Corrigendum: Chromosomal abnormalities and pregnancy outcomes for fetuses with gastrointestinal tract obstructions
 Xiaoqing Wu1,2,3
Xiaoqing Wu1,2,3 Linjuan Su1,2Qingmei Shen1,2Qun Guo1,2Ying Li1,2Shiyi Xu4Na Lin1,2*
Linjuan Su1,2Qingmei Shen1,2Qun Guo1,2Ying Li1,2Shiyi Xu4Na Lin1,2* Hailong Huang1,2*Liangpu Xu1,2*
Hailong Huang1,2*Liangpu Xu1,2*- 1Department of Medical Genetic Diagnosis and Therapy Center, Fujian Maternity and Child Health Hospital, College of Clinical Medicine for Obstetrics and Gynecology and Pediatrics, Fujian Medical University, Fuzhou, China
- 2Fujian Provincial Key Laboratory for Prenatal Diagnosis and Birth Defect, Fuzhou, China
- 3Department of Laboratory Medicine, Fujian Medical University, Fuzhou, China
- 4Department of Pediatrics, Guangxi Medical University, Nanning, China
Fetal gastrointestinal tract obstruction (GITO) is the most frequently encountered gastrointestinal defect in the prenatal period. This study aimed to investigate the genetic disorders and pregnancy outcomes of fetal GITO. We reviewed data from 70 pregnancies that were referred for invasive prenatal testing because of fetal GITO. According to the level of obstruction, they were classified into esophageal atresia/stenosis, duodenal atresia/stenosis, jejunal or ileal atresia/stenosis, or anal atresia. Traditional karyotyping was performed on all the 70 pregnancies, and chromosomal microarray analysis (CMA) was performed on 32 of them in parallel. Traditional karyotyping revealed twelve (17.1%) chromosomal abnormalities, including 11 cases of trisomy 21 (Down syndrome), and one case of a supernumerary marker chromosome related to Cat eye syndrome. According to the absence or presence of other ultrasound anomalies, they were categorized into isolated GITO (n = 36) and non-isolated GITO (n = 34). The rate of chromosomal abnormalities in the non-isolated GITO pregnancies was significantly higher than that in the isolated GITO pregnancies (29.4 vs. 5.5%, p < 0.05); the survival rate in the isolated group was significantly higher than that in the non-isolated group (67.6 vs. 34.4%, p < 0.05). Among the 32 cases where CMA was performed, an additional one (3.1%) copy number variant with clinical significance was noted in a fetus with normal karyotype. The microduplication on 7q12 was considered to be the genetic etiology of duodenal stenosis, although it was inherited from a phenotypically normal mother. Our study supports the strong association between Down syndrome and fetal GITO, especially duodenal stenosis. Our findings suggested that the risk of chromosomal abnormalities was increased when GITO was accompanied by other ultrasound anomalies; thus, chromosomal abnormalities and fetal anatomy should be carefully evaluated for pregnancy management of fetal GITO.
Introduction
Fetal gastrointestinal tract obstruction (GITO) is the most frequently encountered gastrointestinal defect in the prenatal period. It is the result of maldevelopment of the gastrointestinal tract including atresia and stenosis and is reported to affect one–five per 100,00 live births (1–3). It is mainly diagnosed by routine obstetrical ultrasound screening during the second and third trimesters with variable diagnostic accuracy (1, 4, 5). Indirect ultrasound findings such as failure to visualize the stomach, double bubble, and dilated bowel loops are typical signs. The obstruction may occur at any site of the gastrointestinal tract. According to different levels of obstruction, they are generally defined as esophageal atresia/stenosis, duodenal atresia/stenosis, jejunal or ileal atresia/stenosis, or anorectal malformation.
The etiology and pathogenesis of GITO are not completely understood, because genetic, biological, hormonal, and environmental factors all play an important role in it (5–8). Medical care such as surgical correction within the first few days after birth can effectively improve the prognosis; thus, most infants with congenital GITO tend to have an excellent prognosis (9, 10). However, the premise is that there are no other ultrasound anomalies and chromosomal abnormalities. GITO could be isolated or more frequently accompanied by other ultrasound anomalies, which were reported to present in 10%−65% of cases (1, 2, 11, 12), especially in esophageal and anorectal obstruction cases. The association between GITO and chromosomal abnormalities has been explored in several previous reports (13). For instance, trisomy 21, trisomy 18, and trisomy 13 have been observed in cases with esophageal atresia (14, 15). Best et al. (16) reported that 20% of small intestinal atresia cases were associated with chromosomal anomalies. However, most of these data were from traditional karyotyping, whereas the detective efficiency of chromosomal microarray analysis (CMA) in GITO is limited. Here, we presented our experience on prenatal diagnosis of GITO based on genetic disorders by traditional karyotyping and SNP array testing, as well as associated ultrasound abnormalities, to provide more evidence for genetic counseling and pregnancy management.
