 Min Young Park
Min Young Park Carole Le Henaff1
Carole Le Henaff1 Despina Sitara
Despina Sitara
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 23 June 2022
Sec. Pediatric Nephrology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.924915
Renal anemia is a common complication in chronic kidney disease (CKD), associated with decreased production of erythropoietin (EPO) due to loss of kidney function, and subsequent decreased red blood cell (RBC) production. However, many other factors play a critical role in the development of renal anemia, such as iron deficiency, inflammation, and elevated fibroblast growth factor 23 (FGF23) levels. We previously reported that inhibition of FGF23 signaling rescues anemia in mice with CKD. In the present study we sought to investigate whether α-Klotho deficiency present in CKD also contributes to the development of renal anemia. To address this, we administered α-Klotho to mice with CKD induced by an adenine-rich diet. Mice were sacrificed 24 h after α-Klotho injection, and blood and organs were collected immediately post-mortem. Our data show that α-Klotho administration had no beneficial effect in mice with CKD-associated anemia as it did not increase RBC numbers and hemoglobin levels, and it did not stimulate EPO secretion. Moreover, α-Klotho did not improve iron deficiency and inflammation in CKD as it had no effect on iron levels or inflammatory markers. Interestingly, Klotho supplementation significantly reduced the number of erythroid progenitors in the bone marrow and downregulated renal Epo and Hif2α mRNA in mice fed control diet resulting in reduced circulating EPO levels in these mice. In addition, Klotho significantly decreased intestinal absorption of iron in control mice leading to reduced serum iron and transferrin saturation levels. Our findings demonstrate that α-Klotho does not have a direct role in renal anemia and that FGF23 suppresses erythropoiesis in CKD via a Klotho-independent mechanism. However, in physiological conditions α-Klotho appears to have an inhibitory effect on erythropoiesis and iron regulation.
Anemia is a common complication of chronic kidney disease (CKD) associated with poor outcomes such as reduced quality of life and increased cardiovascular disease, hospitalizations, cognitive impairment, and mortality (1). The kidney is the main source of the circulating protein erythropoietin (EPO), which is responsible for stimulating erythropoiesis. As kidney disease progresses, the kidneys are unable to produce sufficient amount of EPO and the prevalence of anemia increases, affecting nearly all patients with stage 5 CKD / end stage renal disease (ESRD) (1). However, the cause of anemia in CKD is multifactorial and includes not only insufficient EPO production but also pro-inflammatory cytokine activation and iron deficiency, among others.
Renal anemia is closely linked to iron-deficiency, which is due to both severely reduced or absent iron stores (absolute iron deficiency) and insufficient iron availability despite adequate iron stores (functional iron deficiency) (2–6). Functional iron deficiency is due to underlying inflammation which stimulates hepatic secretion of hepcidin that inhibits iron absorption in the gut and iron release from body stores (3, 5, 7). Renal anemia is typically normocytic, normochromic, and hypoproliferative due to reduced EPO activity in the bone marrow. However, the presence of iron deficiency in CKD results in microcytic and hypochromic erythrocytes (8–10).
Renal anemia is typically managed by the use of agents that stimulate erythropoiesis (ESAs) to activate Epo receptors (Epo-Rs) on erythroid progenitor cells directly or indirectly. Despite their benefits in anemia management, ESAs are linked to several adverse effects such as worsened hypertension, thromboembolic complications, tumor growth, as well as resistance to Epo therapy itself (1, 3, 11–13). In addition, ESAs do not improve adverse outcomes associated with anemia, such as mortality, cardiovascular events, left ventricular hypertrophy, or progression of kidney disease (1). Thus, there is a need to discover mechanisms that would stimulate erythropoiesis with fewer side effects.
Fibroblast growth factor 23 (FGF23) is a phosphaturic hormone produced by bone that regulates phosphate homeostasis by inducing phosphate excretion through its binding to the Klotho-FGFR complex. CKD is characterized by disordered mineral ion homeostasis, associated with marked elevation of FGF23 and Klotho deficiency (14, 15). In our previous studies we showed that FGF23 is a negative regulator of erythropoiesis and deletion of Fgf23 in mice stimulates erythropoiesis (16). Similarly, disruption of FGF23 signaling by inactivation of Klotho results in increased red blood cell production (17). Importantly, we reported that inhibition of FGF23 signaling by the use of an antagonist rescues anemia, and ameliorates iron deficiency and inflammation in mice with CKD (18).
Growing evidence suggests that α-Klotho is a significant biomarker for CKD (14, 19) and a pathogenic factor in CKD progression (20, 21). Moreover, overexpression of Klotho or addition of exogenous recombinant Klotho increased renal EpoR protein and mRNA (22, 23). A role for Klotho in iron metabolism has also been reported by showing that serum iron overload decreases renal Klotho expression, whereas iron chelation suppresses angiotensin II-induced downregulation of Klotho (24). In the present study, we sought to investigate whether Klotho deficiency contributes to the development of renal anemia and iron deficiency and whether administration of Klotho can improve these conditions.
Six-week-old C57/B6J male mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and housed at New York University (NYU) College of Dentistry Animal Facility, where they were kept on a light/dark (12 h/12 h) cycle at 23°C, and received food (standard chow) and water ad libitum. Upon arrival, mice were acclimatized for 1 week, after which they were randomly assigned to two groups fed either control diet (TD 200790, Harlan Teklad, Madison, WI) or 0.2% adenine diet (TD 200791) for 8 weeks, as described in (25). After 8 weeks, control and adenine-fed mice were randomly assigned to 4 groups to receive intraperitoneally either saline (vehicle) or 10 μg/kg of recombinant mouse α-Klotho protein (R&D systems, Minneapolis, MN, USA). Groups: Control diet + Klotho, Control diet + Vehicle, Adenine diet + Klotho, and Adenine diet + Vehicle. Mice were sacrificed 24 hrs after Klotho or saline injection. The institutional animal care and use committee (IACUC) at New York University approved the animal studies.
Mice were immediately necropsied after euthanasia and blood was collected by cardiac puncture. Whole blood was collected in Microtainer® Blood Collection Tubes with K2EDTA (Becton, Dickinson and Company, Franklin Lakes, NJ) and complete blood count was performed using the VetScan HM5 Hematology Analyzer (ABAXIS, Union City, CA). For serum, blood was obtained in separate Microtainer® serum separator tube (Becton, Dickinson and Company, Franklin Lakes, NJ) and centrifuged at 1,800 x g for 15 min. Liver, spleen, kidneys, intestine, bones (diaphysis), and bone marrow cells obtained from femora and tibiae were collected, snap-frozen in liquid nitrogen, and stored at −80°C until further use.
Serum and urinary phosphorus and creatinine levels were determined by colorimetric measurements using the Stanbio Phosphorus Liqui-UV® and Stanbio Direct Creatinine LiquiColor® Test reagents, respectively (Stanbio Laboratory, Boerne, TX). Serum FGF23 levels were measured using mouse FGF23 Intact and C-terminal ELISA assays (Quidel Corporation/Immutopics International, San Clemente, CA, USA). Serum iron and transferrin saturation were measured using the Iron-TIBC kit from Pointe Scientific (Canton, MI, USA). Tissue iron content was determined by colorimetric iron assay kit (Abcam, Waltham, MA, USA). Serum erythropoietin (EPO) and parathyroid hormone (PTH) levels were measured using the Rat/Mouse EPO Quantikine (R&D Systems, Minneapolis, MN, USA), and MicroVue Mouse PTH 1-84 (Quidel Corporation/Immutopics International, San Clemente, CA, USA) ELISA assays, respectively.
A colony-forming unit assay was performed for bone marrow (BM) cells collected from both tibiae and femoral bones. The cells were plated in methyl cellulose medium (MethoCult M3231, Stemcell Technologies Inc., Vancouver, BC, Canada) at 2 × 105 cells/mL and incubated at 37°C and 5% CO2. After 12 days of incubation, the colonies were scored based on morphology as burst-forming unit-erythoid (BFU-E) under a Nikon TMS inverted phase contrast microscope (Nikon Instruments, Melville, NY).
Total RNA was extracted from kidneys, liver, duodenum, and bone marrow using Trizol (Ambion; Life Technologies, Carlsbad, CA, USA) according to the manufacturer's protocol (Molecular Research Center, Cincinnati, OH, USA). cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit, as described by the manufacturer (Applied Biosystems; Thermo Fisher Scientific, Waltham, MA, USA), and amplified by quantitative PCR (qPCR) using the PerfeCTa SYBR Green SuperMix (Quanta Biosciences, Gaithersburg,MD, USA). All primers used in this study are listed in Supplementary Table 1. mRNA levels were normalized to the housekeeping gene (Hprt) in the same RT sample. The relative transcript expression of a gene is given as ΔCt = Cttarget-Ctreference. The fold change in gene expression, as compared to control mice, was determined as 2ΔΔCt values (ΔΔCt = ΔCttreated-ΔCtcontrol).
