamara JJ, McColley SA, Marigowda G, Liu F, Tian S, Owen CA, et al. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2–5 years with cystic fibrosis homozygous for F508del-CFTR: an open-label phase 3 study. Lancet Respir Med. (2019) 7:325–35. doi: 10.1016/S2213-2600(18)30460-0
17. Rosenfeld M, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2-5years (KLIMB). J Cyst Fibros. (2019) 18:838–43. doi: 10.1016/j.jcf.2019.03.009
18. Walker S, Flume P, McNamara J, Solomon M, Chilvers M, Chmiel J, et al. A phase 3 study of tezacaftor in combination with ivacaftor in children aged 6 through 11years with cystic fibrosis. J Cyst Fibros. (2019) 18:708–13. doi: 10.1016/j.jcf.2019.06.009
19. Davies JC, Wainwright CE, Sawicki GS, Higgins MN, Campbell D, Harris C, et al. Ivacaftor in infants aged 4 to <12 months with cystic fibrosis and a gating mutation. results of a two-part phase 3 clinical trial. Am J Respir Crit Care Med. (2021) 203:585–93. doi: 10.1164/rccm.202008-3177OC
20. Zemanick ET, Taylor-Cousar JL, Davies J, Gibson RL, Mall MA, McKone EF, et al. A phase 3 open-label study of elexacaftor/tezacaftor/ivacaftor in children 6 through 11 years of age with cystic fibrosis and at least one F508del allele. Am J Respir Crit Care Med. (2021) 203:1522–32. doi: 10.1164/rccm.202102-0509OC
21. Sawicki GS, Chilvers M, McNamara J, Naehrlich L, Saunders C, Sermet-Gaudelus I, et al. A phase 3, open-label, 96-week trial to study the safety, tolerability, and efficacy of tezacaftor/ivacaftor in children >/= 6 years of age homozygous for F508del or heterozygous for F508del and a residual function CFTR variant. J Cyst Fibros. (2022) 22:00033–9. doi: 10.1016/j.jcf.2022.02.003 [Epub ahead of print].
22. Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. (2013) 187:1219–25. doi: 10.1164/rccm.201301-0153OC
23. Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR : a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. (2017) 5:557–67. doi: 10.1016/S2213-2600(17)30215-1
24. Davies JC, Sermet-Gaudelus I, Naehrlich L, Harris RS, Campbell D, Ahluwalia N, et al. A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in participants 6 through 11 years of age with cystic fibrosis homozygous for F508del or heterozygous for the F508del-CFTR mutation and a residual function mutation. J Cyst Fibros. (2021) 20:68–77. doi: 10.1016/j.jcf.2020.07.023
25. Wilschanski M, Novak I. The cystic fibrosis of exocrine pancreas. Cold Spring Harbor Perspect Med. (2013) 3:a009746. doi: 10.1101/cshperspect.a009746
26. Kozlowska WJ, Bush A, Wade A, Aurora P, Carr SB, Castle RA, et al. Lung function from infancy to the preschool years after clinical diagnosis of cystic fibrosis. Am J Respir Crit Care Med. (2008) 178:42–9. doi: 10.1164/rccm.200710-1599OC
27. Meoli A, Fainardi V, Deolmi M, Chiopris G, Marinelli F, Caminiti C. State of the art on approved cystic fibrosis transmembrane conductance regulator (CFTR) modulators and triple-combination therapy. Pharmaceuticals (Basel). (2021) 14:928. doi: 10.3390/ph14090928
28. Gustafsson PM, De Jong PA, Tiddens HA, Lindblad A. Multiple-breath inert gas washout and spirometry versus structural lung disease in cystic fibrosis. Thorax. (2008) 63:129–34. doi: 10.1136/thx.2007.077784
29. Owens CM, Aurora P, Stanojevic S, Bush A, Wade A, Oliver C, et al. Lung clearance index and HRCT are complementary markers of lung abnormalities in young children with CF. Thorax. (2011) 66:481–8. doi: 10.1136/thx.2010.150375
30. Veit G, Roldan A, Hancock MA, Da Fonte DF, Xu H, Hussein M, et al. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight. (2020) 5:e139983. doi: 10.1172/jci.insight.139983
31. Becq F, Mirval S, Carrez T, Lévêque M, Billet A. The rescue of F508del-CFTR by elexacaftor/tezacaftor/ivacaftor (Trikafta) in human airway epithelial cells is underestimated due to the presence of ivacaftor. Eur Respir J. (2022) 59:2100671. doi: 10.1183/13993003.00671-2021
Keywords: CFTR (cystic fibrosis transmembrane conductance regulator), cystic fibrosis – CF, corrector, potentiator, cystic fibrosis transmembrane conductance regulator
Citation: Li Q, Liu S, Ma X and Yu J (2022) Effectiveness and Safety of Cystic Fibrosis Transmembrane Conductance Regulator Modulators in Children With Cystic Fibrosis: A Meta-Analysis. Front. Pediatr. 10:937250. doi: 10.3389/fped.2022.937250
Received: 06 May 2022; Accepted: 27 May 2022;
Published: 29 June 2022.
Edited by:
Ron Rubenstein, Washington University in St. Louis, United StatesReviewed by:
Jun Wang, Zhejiang University, ChinaCopyright © 2022 Li, Liu, Ma and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiaping Yu, ZXJrZXl1amlhcGluZ0AxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.