Materials and Methods
Patients and Samples
Between May 2012 and November 2021, a total of 73 singleton pregnancies were referred for invasive prenatal testing because of fetal digestive tract atresia/stenosis at the Medical Genetic Diagnosis and Therapy center of Fujian Maternal and Child Health Hospital, China. Three cases were excluded because GITO was not observed on repeat prenatal ultrasound. As a result, 70 pregnancies were enrolled, including nine cases of esophageal atresia/stenosis, 43 cases of duodenal atresia/stenosis, 16 cases of jejunal or ileal atresia/stenosis, and two cases of anal atresia. All the diagnoses of GITO were made by ultrasonography, and 15 of them were confirmed further by magnetic resonance imaging (MRI). The gestational age at the diagnosis of GITO was 26.3 ± 3.6 weeks. The general information is presented in Table 1.
TABLE 1
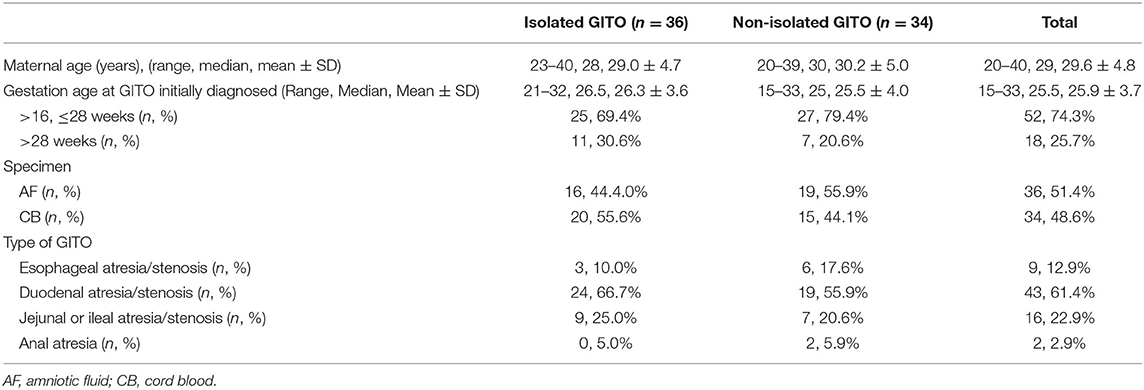
Table 1. Demographic characters for 70 pregnancies with fetal gastrointestinal tract obstruction (GITO).
The enrolled cases were categorized into non-isolated GITO (n = 36) and isolated GITO (n = 34) according to the presence or absence of other ultrasound abnormalities. Considering that polyhydramnios was a frequent development secondary to gastrointestinal obstruction, GITO accompanied with polyhydramnios was categorized as isolated GITO.
Generally, taking into account the risk of invasive procedures and the success rate of cell culture, we offered an amniocentesis to cases with gestational ages within 24 weeks, and cordocentesis to cases with gestational age beyond 24 weeks. As a result, the prenatal specimens included 36 cases of amniotic fluid and 34 cases of cord blood. Traditional karyotyping was performed on all 70 pregnancies, and an SNP array was performed in parallel on 32 of them.
Follow-up assessments were performed via clinical records or telephone calls, and the age at follow-up ranged from 3 months to 10 years. The study was approved by the local Ethics Committee of Fujian Maternity and Child Health Hospital. Written informed consent to participate in the study was obtained from each patient.
Traditional Karyotyping
Traditional karyotyping consisted of cell culture, and G-banded karyotyping was performed on cultured amniotic fluid or fetal cord blood according to the standard protocols in our laboratory. The karyotype was determined at a resolution of 320–500 band level.
CMA Platforms and Data Interpretation
Genomic DNA was extracted from uncultured amniotic fluid or fetal cord blood using a QIAGEN kit (Qiagen, Hilden, Germany) and according to the manufacturer's instructions. A single nucleotide polymorphism (SNP) array was performed using an Affymetrix CytoScan 750K array (Affymetrix Inc., Santa Clara, CA, United States), which included 200,000 probes for SNPs and 550,000 probes for copy number variants (CNVs) distributed across the entire human genome. The Chromosome Analysis Suite software (Affymetrix) and human genome version GRCh37 (hg19) were used. A resolution was generally applied: gains or losses of ≥400 kb and loss of heterozygosity (LOH) ≥10 Mb. All detected CNVs were compared with in-house and national public CNV databases as follows: Database of Genomic Variants (DGV), Database of Chromosome Imbalance and Phenotype in Humans Using Ensemble Resources (DECIPHER), International Standards for Cytogenomic Arrays Consortium, and Online Mendelian Inheritance in Man (OMIM). The CNVs were classified into five groups according to the American College of Medical Genetics (ACMG) definitions (17) and local database: pathogenic, benign, likely pathogenic, likely benign, and variant of unknown significance (VOUS). Pathogenic/likely pathogenic CNVs were considered clinically significant findings. CNV inherited from a phenotypically normal parent was considered to be likely benign.
Statistical Analysis
The data were analyzed with SPSS software v26.0 (SPSS Inc., Chicago, IL, United States). Statistical comparisons were performed by chi-square test, and p < 0.05 was considered statistically significant.