All data were analyzed by two-way analysis of variance (ANOVA) to determine the effect of diet (control vs. adenine diet) and/or treatment (saline vs. recombinant Klotho protein) using GraphPad Prism version 8.0 for Windows (GraphPad Software, San Diego, CA, USA). Outliers were identified and removed using the ROUT method prior to statistical analysis. Bonferroni correction was used to adjust for multiple pairwise comparison. All data were expressed at mean ± SD. P < 0.05 were considered significant.
In the present study, we used a non-surgical mouse model of renal failure induced by a diet rich in adenine. After developing renal failure, adenine-fed and control mice were randomized to receive either Klotho protein or vehicle. A schematic of the experimental design is shown in Figure 1A. Consistent with other studies (26–29), mice fed adenine had significantly reduced body weight and kidney weight in comparison to mice fed control diet (Table 1). However, the total kidney size, as assessed by the kidney weight to body weight ratio, was not different between adenine-fed and control mice (Table 1). Kidney function was appropriately impaired following consumption of adenine, as determined by elevated serum creatinine concentration (Table 2) and decreased phosphate urinary excretion (Supplementary Figure 1). Similar to CKD patients (30, 31), the adenine group also developed significant hyperphosphatemia, secondary hyperparathyroidism, and severely elevated FGF23 (Table 2). Moreover, we found that renal expression of Klotho and the sodium phosphate transporters NaPi2a and NaPi2c was significantly decreased in mice fed adenine diet compared to control mice (Figures 1H–J), in agreement with previous reports (32–37). Survival was identical between the two groups of mice (adenine-fed and control-fed) during our study. Together, these findings clearly demonstrate the development of kidney disease in our mice following consumption of adenine diet.
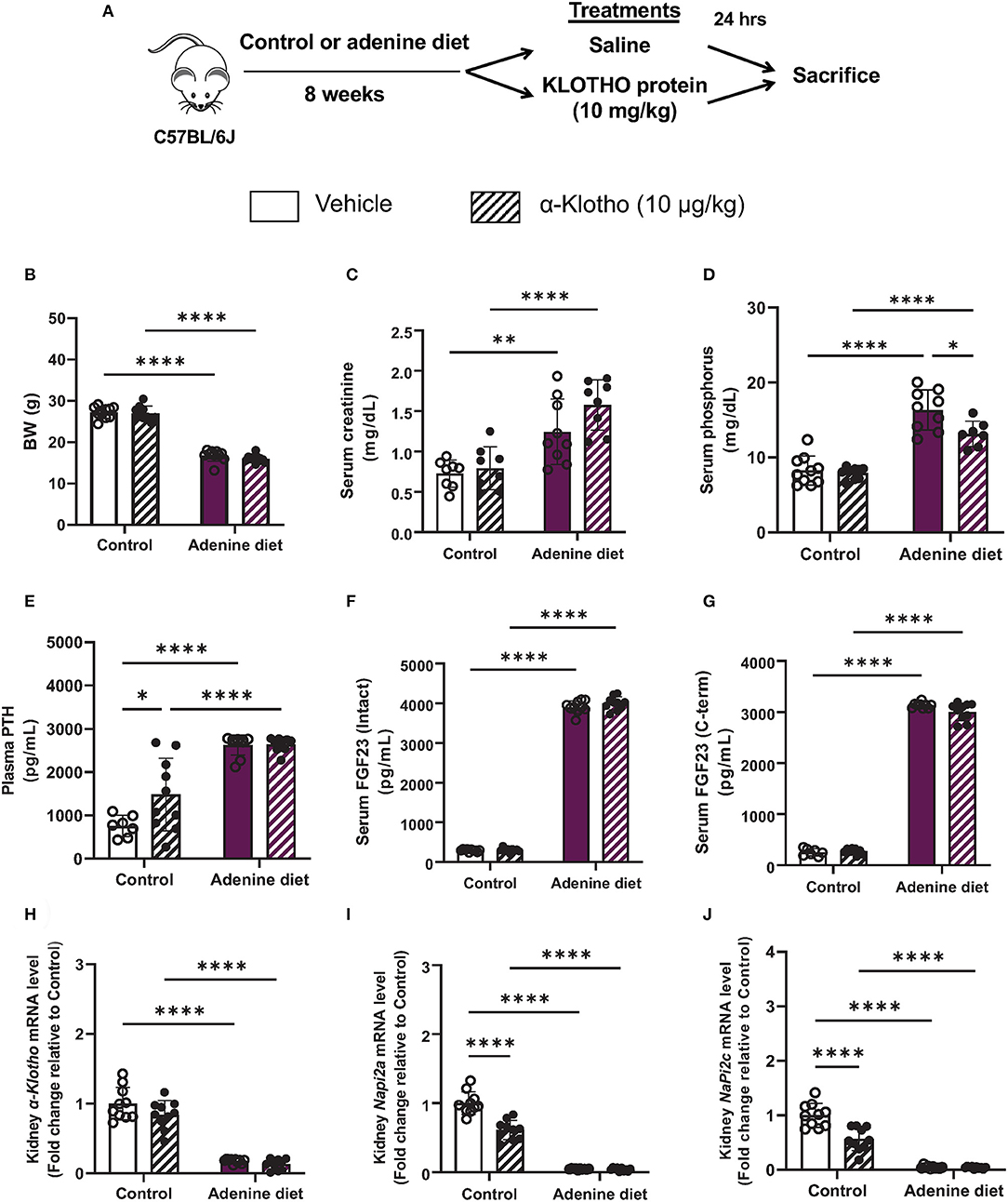
Figure 1. Validation of chronic kidney disease by adenine diet in mice. (A) A schematic of the experimental design. C57BL/6J male mice were fed control or 0.2% adenine diet for 8 weeks. After 8 weeks, mice in each group were further divided into 2 groups and were injected with either saline or mouse recombinant Klotho protein (10 μg/kg) 24 h before sacrifice. (B) Body weight at the end of experiment. (C,D) Serum concentration of (C) creatinine and (D) phosphorus. (E) Plasma PTH levels measured by ELISA. (F) Intact and (G) C-terminal FGF23 levels in serum measured by ELISA. (H–J) Quantitative real-time RT-PCR for renal (H) α-Klotho, (I) Napi2a, and (J) Napi2c expression. Data are expressed as fold change (2−ΔΔCt) relative to housekeeping gene Hprt. Samples were measured in duplicates. Data are represented as mean ± SD. (n = 7–10 per group). *P < 0.05, **P < 0.01, ****P < 0.0001 compared to Control.
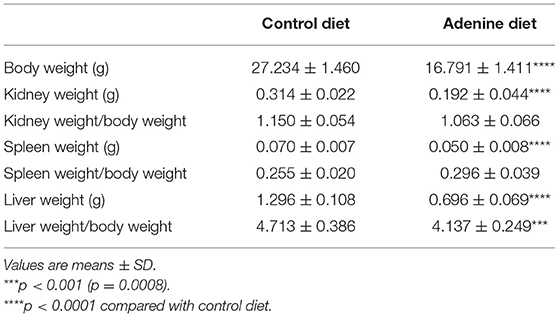
Table 1. Body and organ weights.
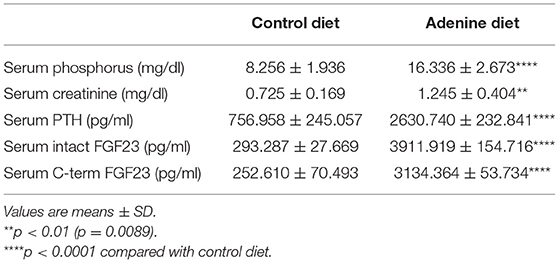
Table 2. Biochemical parameters.
Our data also show that 24 h after Klotho administration, there was no effect of Klotho on body weight (Figure 1B) and organ weight or size (Supplementary Figure 2). Biochemical parameters such as serum creatinine, PTH, and FGF23 were also not affected by Klotho treatment and remained elevated in adenine-fed mice (Figures 1C,E–G). However, the prevailing hyperphosphatemia in these mice was significantly reduced after Klotho treatment (Figure 1D). In control mice, Klotho treatment increased serum PTH (Figure 1E) and it was capable of reducing Napi2a and Napi2c expression (Figures 1I,J), consistent with previous reports showing a direct interaction between circulating Klotho and Npt2a to suppress renal phosphate reabsorption (38, 39).
Anemia is a common complication of CKD that develops primarily due to insufficient secretion of EPO by the diseased kidneys. Moreover, low levels of hemoglobin and hematocrit have been reported in both CKD patients and mice with 5/6 nephrectomy (40–46). Similarly, we found that mice fed adenine diet exhibited anemia, as it was determined by low hemoglobin, hematocrit, and serum EPO levels (Table 3), compared to mice fed control diet. Furthermore, we assessed the presence of erythroid progenitors in the bone marrow of control and adenine-fed mice by colony-forming unit (CFU) assay. Erythroid progenitors, termed burst-forming unit-erythroid (BFU-E), are detected by the formation of discrete erythroid colonies in vitro. As shown in Figure 2D, bone marrow (BM) cells from adenine-fed mice generated significantly less BFU-E colonies in vitro compared to BM cells from the control group, suggesting that BM cells from adenine-fed mice contain less functional erythroid progenitors resulting in anemia.