Results
Results of Traditional Karyotyping
The traditional karyotyping revealed 12 (17.1%) chromosomal abnormalities from a total 70 cases (Table 2). The detection rates in pregnancies of esophageal atresia/stenosis, duodenal atresia/stenosis, jejunal or ileal atresia/stenosis, and anorectal malformation were 11.1, 23.3, 0, and 50%, respectively (Table 3). The aberrations included 11 cases of trisomy 21 (T21) and 1 case of supernumerary marker chromosome (sSMC). Among them, T21 was noted in 10 out of the 43 (23.3%) cases of duodenal atresia/stenosis (cases 1–2 and 4–11, Table 2) and in one out of the nine (11.1%) cases of esophageal atresia/stenosis (case 12). For sSMC analysis (case 13), the SNP array demonstrated four copies of the gene dosage encompassing the 22q11.1q11.21 region (16,888,899–18,649,190). FISH studies were subsequently performed using a chromosome 22-specific DNA probe and showed that the marker had two 22q11.2 signals. Therefore, the karyotype was finally confirmed to be 47, XY,+psuidic(22) (q11.2), harboring additional copies of cat eye syndrome (CES, OMIM # 115470) critical region genes. The affected fetus showed suspected anal atresia and multiple malformations. The parents declined to terminate the pregnancy, and the fetus was delivered at 38 gestational weeks but died 10 days after birth.
TABLE 2
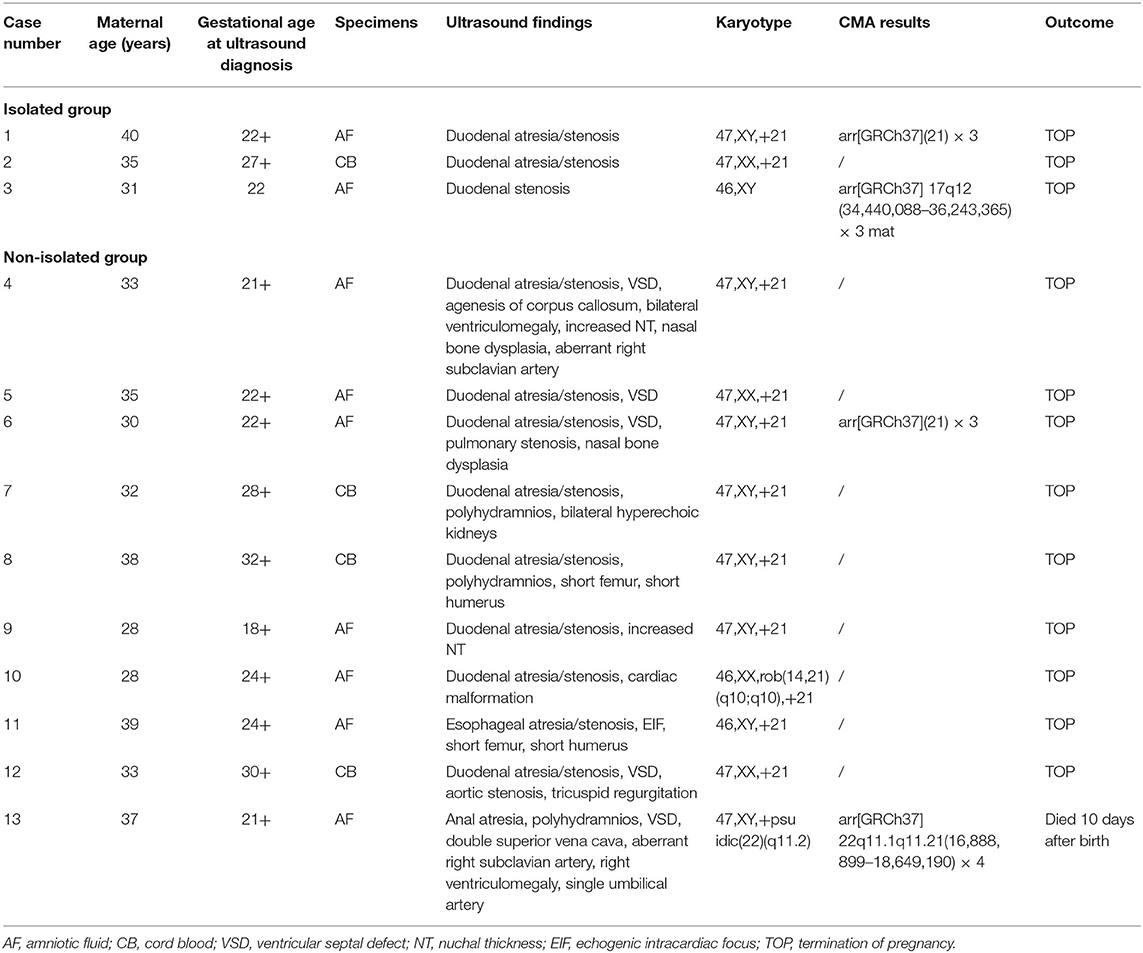
Table 2. Details of 12 abnormal karyotypes and one copy number variants.