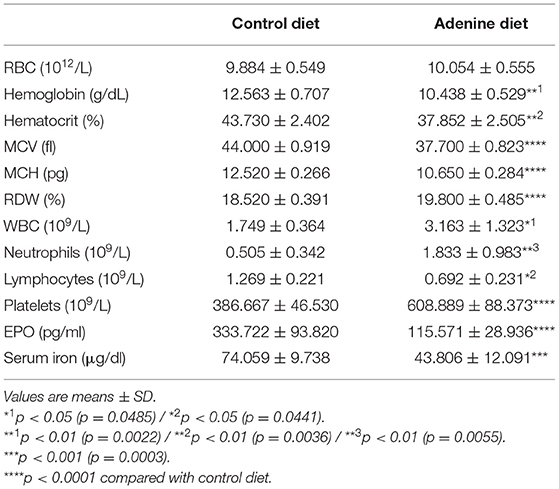
Table 3. Hematological parameters.
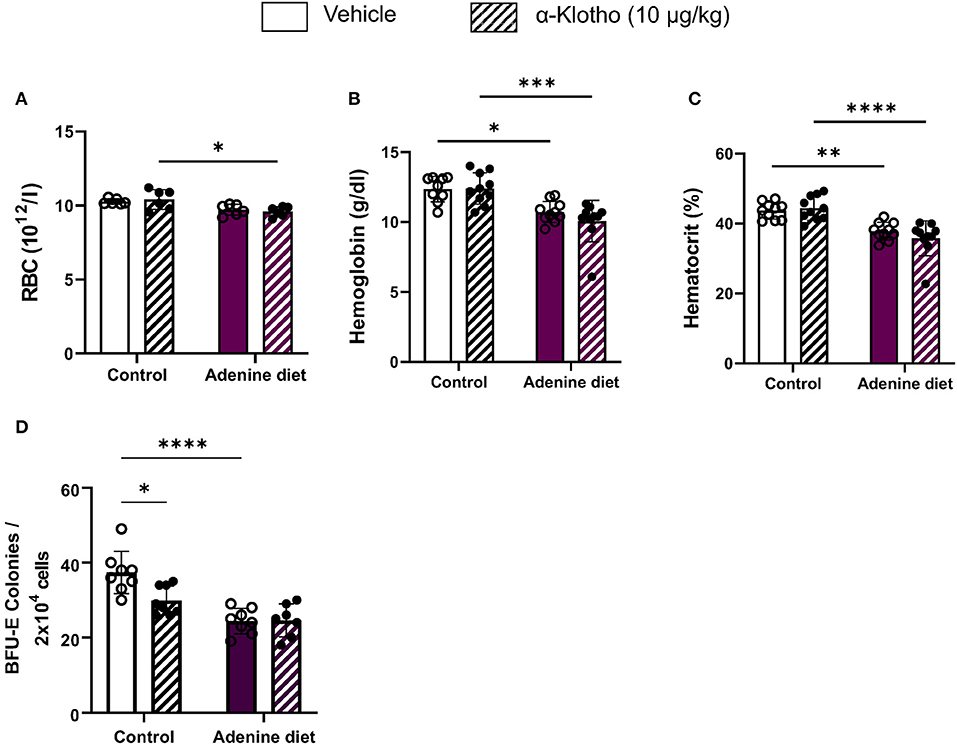
Figure 2. Effect of adenine diet and Klotho administration on circulating red blood cell parameters and bone marrow erythroid progenitor cells. (A–C) Whole blood was collected and analyzed by complete blood count for circulating red blood cell parameters. (A) Red Blood Cells (RBCs), (B) Hemoglobin (Hgb), (C) Hematocrit (Hct). (D) Femoral and tibial bone marrow cells were isolated and cultured for 12 days on methyl cellulose medium and counted for erythroid progenitor cells, burst forming unit erythroid (BFU-E). Data are represented as mean ± SD (n = 7–10 per group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to Control.
Work from our lab has shown that inhibition of FGF23 signaling rescues renal anemia in the 5/6 nephrectomy mouse model (18). Here, we investigated whether amelioration of Klotho deficiency by Klotho protein administration can improve anemia in mice with adenine-induced CKD. Our data show that a single injection of Klotho protein did not have any effect on red blood cells (RBCs) (Figure 2A), hemoglobin (Figure 2B), or hematocrit levels (Figure 2C) in either adenine-fed or control mice. Furthermore, treatment with Klotho did not affect the number of BFU-E colonies formed in vitro in adenine-fed mice (Figure 2D). However, in control mice Klotho reduced the number of erythroid progenitors (Figure 2D).
Consistent with the decrease in circulating EPO levels, renal Epo mRNA expression was significantly suppressed in adenine-fed mice (Figure 3B) due to significant down-regulation of the hypoxia-inducible transcription factor (HIF) Hif2α (Figure 3C). We further evaluated the effect of Klotho administration on erythropoietin production in mice with adenine-induced CKD compared to mice fed control diet. Klotho treatment did not affect Epo secretion in adenine-fed mice, as it was evident by the unchanged levels of serum EPO (Figure 3A), as well as Epo and Hif2α expression in the kidneys (Figures 3B,C). Conversely, circulating EPO levels and renal Epo mRNA expression were significantly reduced by Klotho in control mice (Figures 3A,B). This reduction in Epo was due to decreased Hif2α mRNA expression in the kidneys (Figure 3C). Taken together, our data suggest that Klotho administration does not rescue renal anemia but in physiological conditions Klotho appears to suppress early stages of erythropoiesis.
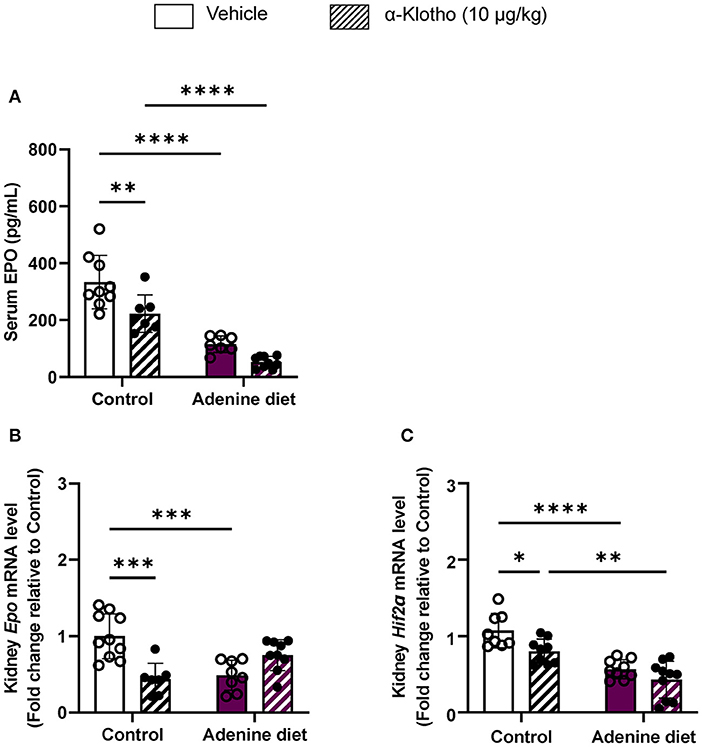
Figure 3. Regulation of erythropoietin production by Klotho administration. C57BL/6J male mice were fed control diet or 0.2% adenine diet for 8 weeks, and further divided into 2 groups for administration of either saline or mouse recombinant Klotho protein (10 μg/kg) 24 h prior to sacrifice. (A) Serum erythropoietin (EPO) levels measured by ELISA. (B,C) Quantitative real-time RT-PCR for expression of (B) renal Epo, and (C) renal Hif-2α. Data are expressed as fold change (2−ΔΔCt) relative to housekeeping gene Hprt. Data are represented as mean ± SD (n = 7–10 per group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to Control.
Iron is an essential element for hemoglobin synthesis and normal differentiation and proliferation of erythroid progenitor cells. Iron homeostasis is achieved by tight regulation between duodenal iron absorption and macrophage iron recycling. Hepcidin, a liver-secreted protein, is a master regulator of iron metabolism controlling the release of iron from the stores into the circulation (47, 48). Hepcidin is upregulated during inflammation, particularly in chronic inflammatory conditions such as CKD, leading to anemia, also known as anemia of chronic disease or anemia of inflammation (49, 50). Patients with CKD have elevated levels of pro-inflammatory cytokines (e.g., IL-6, IL-1β, TNFα, etc.) leading to upregulation of hepcidin (51). Consistent with the patient data, inflammatory markers, TNFα and IL-6, were significantly increased in the liver of adenine-fed mice (Figures 4A,B) compared to control mice, resulting in upregulation of hepcidin (Hamp) mRNA expression (Figure 4C). Klotho administration did not have any effect on these inflammatory markers or hepcidin expression (Figures 4A–C) in either adenine or control groups.
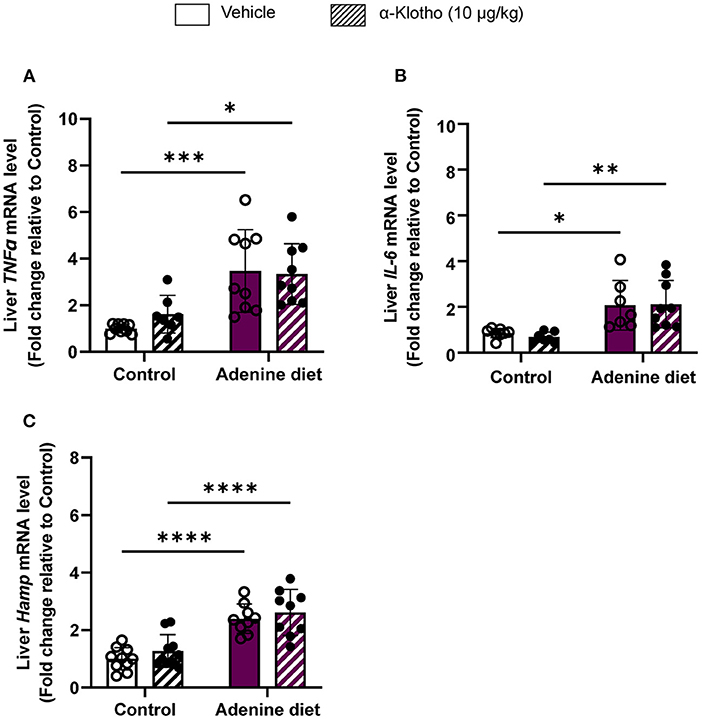
Figure 4. Effect of Klotho administration on inflammation in CKD. C57BL/6J male mice were fed control diet or 0.2% adenine diet. After 8 weeks, mice in each group were further divided into 2 groups and were injected with either saline or mouse recombinant Klotho protein (10 μg/kg) 24 h prior to sacrifice. Liver tissue samples were collected at the end of the experiment and relative mRNA expression was determined by quantitative real-time RT-PCR for (A) TNFα, (B) IL-6, and (C) hepcidin (Hamp). Data are represented as mean ± SD (n = 7–10 per group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to Control.
Hepcidin inhibits intestinal iron absorption and release of stored iron, thereby limiting available iron to be transported to the circulation and resulting in iron deficiency (51, 52). Iron deficiency is common among CKD patients with anemia characterized by microcytic hypochromic red blood cells (5). Our study confirms the presence of iron deficiency in adenine-fed mice as determined by low serum iron levels (Figure 5A; Table 3) and transferrin saturation (Figure 5B, compared to control mice. Moreover, red blood cells were microcytic and hypochromic, as it was evident by the low mean corpuscular volume (MCV) (Figure 5C; Table 3) and mean corpuscular hemoglobin (MCH) (Figure 5D; Table 3), representing RBC size and amount of hemoglobin per RBC, respectively. Klotho treatment did not affect serum iron or transferrin saturation in adenine-fed mice but it reduced these parameters in control mice (Figures 5A,B). Moreover, Klotho did not affect the size of RBCs or the amount of hemoglobin present in RBCs in either adenine-fed or control mice (Figures 5C,D).
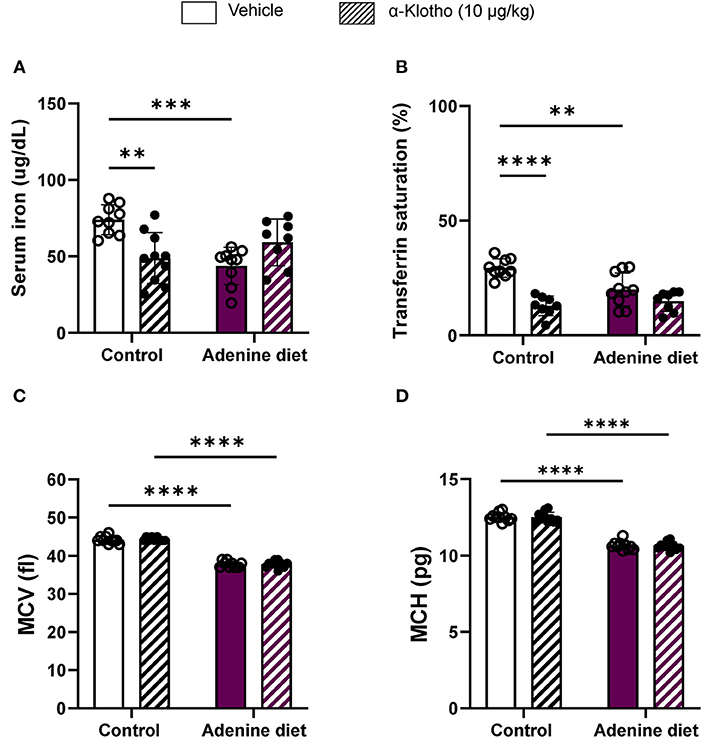
Figure 5. Iron homeostasis in response to CKD and klotho supplementation. C57BL/6J male mice were fed control diet or 0.2% adenine diet. After 8 weeks, mice in each group were further divided into 2 groups and were injected with either saline or mouse recombinant Klotho protein (10 μg/kg) 24 h prior to sacrifice. Serum samples were obtained and iron concentration was determined by colorimetric method. (A) Serum iron, (B) transferrin saturation. Whole blood was collected separately at sacrifice and quantified for MCV and MCH using Hematology analyzer. (C) Mean corpuscular volume (MCV), (D) mean corpuscular hemoglobin (MCH). Data are represented as mean ± SD (n = 8–10 per group). **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to Control.
Iron uptake is facilitated by divalent metal transporter 1 (DMT1), whereas iron export is achieved by ferroportin 1 (FPN1). The action of hepcidin is tissue-specific. Studies have shown that hepcidin decreases dietary iron absorption by inhibiting iron transport in enterocytes through downregulation of DMT1 and suppression of ferroportin activity (53–55), whereas in hepatocytes it prevents the mobilization of hepatic iron stores resulting in iron overload (52). In agreement with these data, we found that DMT1 and FPN1 are significantly suppressed in the duodenum of adenine-fed mice but did not decrease any further by Klotho (Figures 6A,B). However, Klotho significantly decreased duodenal DMT1 in control mice (Figure 6A). The decrease in serum iron in adenine-fed mice was associated with increased liver iron retention (Figure 6C), which correlated with increased hepatic expression of the iron storage protein ferritin H (Fth) (Figure 6D) and the iron sequester lipocalin 2 (Lcn2) (Figure 6E). The number of circulating neutrophils, which are the main source of lipocalin secretion, was also increased in adenine-fed mice (Table 3; Supplementary Figure 3). Klotho had no effect in liver iron content (Figure 6C), ferritin (Figure 6D), lipocalin (Figure 6E), or neutrophil number (Supplementary Figure 3) in either CKD or control mice. Moreover, we assessed expression of erythroferrone (Erfe), a hormone produced by bone marrow erythroblasts in response to increased renal EPO production that acts on hepatocytes to suppress hepcidin synthesis, thereby mobilizing iron from stores (56). Our data show that reduced EPO secretion in adenine-fed mice (Figures 3A,B) led to decreased Erfe expression in the bone marrow of adenine-fed mice compared to control mice (Figure 6F). Klotho did not affect Erfe expression in control or CKD mice (Figure 6F). Taken together, our data demonstrate that mice fed adenine diet show consistent characteristics of renal anemia and iron deficiency, and suggest that iron deficiency in adenine-induced CKD is due to upregulation of hepcidin by inflammation resulting in inhibition of iron absorption and sequestration of iron in liver. Furthermore, Klotho administration did not improve iron deficiency in CKD mice but it decreased intestinal absorption of iron in control mice leading to reduced serum iron and transferrin saturation levels.