TABLE 3
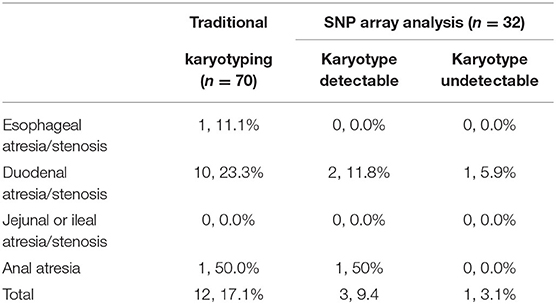
Table 3. Distribution of chromosomal abnormalities in different types of GITO.
The rate of chromosomal abnormalities determined by traditional karyotyping in isolated GITO was 5.5% (2/34), which is significantly lower than the 29.4% (10/24) in the non-isolated group (p < 0.05). Polyhydramnios was the most common accompanying abnormality that was noted in 41.4% (29/70) of the cases, followed by cardiovascular malformations (11.4%, 8/70) including atrioventricular septal defect, aortic stenosis, pulmonic stenosis, and persistent left superior vena cava.
CMA Results of 32 Fetuses
Among the 32 cases that underwent traditional karyotyping and CMA in parallel, an additional pathogenic aberration (case 3, Table 2) was identified in a fetus with duodenal atresia/stenosis (Figure 1) and normal karyotype, contributing to an incremental detection yield of 5.9% (1/17) for fetuses with duodenal atresia. The fetus had a 1.8-Mb duplication in the 17q12 region (Figure 2), which is responsible for17q12 duplication syndrome (OMIM # 614526). The overall penetrance of the syndrome for developing any disorder was reported to be nearly 21.1%. Here, the CNVs were categorized as “pathogenic,” although it was inherited from a phenotypically normal mother. The pregnancy was terminated. The detailed ultrasound findings, CMA results, and pregnancy outcomes are summarized in Supplementary Table 1.
FIGURE 1
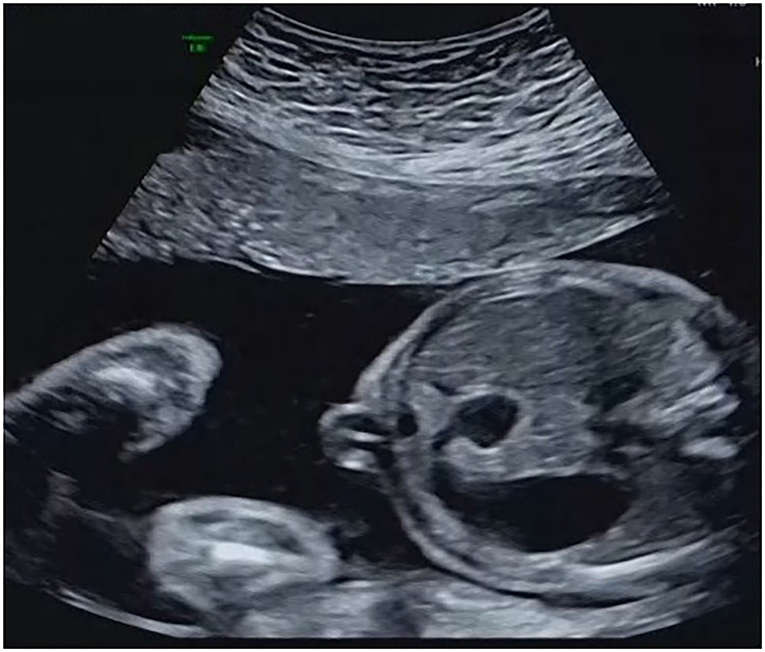
Figure 1. The presence of the “double-bubble” sign.
FIGURE 2
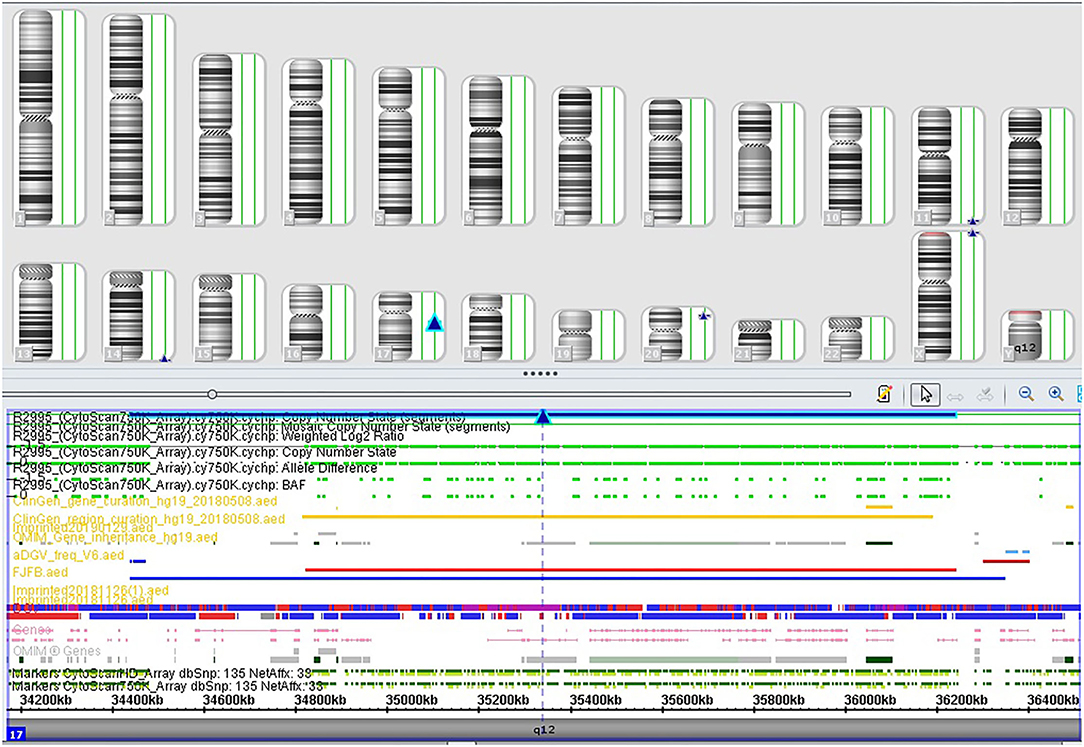
Figure 2. SNP array profile of Case 3. The image indicates a 1.8 Mb duplication on 17q12 (34,440,088_36,243,365).
Clinical Follow-Up
Follow-up information was obtained for 66 (94.3%) of the cases. The overall survival rate and TOP rates were 51.3 and 33.3%, respectively. Twenty-two of the cases ended in pregnancy termination, and three were stillbirth. Among the 41 retained pregnancies, three cases had normal MRI and no gastrointestinal obstruction syndrome after birth; 37 neonates underwent surgery operation, but three of them died within 6 months because of other complications, and 34 of them had normal development during the follow-up period; the remaining case (case 13) died 10 days after birth owing to anal atresia and cardiac malformation. The details are presented in Table 4. The survival rate in the isolated group was significantly higher than that in the non-isolated group (67.6 vs. 34.4%, p < 0.05), and duodenal atresia/stenosis had the highest survival rate (61.9%) among the other levels of GITO.
TABLE 4

Table 4. Follow-up information for 66 pregnancies with GITO.
Discussion
Consistent with most studies (18), almost all cases of fetal digestive tract atresia/stenosis were diagnosed in the second and third trimesters. Owing to late gestational age at diagnosis, GITO is a difficult condition for clinicians to treat. In general, once fetal GITO is suspected, chromosomal analysis and thorough ultrasound examination are strongly recommended for fetal prognosis assessment and pregnancy management.
Most previous reports focused on cytogenetic aberrations, with frequencies ranging from 5.4 to 20% (1, 2, 5, 15, 16, 19, 20). The association between GITO and aneuploidies, such as T21 and T18, has been well-established. In our study, the traditional karyotyping identified chromosomal abnormalities in 21.4% of the cases, including11 cases of T21 and a single case of 22q11.2 partial tetrasomy. Among them, 10 cases of T21 were observed in fetuses with duodenal stenosis, accounting for 23.3% of all the pregnancies with duodenal stenosis, which is considerably lower than the proportion reported by Tonni et al. (5) and Bethell et al. (3). This is probably attributed to improvements in the quality of ultrasound evaluation, indicating T21, and non-invasive prenatal testing (NIPT) that screen T21 with high specificity and sensitivity and detect it earlier. Consequently, more cases of pregnancy with identified T21 have been terminated before GITO could be detected by ultrasound scans. Esophageal atresia/stenosis is more frequently reported to be associated with T18 (2, 11), and this association may be explained by the decreased cholesterol synthesis observed in a series of neonates and fetuses with T18 (21, 22). Owing to the small size of the sample, our data could not confirm this relationship, but only one case of T21 was noted in a fetus with esophageal atresia/stenosis that was accompanied by soft markers. Among the different levels of GITO, jejunum and ileum atresia/stenosis is known to have the lowest risk of chromosomal anomalies (1, 23). In this study, no abnormalities were detected in the jejunum or ileum atresia/stenosis. Overall, our results further support the close association of DS and GITO, especially in cases of duodenal and esophageal stenoses. Efforts have been made to identify the contributing factors of congenital defects among Down Syndrome fetuses (24, 25), but the exact mechanism has yet to be explained. In addition to T21, a rare aberration was noted in a fetus with anal atresia, which has been described in our previous case report (26). The CES derived from duplicated regions of 22pter-22q11.2 is characterized clinically by the combination of coloboma of the iris and multiple malformations. Anal atresia is one of the most common gastrointestinal malformations, occurring in approximately 73%−81% of CES cases (27), but it is rarely detected in fetuses with anal atresia probably because of the difficulty in identifying prenatal anal atresia. Confirmation of CES requires the assistance of molecular methodologies such as FISH or CMA.