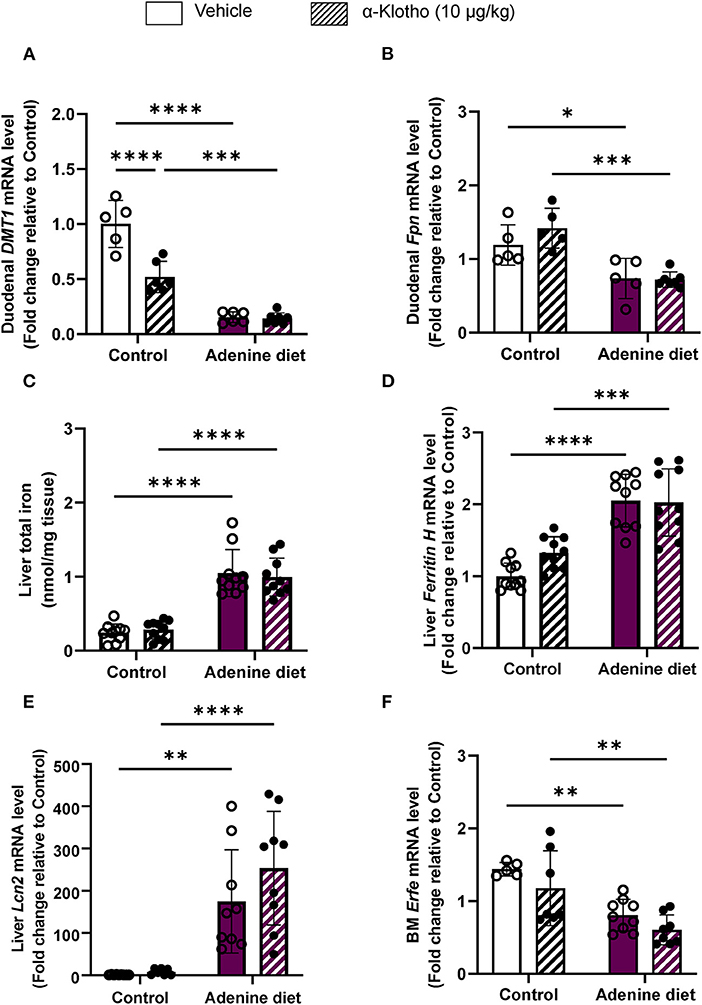
Figure 6. Dysregulation of iron transport and storage in CKD. C57BL/6J male mice fed control or 0.2% adenine diet for 8 weeks were administered with either saline or mouse recombinant Klotho protein (10 ug/kg) 24 h before sacrifice. Quantitative real-time RT-PCR for duodenal (A) Dmt1 and (B) ferroportin (Fpn) mRNA levels (n = 5–7 per group). (C) Total iron content in the liver by colorimetric assay. Quantitative real-time RT-PCR for (D) hepatic ferritin H (E) hepatic Lipocalin 2 (Lcn2) and (F) bone marrow erythroferrone (Erfe) mRNA expression. qRT-PCR data are expressed as fold change (2ΔΔCt) relative to housekeeping gene Hprt. Data are represented as mean ± SD (n = 8–10 per group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to Control.
Chronic kidney disease (CKD) is an inflammatory condition characterized by impaired excretory capacity and a gradual loss of renal function, putting patients with the condition at a higher risk for cardiovascular disease and mortality. There is no treatment to cure CKD, only to slow it down. Treatment options include medications that help manage symptoms, and in later stages, dialysis or kidney transplant. Complications such as anemia and iron deficiency are associated with poor outcomes such as reduced quality of life and increased cardiovascular disease, hospitalizations, cognitive impairment, and mortality (1). The cause of renal anemia is multifactorial and includes impaired EPO production due to renal dysfunction, pro-inflammatory cytokine activation, and inhibition of the renin-angiotensin system (RAS) by the use of angiotensin converting enzyme (ACE) inhibitors (3). To add to these causes, we previously reported that elevated FGF23 in CKD is significantly contributing to the development of renal anemia (18).
FGF23 signal transduction is achieved by binding of FGF23 to the Klotho-FGFR complex, with Klotho being an obligatory co-receptor for FGF23. Klotho is primarily expressed in the kidney and its expression is significantly reduced in CKD (57). Deletion of Klotho in mice results in a phenotype that resembles CKD, including hyperphosphatemia, elevated FGF23, ectopic soft tissue calcifications, and decreased plasma and renal Klotho (57, 58). Current treatment for renal anemia is the use of ESAs, which they are associated with a number of adverse outcomes such as worsened hypertension and cardiovascular disease, thromboembolism, tumor growth, and resistance to Epo therapy itself (1, 3, 11–13, 59). In search for treatment options with less adverse effects, we previously reported that inhibition of FGF23 signaling by the use of an FGF23 antagonist successfully rescues renal anemia and attenuates iron deficiency and inflammation in a CKD mouse model (18). Previous studies have reported an association of α-Klotho with several diseases, such as kidney disease (60), cardiovascular disease (61), and periodontitis (62). However, its role in hematological disorders, including anemia, remains unclear. Based on prior studies demonstrating beneficial and therapeutic effects of Klotho administration in CKD (23, 63–65) and our own work showing that disruption of FGF23 signaling by genetic ablation of Fgf23 or Klotho results in increased erythropoiesis (16, 17), in the present study we investigated the effect of exogenous α-Klotho in the development of renal anemia.
Our data show that α-Klotho administration had no beneficial effect in mice with CKD-associated anemia as it did not increase RBC numbers and hemoglobin levels, and it did not stimulate EPO secretion. Moreover, α-Klotho did not improve iron deficiency and inflammation in CKD as it had no effect on iron levels or inflammatory markers. These unexpected data suggest that one bolus injection of 10μg/Kg of recombinant α-Klotho was not sufficient to improve established anemia and iron deficiency in a CKD mouse model. Our choice of the given dose of Klotho (10μg/Kg) was based on published data showing that this dose was sufficient to prevent or delay AKI to CKD progression and uremic cardiomyopathy in mice (63) and resulted in less kidney damage in mice with AKI (32).
However, in agreement with our previous work (17), Klotho significantly decreased the number of bone marrow erythroid progenitors (BFU-E), in mice fed control diet, through downregulation of renal Hif2α and subsequent decrease in synthesis of EPO in the kidney and its secretion into the circulation. It also decreased serum iron and transferrin saturation by reducing duodenal iron transport through downregulation of DMT1 in control mice. Thus, although α-Klotho does not rescue renal anemia and associated iron deficiency, in physiological conditions α-Klotho appears to have an inhibitory effect on erythropoiesis and iron regulation. Our data are consistent with other reports that demonstrated a negative correlation between Klotho and iron and reported that serum iron overload decreases renal mRNA and protein levels of Klotho, whereas iron chelation suppresses angiotensin II-induced downregulation of Klotho (24, 66). Furthermore, in the same study, it was shown that a free radical scavenger suppressed the angiotensin II-induced downregulation of klotho, suggesting a role for reactive oxygen species (ROS) production in this process.
Iron deficiency in CKD is due to decreased iron absorption and increased iron loss caused by GI bleeding or platelet dysfunction (5). Moreover, studies have shown that iron deficiency is associated with elevated number of platelets as it was determined by a negative correlation between platelet count and transferrin saturation in CKD patients (67, 68). Conversely, it has been shown that iron repletion treatment reduces platelet numbers in patients with CKD (67–71). Consistent with these studies, our data show a significantly elevated platelet count in adenine fed mice (Table 3; Figure 7). However, Klotho did not have any effect on platelet numbers in either group (control or adenine) of our mice. The mechanism of platelet elevation in response to iron deficiency is not determined. Previous reports suggest that iron may inhibit platelet maturation (67, 72), whereas iron deficiency may affect platelet production through EPO (67, 73, 74) or have a direct effect on megakaryopoiesis by resulting in megakaryocyte progenitor expansion (67, 75). Since Klotho reduced serum iron and transferrin saturation in our control mice, we expected to see increase in platelet count in these mice. Lack of platelet count elevation in our control mice suggests that iron deficiency stimulates platelet production in the presence of inflammation.
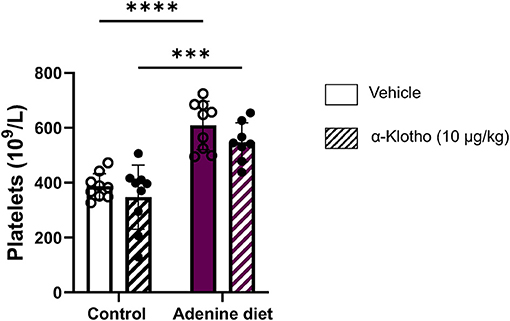
Figure 7. Effect of adenine diet and Klotho administration on platelets. C57BL/6J male mice fed control or 0.2% adenine diet for 8 weeks were administered with either saline or mouse recombinant Klotho protein (10 μg/kg) 24 h before sacrifice. Whole blood was collected and platelet concentration was quantified using Hematology Analyzer. Data are represented as mean ± SD (n = 8–9 per group). ***P < 0.001, ****P < 0.0001 compared to Control.