With the wide application of CMA in prenatal diagnosis, some pathogenic CNVs have been identified in fetal GITO (28, 29). It is well-known that CMA produces a significant yield of clinically relevant abnormalities in pregnancies with various ultrasonographic anomalies and normal karyotype (30, 31). In this study, we observed an additional 5.9% of pathogenic aberration by SNP array in pregnancies with duodenal atresia/stenosis, which is lower than that reported by Bishop et al. (32) and Zhang et al. (33). The divergence might be attributed to different sample sizes. The only CNV in our study showed a 1.8-Mb microduplication at 17q12 that overlapped with the critical region of 17q12 duplication syndrome (OMIM # 614526). The HNF1B gene in 17q12 is a transcription factor that is expressed during endoderm and mesoderm development, and it plays an important role in organ differentiation of the genitourinary and gastrointestinal tracts. Mutation in the HNF1B gene might be responsible for the range of phenotypes because of its essential role in early human GI development (34, 35). The syndrome has been regarded as a pathogenic variant with an incomplete penetrance of 22.1% (36, 37), which explained why the mother harbored the same aberration but had a normal phenotype.
Fetal GITO often coexists with other ultrasound abnormalities. Polyhydramnios is considered a common development secondary to gastrointestinal obstruction, and it may be the result of impaired swallowing. In our cohort, polyhydramnios was noted in 41.4% of the cases, much lower than the frequency reported in most previous publications. This difference may be explained by the different timing of ultrasound scans. Similar to that reported by Orgul et al. (15) and Haeusler et al. (1), associated structural abnormalities were observed in 21.4% of the cohort. Among all the associated malformations, cardiac anomalies were believed to be the primary cause of mortality post-birth (38). In our study, eight fetuses had cardiac anomalies; of them, six revealed chromosomal abnormalities (case 4–6, 10, and 12–13, Table 3) and ended in TOP, one fetus with normal karyotype died 6 months after the surgery procedure, and for the rest, one with normal karyotype was terminated because of multiple cardiac malformations. Therefore, in addition to genetic evaluation, a detailed ultrasound scan is of great impact on pregnancy outcome.
In terms of pregnancy outcomes, consistent with most previous research studies, isolated GITO had a lower rate of TOP and a higher rate of normal development and survival than non-isolated GITO. The survival rates of fetuses with GITO differed in different studies (9, 15). We observed that 51.5% of the fetuses had normal development after surgery operations, similar to that found in the report by Gokcen et al. (15). It is generally acknowledged that prenatal detection of GITO usually depends on indirect ultrasound and/or MRI findings, but with inaccuracy in some cases. In our study, three cases were healthy at birth, suggesting misdiagnosis during the prenatal period. It is worth noting that the pregnancies with duodenal atresia/stenosis showed the highest rate of chromosomal abnormalities in this study, but that they also had the highest survival rate. It is mainly because the majority of them were cases of isolated duodenal atresia/stenosis, and this further highlighted the importance of associated ultrasound findings on the pregnancy outcome of GITO.
The limitation of our study is the small sample size and the fact that CMA was not performed on all the fetuses. Thus, the data may not be convincing enough. Future multiple center studies with a large sample size will hopefully lead to a better understanding of the genetic disorders and outcomes of GITO.
In conclusion, our experience confirmed the strong association between Down syndrome and fetal GITO, especially duodenal stenosis. CMA can effectively increase the detection rate of chromosomal aberrations. When fetal GITO is suspected, associated chromosomal abnormalities and fetal anatomy should be carefully evaluated for pregnancy management.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: accession number GSE203126, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE203126.
Ethics Statement
The studies involving human participants were reviewed and approved by Protection of Human Ethics Committee of Fujian Provincial Maternity and Children's Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
XW and LS prepared the original draft. QS, QG, and YL prepared the experiment. SX and NL performed data analysis. LX and HH revised the initial manuscript. All authors have reviewed and approved the final article.
Funding
This study was supported by the Science Foundation of the Fujian Province, China (Grant No. 2021J01413) and Joint Funds for The Innovation of Science and Technology, Fujian Province (Grant Nos. 2020Y9135 and 2020Y9159).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Xiaorui Xie, Meiying Cai, and Bin Liang for their technical support on the SNP-array analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.918130/full#supplementary-material
References
1. Haeusler MC, Berghold A, Stoll C, Barisic I, Clementi M, Group ES. Prenatal ultrasonographic detection of gastrointestinal obstruction: results from 18 European congenital anomaly registries. Prenat Diagn. (2002) 22:616–23. doi: 10.1002/pd.341
2. Lupo PJ, Isenburg JL, Salemi JL, Mai CT, Liberman RF, Canfield MA, et al. Population-based birth defects data in the United States, 2010-2014: a focus on gastrointestinal defects. Birth Defects Res. (2017) 109:1504–14. doi: 10.1002/bdr2.1145
3. Bethell GS, Long AM, Knight M, Hall NJ. Congenital duodenal obstruction in the UK: a population-based study. Arch Dis Child Fetal Neonatal Ed. (2020) 105:178–83. doi: 10.1136/archdischild-2019-317085
4. Virgone C, D'antonio F, Khalil A, Jonh R, Manzoli L, Giuliani S. Accuracy of prenatal ultrasound in detecting jejunal and ileal atresia: systematic review and meta-analysis. Ultrasound Obstet Gynecol. (2015) 45:523–9. doi: 10.1002/uog.14651
5. Tonni G, Grisolia G, Granese R, Giacobbe A, Napolitano M, Passos JP, et al. Prenatal diagnosis of gastric and small bowel atresia: a case series and review of the literature. J Matern Fetal Neonatal Med. (2016) 29:2753–61. doi: 10.3109/14767058.2015.1107902
6. Nichol PF, Reeder A, Botham R. Humans, mice, and mechanisms of intestinal atresias: a window into understanding early intestinal development. J Gastrointest Surg. (2011) 15:694–700. doi: 10.1007/s11605-010-1400-y
7. Kirillova IA, Kulazhenko VP, Lurie IW, Ljazjuk GI. Atresia, stenosis and duplication of the gastro-intestinal tract: consideration of their origin. Acta Morphol Hung. (1984) 32:9–21.
8. Jong E, Felix JF, Klein AD, Tibboel D. Etiology of esophageal atresia and tracheoesophageal fistula: “mind the gap”. Curr Gastroenterol Rep. (2010). 12:215–22. doi: 10.1007/s11894-010-0108-1
9. Lau PE, Cruz S, Cassady CI, Mehollin-Ray AR, Ruano R, Keswani S, et al. Prenatal diagnosis and outcome of fetal gastrointestinal obstruction. J Pediatr Surg. (2017) 52:722–5. doi: 10.1016/j.jpedsurg.2017.01.028
10. Lawrence MJ, Ford WD, Furness ME, Hayward T, Wilson T. Congenital duodenal obstruction: early antenatal ultrasound diagnosis. Pediatr Surg Int. (2000) 16:342–5. doi: 10.1007/s003839900322
11. Brantberg A, Blaas HG, Haugen SE, Eik-Nes SH. Esophageal obstruction-prenatal detection rate and outcome. Ultrasound Obstet Gynecol. (2007) 30:180–7. doi: 10.1002/uog.4056
12. Schwarz KB, Haber BH, Rosenthal P, Mack CL, Magee JC. Extrahepatic anomalies in infants with biliary atresia: results of a large prospective North American multicenter study. Hepatology. (2013) 58:1724–31. doi: 10.1002/hep.26512
13. Brosens E, De Jong EM, Barakat TS, Eussen BH, D' Haene B, De Baere E, et al. Structural and numerical changes of chromosome X in patients with esophageal atresia. Eur J Hum Genet. (2014) 22:1077–84. doi: 10.1038/ejhg.2013.295
14. Fe Lix JF, Tibboel D, Klein AD. Chromosomal anomalies in the aetiology of oesophageal atresia and tracheo-oesophageal fistula. Eur J Med Genet. (2007) 50:163–75. doi: 10.1016/j.ejmg.2006.12.004
15. Orgul G, Soyer T, Yur Da Kok M, Beksac M. Evaluation of pre- and postnatally diagnosed gastrointestinal tract obstructions. J Matern Fetal Neonatal Med. (2019) 32:3215–20. doi: 10.1080/14767058.2018.1460350
16. Best KE, Tennant P, Addor MC, Bianchi F, Rankin J. Epidemiology of small intestinal atresia in Europe: a register-based study. Arch Dis Child Fetal Neonatal Ed. (2012). 97:F353–8. doi: 10.1136/fetalneonatal-2011-300631
17. South ST, Lee C, Lamb AN, Higgins AW, Kearney HM. ACMG standards and guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: revision 2013. Genet Med. (2013) 15:901–9. doi: 10.1038/gim.2013.129
18. Tsukerman GL, Krapiva GA, Kirillova IA. First-trimester diagnosis of duodenal stenosis associated with oesophageal atresia. Prenat Diagn. (2010) 13:371–6. doi: 10.1002/pd.1970130509
19. Galani A, Zikopoulos A, Papandreou L, Mastora E, Zikopoulos K, Makrydimas G. Prenatal diagnosis of fetal jejunal atresia: a case report. Cureus. (2021) 13:e18947. doi: 10.7759/cureus.18947
20. Bethell GS, Long AM, Knight M, Hall NJ, Baps C. The impact of trisomy 21 on epidemiology, management, and outcomes of congenital duodenal obstruction: a population-based study. Pediatr Surg Int. (2020) 36:477–83. doi: 10.1007/s00383-020-04628-w