Activation of inducible nitric oxide synthase (iNOS; also referred to as NOS2) is associated with inflammatory conditions such as CKD and results in high levels of nitric oxide (NO) that lead to enhanced oxidative damage by increased generation of ROS (76–79). Thus, high levels of NO produced by iNOS may contribute to the progression of renal disease (79). It has been shown that the use of iNOS inhibitors may block the activation of pro-inflammatory cytokines produced during inflammation, resulting in translocation of NF-κB from cytoplasm to the nucleus and increase of iNOS expression (80). In agreement with these reports, our data show increased iNOS expression in adenine fed mice compared to control mice (Figure 8). Klotho exerts anti-inflammatory effects and negatively regulates the production of NF-κB-associated inflammatory proteins (81). Moreover, in CKD Klotho may mitigate activation of NF-κB by downregulating the expression of toll-like receptor 4 (TLR4) (82, 83). Conversely, it has been reported that NF-κB suppresses the activity of Klotho by possibly promoting ROS production (84). In agreement with its anti-inflammatory role, Klotho treatment significantly reduced iNOS expression in the kidney in both control- and adenine-fed mice (Figure 8).
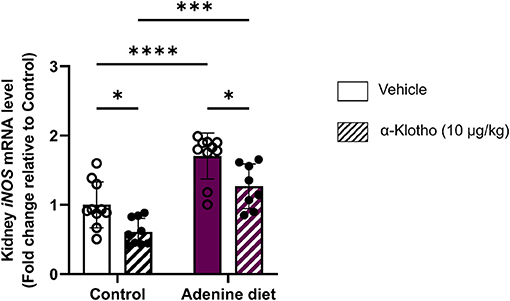
Figure 8. Response of renal iNOS mRNA expression in CKD and klotho administration. C57BL/6J male mice were fed control diet or 0.2% adenine diet. After 8 weeks, mice in each group were further divided into 2 groups and were injected with either saline or mouse recombinant Klotho protein (10 μg/kg) 24 h prior to sacrifice. Quantitative real-time RT-PCR for renal inducible nitric oxide synthase (iNOS). Data are expressed as fold change (2ΔΔCt) relative to housekeeping gene Hprt. Samples were measured in duplicates. Data are represented as mean ± SD (n = 8–9 per group). *P < 0.05, ***P < 0.001, ****P < 0.0001 compared to Control.
Our study has a number of limitations. First, the lack of a standardized assay to measure plasma Klotho, rendered us unable to confirm reduced Klotho levels in the adenine mice and assess the degree of circulating Klotho increase after α-Klotho administration. Second, our choice to use the adenine CKD mouse model vs. the 5/6 nephrectomy (Nx) mouse model may have played a role in the outcome of our study. Although the duration of disease progression is similar between the two mouse models (adenine and 5/6 Nx), the adenine diet model produces rapid-onset kidney disease with extensive tubulointerstitial fibrosis and tubular atrophy, whereas in the 5/6 Nx model the remaining kidney is functional and assumes a heavy-duty filtration function resulting in eventual nephron damage and occurrence of CKD. In addition, we previously showed that inhibition of FGF23 signaling rescues renal anemia and iron deficiency in mice with CKD induced by 5/6 nephrectomy (18). One important difference between our published study (18) and the present adenine study is the degree of increase in FGF23 levels. In the 5/6 Nx model, FGF23 levels were increased ~2.5 times above normal levels (306.3 vs. 801.2 pg/ml) (18), whereas in the present adenine study, FGF23 levels were massively increased ~13 times above normal levels (293.287 vs. 3911.919 pg/ml). Thus, the difference in circulating FGF23 and in the form of decreased renal function may have had important impact on whether Klotho injections alter FGF23-mediated changes on red blood cell production and iron regulation. Third, it is possible that we did not see any effect of Klotho in adenine-fed mice due to the chosen dose and duration of Klotho injection. In the present study, we injected mice with 10 μg/Kg of recombinant α-Klotho, a dose similar to what other investigators used in published reports (32, 63). In these published studies, investigators showed that this dose is sufficient to prevent or delay AKI to CKD progression and uremic cardiomyopathy in mice (63) and results in less kidney damage in mice with AKI (32). Our decision to use 10μg/Kg of recombinant α-Klotho was also based on our previous published work where we injected the same dose and we observed significant effects on the hematopoietic phenotype of wild-type mice (17). However, it is possible that one bolus injection at this dose was not sufficient to see an effect in adenine-fed mice in which Klotho levels are already low. Consideration should be given to assessing the effect of klotho after 3–4 daily injections (acute effect) or using minipumps delivering Klotho over a few weeks for a chronic effect, as described by Hu et al. (63). A dose and time course would be necessary to assess the effects of exogenous Klotho on hematological and iron parameters. One other aspect to consider, is the half-life of circulating Klotho. Hu et al. showed that although the levels of circulating exogenous Klotho immediately after injection were similar between normal mice and mice with CKD, the half-life of circulating exogenous Klotho (10 μg/Kg) in mice with CKD was much longer than in normal mice (25 h vs. 7.2 h) and similar to the levels of endogenous Klotho in mice with CKD (26.6 h) (14). These results show that normal healthy kidneys clear circulating Klotho faster and also suggest that the reason we did not see any increase on renal Klotho expression after administration of exogenous Klotho is because excess Klotho was already cleared by the kidneys after 24 h.
Because of the adverse effects ESAs have, it is clear that there is a need for treatment options that have fewer side effects. Recently, Hanudel et al. showed that Vadadustat, a hypoxia-inducible factor-prolyl hydroxylase inhibitor (HIF-PHI), ameliorates CKD-associated anemia and improves iron levels, as well as reducing the loss of kidney function and lowering FGF23 (85). Our previous work also clearly demonstrated the efficiency of inhibiting FGF23 signaling in stimulating erythropoiesis and rescuing anemia, iron deficiency and inflammation (18). Moreover, we found that blocking FGF23 signaling significantly increased renal Klotho expression in mice with CKD induced by 5/6 nephrectomy. Therefore, FGF23 antagonists are strong candidates for the treatment of renal anemia, iron deficiency and associated inflammation, and have the potential to attenuate Klotho deficiency. Although in our study one bolus injection of recombinant Klotho at the chosen dose did not improve anemia and iron deficiency in CKD, it does not exclude the possibility that suppression of Klotho levels deteriorates CKD-associated anemia and, thus, optimization of the treatment protocol may provide evidence that Klotho may still be a potential therapeutic target for renal anemia.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by New York University IACUC.
MP performed research, analyzed data, assisted with data interpretation, and manuscript preparation. CLH assisted with experimental procedures. DS designed and performed research, analyzed data, wrote the manuscript, and oversaw the study. All authors contributed to the article and approved the submitted version.
This work was supported by funds from the US Department of Defense (W81XWH-16-1-0598) to DS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.924915/full#supplementary-material
1. KDOQI National Kidney Foundation. KDOQI clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am J Kidney Dis. (2006) 47(5 Suppl 3):S11–145. doi: 10.1053/j.ajkd.2006.03.011
2. Fishbane S, Pollack S, Feldman HI, Joffe MM. Iron indices in chronic kidney disease in the National Health and Nutritional Examination Survey 1988–2004. Clin J Am Soc Nephrol. (2009) 4:57–61. doi: 10.2215/CJN.01670408
3. Babitt JL, Lin HY. Mechanisms of anemia in CKD. J Am Soc Nephrol. (2012) 23:1631–4. doi: 10.1681/ASN.2011111078
4. Batchelor EK, Kapitsinou P, Pergola PE, Kovesdy CP, Jalal DI. Iron deficiency in chronic kidney disease: updates on pathophysiology, diagnosis, and treatment. J Am Soc Nephrol. (2020) 31:456–68. doi: 10.1681/ASN.2019020213
5. Gafter-Gvili A, Schechter A, Rozen-Zvi B. Iron deficiency anemia in chronic kidney disease. Acta Haematol. (2019) 142:44–50. doi: 10.1159/000496492
6. Auerbach M, Adamson JW. How we diagnose and treat iron deficiency anemia. Am J Hematol. (2016) 91:31–8. doi: 10.1002/ajh.24201
8. Fertrin KY. Diagnosis and management of iron deficiency in chronic inflammatory conditions (CIC): is too little iron making your patient sick? Hematology Am Soc Hematol Educ Program. (2020) 2020:478–86. doi: 10.1182/hematology.2020000132
9. Joanne M, Bargman JM, Skorecki K. Harrison's Principle and Practice of Internal Medicine. New York: McGraw-Hill (2008). p. 1761–71.