21. Cooper MK, Wassif CA, Krakowiak PA, Taipale J, Gong R, Kelley RI, et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet. (2003) 33:508–13. doi: 10.1038/ng1134
22. Lam WW, Kirk J, Manning N, Reardon W, Kelley RI, Fitzpatrick D. Decreased cholesterol synthesis as a possible aetiological factor in malformations of trisomy 18. Eur J Med Genet. (2006) 49:195–9. doi: 10.1016/j.ejmg.2005.05.011
23. Forrester MB, Merz RD. Population-based study of small intestinal atresia and stenosis, Hawaii, 1986-2000. Public Health. (2004) 118:434–8. doi: 10.1016/j.puhe.2003.12.017
24. Patterson D. Genetic mechanisms involved in the phenotype of Down syndrome. Ment Retard Dev Disabil Res Rev. (2007) 13:199–206. doi: 10.1002/mrdd.20162
25. Pritchard M, Reeves RH, Dierssen M, Patterson D, Gardiner KJ. Down syndrome and the genes of human chromosome 21: current knowledge and future potentials. Report on the Expert workshop on the biology of chromosome 21 genes: towards gene-phenotype correlations in Down syndrome. Washington D.C., September 28-October 1, 2007. Cytogenet Genome Res. (2008) 121:67–77. doi: 10.1159/000124384
26. Wu X, An G, He D, Shen Q, Cai M, Huang H, et al. [Prenatal diagnosis and clinical analysis of two fetuses with Cat-eye syndrome]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2019) 36:498–501. doi: 10.3760/cma.j.issn.1003-9406.2019.05.021
27. Rosias PR, Sijstermans JM, Theunissen PM, Pulles-Heintzberger CF, De Die-Smulders CE, Engelen JJ, et al. Phenotypic variability of the cat eye syndrome. Case report and review of the literature. Genet Couns. (2001) 12:273–82.
28. Meng X, Jiang L. Prenatal detection of chromosomal abnormalities and copy number variants in fetuses with congenital gastrointestinal obstruction. BMC Pregnancy Childbirth. (2022) 22:50. doi: 10.1186/s12884-022-04401-y
29. Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, et al. Genomic and genic deletions of the FOX gene cluster on 16q241 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. (2009) 84:780–91. doi: 10.1016/j.ajhg.2009.05.005
30. Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. (2012) 367:2175–84. doi: 10.1056/NEJMoa1203382
31. Jelin A, Sagaser K, Lawson C, Forster KR, Leppert K, Hertenstein C, Blakemore K., et al. Chromosomal microarray analysis results from pregnancies with various ultrasonographic anomalies. Obstet Gynecol. (2019) 133:827–8. doi: 10.1097/AOG.0000000000003207
32. Ther FD, Bishop JC, Mccormick B, Johnson CT, Miller J, Jelin E, et al. The double bubble sign: duodenal atresia and associated genetic etiologies. Fetal Diagn Ther. (2019) 47:1–6. doi: 10.1159/000500471
33. Zhang W, Lei T, Fu F, Deng Q, Li R, Wang D, et al. Microarray analysis in fetuses with duodenal obstruction: it is not just trisomy 21. Prenat Diagn. (2021) 41:316–22. doi: 10.1002/pd.5834
34. Zhang W, Du K, Fu F, Li R, Zhang Y, Jing X, et al. [Value of chromosomal microarray analysis for fetuses with duodenal obstruction]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2021) 38:210–3.
35. Faguer S, Chassaing N, Bandin F, Prouheze C, Arveiler B, Rooryck C, et al. A 17q12 chromosomal duplication associated with renal disease and esophageal atresia. Eur J Med Genet. (2011) 54:e437–40. doi: 10.1016/j.ejmg.2011.03.010
36. Bierhals T, Maddukuri SB, Kutsche K, Girisha KM. Expanding the phenotype associated with 17q12 duplication: case report and review of the literature. Am J Med Genet A. (2013) 161A:352–9. doi: 10.1002/ajmg.a.35730
37. Rosenfeld JA, Coe BP, Eichler EE, Cuckle H, Shaffer LG. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet Med. (2013) 15:478–81. doi: 10.1038/gim.2012.164
Keywords: gastrointestinal tract obstructions, traditional karyotyping, chromosomal abnormalities, chromosomal microarray analysis, Down syndrome, copy number variants
Citation: Wu X, Su L, Shen Q, Guo Q, Li Y, Xu S, Lin N, Huang H and Xu L (2022) Chromosomal Abnormalities and Pregnancy Outcomes for Fetuses With Gastrointestinal Tract Obstructions. Front. Pediatr. 10:918130. doi: 10.3389/fped.2022.918130
Received: 12 April 2022; Accepted: 09 May 2022;
Published: 06 June 2022.
Edited by:
Antonio Percesepe, University of Parma, ItalyReviewed by:
Gabriele Tonni, IRCCS Local Health Authority of Reggio Emilia, ItalyEmilia Severin, Carol Davila University of Medicine and Pharmacy, Romania
Copyright © 2022 Wu, Su, Shen, Guo, Li, Xu, Lin, Huang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liangpu Xu, eGlsaWFuZ3B1QGZqbXUuZWR1LmNu; Hailong Huang, aHVhbmdoYWlsb25nQGZqbXUuZWR1LmNu; Na Lin, ODQ2NTE5NDY1QHFxLmNvbQ==