10. Shastry I, Belurkar S. The spectrum of red blood cell parameters in chronic kidney disease: a study of 300 cases. J Appl Hematol. (2019) 10:61–6. doi: 10.4103/joah.joah_13_19
11. Bennett CL, Becker PS, Kraut EH, Samaras AT, West DP. Intersecting guidelines: administering erythropoiesis-stimulating agents to chronic kidney disease patients with cancer. Semin Dial. (2009) 22:1–4. doi: 10.1111/j.1525-139X.2008.00524.x
12. Eschbach JW, Egrie JC, Downing MR, Browne JK, Adamson JW. Correction of the anemia of end-stage renal disease with recombinant human erythropoietin. Results of a combined phase I and II clinical trial. N Engl J Med. (1987) 316:73–8. doi: 10.1056/NEJM198701083160203
13. Winearls CG, Oliver DO, Pippard MJ, Reid C, Downing MR, Cotes PM. Effect of human erythropoietin derived from recombinant DNA on the anaemia of patients maintained by chronic haemodialysis. Lancet. (1986) 2:1175–8. doi: 10.1016/S0140-6736(86)92192-6
14. Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL, et al. Renal production, uptake, and handling of circulating alphaKlotho. J Am Soc Nephrol. (2016) 27:79–90. doi: 10.1681/ASN.2014101030
15. Olauson H, Larsson TE. FGF23 and Klotho in chronic kidney disease. Curr Opin Nephrol Hypertens. (2013) 22:397–404. doi: 10.1097/MNH.0b013e32836213ee
16. Coe LM, Madathil SV, Casu C, Lanske B, Rivella S, Sitara D. FGF-23 is a negative regulator of prenatal and postnatal erythropoiesis. J Biol Chem. (2014) 289:9795–810. doi: 10.1074/jbc.M113.527150
17. Vadakke Madathil S, Coe LM, Casu C, Sitara D. Klotho deficiency disrupts hematopoietic stem cell development and erythropoiesis. Am J Pathol. (2014) 184:827–41. doi: 10.1016/j.ajpath.2013.11.016
18. Agoro R, Montagna A, Goetz R, Aligbe O, Singh G, Coe LM, et al. Inhibition of fibroblast growth factor 23 (FGF23) signaling rescues renal anemia. FASEB J. (2018) 32:3752–64. doi: 10.1096/fj.201700667R
19. Zhou L, Mo H, Miao J, Zhou D, Tan RJ, Hou FF, et al. Klotho ameliorates kidney injury and fibrosis and normalizes blood pressure by targeting the renin-angiotensin system. Am J Pathol. (2015) 185:3211–23. doi: 10.1016/j.ajpath.2015.08.004
20. Kadoya H, Satoh M, Haruna Y, Sasaki T, Kashihara N. Klotho attenuates renal hypertrophy and glomerular injury in Ins2Akita diabetic mice. Clin Exp Nephrol. (2016) 20:671–8. doi: 10.1007/s10157-015-1202-3
21. Xie J, Yoon J, An SW, Kuro-o M, Huang CL. Soluble klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. J Am Soc Nephrol. (2015) 26:1150–60. doi: 10.1681/ASN.2014040325
22. Kempe DS, Ackermann TF, Fischer SS, Koka S, Boini KM, Mahmud H, et al. Accelerated suicidal erythrocyte death in Klotho-deficient mice. Pflugers Arch. (2009) 458:503–12. doi: 10.1007/s00424-009-0636-4
23. Hu MC, Shi M, Cho HJ, Zhang J, Pavlenco A, Liu S, et al. The erythropoietin receptor is a downstream effector of Klotho-induced cytoprotection. Kidney Int. (2013) 84:468–81. doi: 10.1038/ki.2013.149
24. Saito K, Ishizaka N, Mitani H, Ohno M, Nagai R. Iron chelation and a free radical scavenger suppress angiotensin II-induced downregulation of klotho, an anti-aging gene, in rat. FEBS Lett. (2003) 551:58–62. doi: 10.1016/S0014-5793(03)00894-9
25. Clinkenbeard EL, Noonan ML, Thomas JC, Ni P, Hum JM, Aref M, et al. Increased FGF23 protects against detrimental cardio-renal consequences during elevated blood phosphate in CKD. JCI Insight. (2019) 4:e123817. doi: 10.1172/jci.insight.123817
26. Jia T, Olauson H, Lindberg K, Amin R, Edvardsson K, Lindholm B, et al. A novel model of adenine-induced tubulointerstitial nephropathy in mice. BMC Nephrol. (2013) 14:116. doi: 10.1186/1471-2369-14-116
27. Santana AC, Degaspari S, Catanozi S, Delle H, de Sa Lima L, Silva C, et al. Thalidomide suppresses inflammation in adenine-induced CKD with uraemia in mice. Nephrol Dial Transplant. (2013) 28:1140–9. doi: 10.1093/ndt/gfs569
28. Ali BH, Al-Salam S, Al Za'abi M, Waly MI, Ramkumar A, Beegam S, et al. New model for adenine-induced chronic renal failure in mice, and the effect of gum acacia treatment thereon: comparison with rats. J Pharmacol Toxicol Methods. (2013) 68:384–93. doi: 10.1016/j.vascn.2013.05.001
29. Mori-Kawabe M, Yasuda Y, Ito M, Matsuo S. Reduction of NO-mediated relaxing effects in the thoracic aorta in an experimental chronic kidney disease mouse model. J Atheroscler Thromb. (2015) 22:845–53. doi: 10.5551/jat.28191
30. Gutierrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. (2005) 16:2205–15. doi: 10.1681/ASN.2005010052
31. Komaba H, Goto S, Fujii H, Hamada Y, Kobayashi A, Shibuya K, et al. Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int. (2010) 77:232–8. doi: 10.1038/ki.2009.414
32. Hu MC, Shi M, Zhang J, Quinones H. Kuro-o M, Moe OW. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. (2010) 78:1240–51. doi: 10.1038/ki.2010.328
33. Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. (2013) 75:503–33. doi: 10.1146/annurev-physiol-030212-183727
34. Pavik I, Jaeger P, Ebner L, Wagner CA, Petzold K, Spichtig D, et al. Secreted Klotho and FGF23 in chronic kidney disease Stage 1 to 5: a sequence suggested from a cross-sectional study. Nephrol Dial Transplant. (2013) 28:352–9. doi: 10.1093/ndt/gfs460
35. Eto N, Miyata Y, Ohno H, Yamashita T. Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol Dial Transplant. (2005) 20:1378–84. doi: 10.1093/ndt/gfh781
36. Pulskens WP, Verkaik M, Sheedfar F, van Loon EP, van de Sluis B, Vervloet MG, et al. Deregulated renal calcium and phosphate transport during experimental kidney failure. PLoS ONE. (2015) 10:e0142510. doi: 10.1371/journal.pone.0142510
37. Kaludjerovic J, Komaba H, Sato T, Erben RG, Baron R, Olauson H, et al. Klotho expression in long bones regulates FGF23 production during renal failure. FASEB J. (2017) 31:2050–64. doi: 10.1096/fj.201601036R
38. Hum JM, O'Bryan LM, Tatiparthi AK, Cass TA, Clinkenbeard EL, Cramer MS, et al. Chronic hyperphosphatemia and vascular calcification are reduced by stable delivery of soluble klotho. J Am Soc Nephrol. (2017) 28:1162–74. doi: 10.1681/ASN.2015111266
39. Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. (2010) 24:3438–50. doi: 10.1096/fj.10-154765
40. Silverberg D, Wexler D, Blum M, Wollman Y, Iaina A. The cardio-renal anaemia syndrome: does it exist? Nephrol Dial Transplant. (2003) 18(Suppl 8):viii7–12. doi: 10.1093/ndt/gfg1084
41. Cole J, Ertoy D, Lin H, Sutliff RL, Ezan E, Guyene TT, et al. Lack of angiotensin II-facilitated erythropoiesis causes anemia in angiotensin-converting enzyme-deficient mice. J Clin Invest. (2000) 106:1391–8. doi: 10.1172/JCI10557
42. Kalantar-Zadeh K, Rodriguez RA, Humphreys MH. Association between serum ferritin and measures of inflammation, nutrition and iron in haemodialysis patients. Nephrol Dial Transplant. (2004) 19:141–9. doi: 10.1093/ndt/gfg493
43. Lipkin GW, Kendall RG, Russon LJ, Turney JH, Norfolk DR, Brownjohn AM. Erythropoietin deficiency in acute renal failure. Nephrol Dial Transplant. (1990) 5:920–2. doi: 10.1093/ndt/5.11.920
44. Leelahavanichkul A, Yan Q, Hu X, Eisner C, Huang Y, Chen R, et al. Angiotensin II overcomes strain-dependent resistance of rapid CKD progression in a new remnant kidney mouse model. Kidney Int. (2010) 78:1136–53. doi: 10.1038/ki.2010.287
45. Querbes W, Bogorad RL, Moslehi J, Wong J, Chan AY, Bulgakova E, et al. Treatment of erythropoietin deficiency in mice with systemically administered siRNA. Blood. (2012) 120:1916–22. doi: 10.1182/blood-2012-04-423715
46. Zhang F, Laneuville P, Gagnon RF, Morin B, Brox AG. Effect of chronic renal failure on the expression of erythropoietin message in a murine model. Exp Hematol. (1996) 24:1469–74.
47. Ganz T. Macrophages and systemic iron homeostasis. J Innate Immun. (2012) 4:446–53. doi: 10.1159/000336423
48. Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. (2012) 1823:1434–43. doi: 10.1016/j.bbamcr.2012.01.014
49. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. (2003) 102:783–8. doi: 10.1182/blood-2003-03-0672
50. Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. (2004) 113:1271–6. doi: 10.1172/JCI200420945
51. Barreto DV, Barreto FC, Liabeuf S, Temmar M, Lemke HD, Tribouilloy C, et al. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney Int. (2010) 77:550–6. doi: 10.1038/ki.2009.503
52. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. (2004) 306:2090–3. doi: 10.1126/science.1104742
53. Brasse-Lagnel C, Karim Z, Letteron P, Bekri S, Bado A, Beaumont C. Intestinal DMT1 cotransporter is down-regulated by hepcidin via proteasome internalization and degradation. Gastroenterology. (2011) 140:1261–71.e1. doi: 10.1053/j.gastro.2010.12.037
54. Chaston T, Chung B, Mascarenhas M, Marks J, Patel B, Srai SK, et al. Evidence for differential effects of hepcidin in macrophages and intestinal epithelial cells. Gut. (2008) 57:374–82. doi: 10.1136/gut.2007.131722
55. Mena NP, Esparza A, Tapia V, Valdes P, Nunez MT. Hepcidin inhibits apical iron uptake in intestinal cells. Am J Physiol Gastrointest Liver Physiol. (2008) 294:G192–8. doi: 10.1152/ajpgi.00122.2007
56. Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. (2014) 46:678–84. doi: 10.1038/ng.2996
57. Hu MC, Kuro-o M, Moe OW. Klotho and chronic kidney disease. Contrib Nephrol. (2013) 180:47–63. doi: 10.1159/000346778
58. Hu MC, Shi M, Zhang J, Quinones H, Griffith C, Kuro-o M, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. (2011) 22:124–36. doi: 10.1681/ASN.2009121311
59. Besarab A, Bolton WK, Browne JK, Egrie JC, Nissenson AR, Okamoto DM, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. (1998) 339:584–90. doi: 10.1056/NEJM199808273390903
60. Olauson H, Lindberg K, Amin R, Jia T, Wernerson A, Andersson G, et al. Targeted deletion of Klotho in kidney distal tubule disrupts mineral metabolism. J Am Soc Nephrol. (2012) 23:1641–51. doi: 10.1681/ASN.2012010048
61. Chen J, Lin Y, Sun Z. Deficiency in the anti-aging gene Klotho promotes aortic valve fibrosis through AMPKalpha-mediated activation of RUNX2. Aging Cell. (2016) 15:853–60. doi: 10.1111/acel.12494
62. Liu Y, Zhang Q. Periodontitis aggravated pancreatic beta-cell dysfunction in diabetic mice through interleukin-12 regulation on Klotho. J Diabetes Investig. (2016) 7:303–11. doi: 10.1111/jdi.12410
63. Hu MC, Shi M, Gillings N, Flores B, Takahashi M, Kuro OM, et al. Recombinant alpha-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int. (2017) 91:1104–14. doi: 10.1016/j.kint.2016.10.034
64. Panesso MC, Shi M, Cho HJ, Paek J, Ye J, Moe OW, et al. Klotho has dual protective effects on cisplatin-induced acute kidney injury. Kidney Int. (2014) 85:855–70. doi: 10.1038/ki.2013.489
65. Yamamoto M, Clark JD, Pastor JV, Gurnani P, Nandi A, Kurosu H, et al. Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem. (2005) 280:38029–34. doi: 10.1074/jbc.M509039200
66. Xu Y, Peng H, Ke B. alpha-klotho and anemia in patients with chronic kidney disease patients: A new perspective. Exp Ther Med. (2017) 14:5691–5. doi: 10.3892/etm.2017.5287
67. Liao R, Zhou X, Ma D, Tang J, Zhong H. Iron deficiency is associated with platelet count elevation in patients with dialysis-dependent chronic kidney disease. J Ren Nutr. (2022). doi: 10.1053/j.jrn.2021.09.004. [Epub ahead of print].
68. Yessayan L, Yee J, Zasuwa G, Frinak S, Besarab A. Iron repletion is associated with reduction in platelet counts in non-dialysis chronic kidney disease patients independent of erythropoiesis-stimulating agent use: a retrospective cohort study. BMC Nephrol. (2014) 15:119. doi: 10.1186/1471-2369-15-119
69. Coyne DW, Kapoian T, Suki W, Singh AK, Moran JE, Dahl NV, et al. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients' Response to IV Iron with Elevated Ferritin (DRIVE) Study. J Am Soc Nephrol. (2007) 18:975–84. doi: 10.1681/ASN.2006091034
70. Huscenot T, Darnige L, Wagner-Ballon O, Ronchetti AM, Lousteau V, Limal N, et al. Iron deficiency, an unusual cause of thrombocytopenia: results from a multicenter retrospective case-controlled study. Ann Hematol. (2019) 98:2299–302. doi: 10.1007/s00277-019-03757-0
71. Kuku I, Kaya E, Yologlu S, Gokdeniz R, Baydin A. Platelet counts in adults with iron deficiency anemia. Platelets. (2009) 20:401–5. doi: 10.1080/09537100903137306
72. Choi SI, Simone JV, Jackson CW. Megakaryocytopoiesis in experimental iron deficiency anemia. Blood. (1974) 43:111–20. doi: 10.1182/blood.V43.1.111.111
73. Kaupke CJ, Butler GC, Vaziri ND. Effect of recombinant human erythropoietin on platelet production in dialysis patients. J Am Soc Nephrol. (1993) 3:1672–9. doi: 10.1681/ASN.V3101672
74. Bilic E, Bilic E. Amino acid sequence homology of thrombopoietin and erythropoietin may explain thrombocytosis in children with iron deficiency anemia. J Pediatr Hematol Oncol. (2003) 25:675–6. doi: 10.1097/00043426-200308000-00023
75. Evstatiev R, Bukaty A, Jimenez K, Kulnigg-Dabsch S, Surman L, Schmid W, et al. Iron deficiency alters megakaryopoiesis and platelet phenotype independent of thrombopoietin. Am J Hematol. (2014) 89:524–9. doi: 10.1002/ajh.23682
76. Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD, et al. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science. (1992) 256:225–8. doi: 10.1126/science.1373522
77. Chartrain NA, Geller DA, Koty PP, Sitrin NF, Nussler AK, Hoffman EP, et al. Molecular cloning, structure, and chromosomal localization of the human inducible nitric oxide synthase gene. J Biol Chem. (1994) 269:6765–72. doi: 10.1016/S0021-9258(17)37441-0
78. Zamora R, Vodovotz Y, Billiar TR. Inducible nitric oxide synthase and inflammatory diseases. Mol Med. (2000) 6:347–73. doi: 10.1007/BF03401781
79. Bautista-Garcia P, Sanchez-Lozada LG, Cristobal-Garcia M, Tapia E, Soto V, Avila-Casado MC, et al. Chronic inhibition of NOS-2 ameliorates renal injury, as well as COX-2 and TGF-beta 1 overexpression in 5/6 nephrectomized rats. Nephrol Dial Transplant. (2006) 21:3074–81. doi: 10.1093/ndt/gfl444
80. Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci. (2004) 75:639–53. doi: 10.1016/j.lfs.2003.10.042
81. Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, et al. Suppression of aging in mice by the hormone Klotho. Science. (2005) 309:1829–33. doi: 10.1126/science.1112766
82. Wu C, Lv C, Chen F, Ma X, Shao Y, Wang Q. The function of miR-199a-5p/Klotho regulating TLR4/NF-kappaB p65/NGAL pathways in rat mesangial cells cultured with high glucose and the mechanism. Mol Cell Endocrinol. (2015) 417:84–93. doi: 10.1016/j.mce.2015.09.024
83. Shao Y, Sha M, Chen L, Li D, Lu J, Xia S. HMGB1/TLR4 signaling induces an inflammatory response following high-pressure renal pelvic perfusion in a porcine model. Am J Physiol Renal Physiol. (2016) 311:F915–F25. doi: 10.1152/ajprenal.00480.2015
84. Yang K, Nie L, Huang Y, Zhang J, Xiao T, Guan X, et al. Amelioration of uremic toxin indoxyl sulfate-induced endothelial cell dysfunction by Klotho protein. Toxicol Lett. (2012) 215:77–83. doi: 10.1016/j.toxlet.2012.10.004
Keywords: chronic kidney disease, Klotho, anemia, iron deficiency (anemia), inflammation
Citation: Park MY, Le Henaff C and Sitara D (2022) Administration of α-Klotho Does Not Rescue Renal Anemia in Mice. Front. Pediatr. 10:924915. doi: 10.3389/fped.2022.924915
Received: 20 April 2022; Accepted: 27 May 2022;
Published: 23 June 2022.
Edited by:
Mark Hanudel, University of California, Los Angeles, United StatesReviewed by:
Erica Clinkenbeard, Purdue University Indianapolis, United StatesCopyright © 2022 Park, Le Henaff and Sitara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Despina Sitara, ZHMxOTlAbnl1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.