
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 07 July 2022
Sec. Pediatric Nephrology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.915174
 Rachel K. Cason1
Rachel K. Cason1 Anna Williams1
Anna Williams1 Megan Chryst-Stangl1
Megan Chryst-Stangl1 Guanghong Wu1Kinsie Huggins1Kaye E. Brathwaite2
Guanghong Wu1Kinsie Huggins1Kaye E. Brathwaite2 Brandon M. Lane1Larry A. Greenbaum3Vivette D. D’Agati4
Brandon M. Lane1Larry A. Greenbaum3Vivette D. D’Agati4 Rasheed A. Gbadegesin1*
Rasheed A. Gbadegesin1*Background: Focal segmental glomerulosclerosis (FSGS) is a major cause of end stage kidney disease, with the collapsing form having the worst prognosis. Study of families with hereditary FSGS has provided insight into disease mechanisms.
Methods: In this report, we describe a sibling pair with NUP93 mutations and collapsing FSGS (cFSGS). For each brother, we performed next generation sequencing and segregation analysis by direct sequencing. To determine if the variants found in the index family are a common cause of cFSGS, we screened 7 patients with cFSGS, gleaned from our cohort of 200 patients with FSGS, for variants in NUP93 as well as for APOL1 high-risk genotypes.
Results: We identified segregating compound heterozygous NUP93 variants (1) c.1772G > T p.G591V, 2) c.2084T > C p.L695S) in the two brothers. We did not find any pathogenic variants in the seven patients with cFSGS from our cohort, and as expected five of these seven patients carried the APOL1 high-risk genotype.
Conclusion: To the best of our knowledge, this is the first report of cFSGS in patients with NUP93 mutations, based on this report, mutations in NUP93 and other nucleoporin genes should be considered when evaluating a child with familial cFSGS. Determining the mechanisms by which these variants cause cFSGS may provide insight into the pathogenesis of the more common primary and virus-mediated forms of cFSGS.
Nephrotic syndrome (NS) is a major cause of chronic kidney disease (CKD) in children, and it is the most common glomerular disease encountered in children with an overall incidence of 2.92 per 100,000 children (1). Steroid Resistant NS (SRNS), defined as failure to achieve remission following 4–6 weeks of standard corticosteroid therapy, is seen in about 20% of all cases of childhood NS (2). SRNS is the most severe form of childhood NS, and it is a major cause of end stage kidney disease (ESKD) in children. The cause of SRNS is often unknown; however genetic studies have shown that 10–30% of SRNS cases are monogenic disease due to pathogenic variants in one of more than 60 genes (3). All of these causative genes are expressed in the podocyte, leading to podocyte dysfunction; hence the term “podocytopathy” has been applied to these disorders.
Nucleoporins (Nups) are proteins associated with the nuclear pore complex (NPC) in a variety of kidney cells. They are important in cellular molecular trafficking of proteins, RNA, and other molecules between the cytoplasm and the nucleoplasm (4). Pathogenic variants in NUP genes have been reported as causes of SRNS, with histologic changes including minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS) (4). While pathogenic variants have been reported to cause different morphologic types of FSGS, to the best of our knowledge, mutations in NUP93 and other nucleoporin genes have not been reported previously as a cause of collapsing FSGS (cFSGS) (5). Other genes that have been identified as cause of cFSGS include ACTN4, COQ2, SCARB2, and TRPC6 (6–9). Collapsing FSGS is characterized by retraction and collapse of the glomerular capillaries associated with hypertrophy and hyperplasia of the overlying glomerular epithelial cells; it portends the worst prognosis among histologic subtypes of FSGS (10, 11). This subtype was first described in patients with HIV-associated nephropathy and has been seen in association with other viral infections such as SARS-CoV-2, parvovirus B19 and cytomegalovirus (12–15). cFSGS has also been documented to occur after treatment with certain medications, such as bisphosphonates and interferon, and in association with systemic diseases, such as systemic lupus erythematosus and thrombotic microangiopathy (16–19). Here, we have identified a white sibling pair with cFSGS due to compound heterozygous mutations in NUP93. We describe their clinical course and screen other patients with cFSGS in our cohort for these variants. We also determine the impact of the pathogenic variants on the three-dimensional (3D) structure of the nucleoporin 93 (NUP93) protein and the potential implications of the changes for the more common primary and infection-mediated cFSGS.
The Institutional Review Board of the Duke University Medical Center (Durham, NC, United States) approved the study. Written informed consent/assent was obtained from all participants prior to inclusion in the study. The methods of subject recruitment have been previously reported (15). Inclusion criteria for the study were (1) proteinuria, (2) biopsy proven MCD or FSGS. Clinical data obtained included family history, age at onset of disease, age at end stage kidney disease, age at kidney transplantation and history of disease recurrence following kidney transplantation.
We obtained DNA from the affected brothers and their unaffected parents and performed whole genome sequencing in the two affected siblings. Whole genome sequencing was performed by Veritas Genetics (Danvers, MA). Samples were sequenced on the HiSeq X with a 2 × 150 bp paired-end sequencing configuration. Variant calling, annotation, and variants filtering were carried out using previously published standard methods (15). All variants of interest including APOL1 G0, G1, and G2 were confirmed by direct sequencing. In silico 3D modeling of NUP93 peptides was carried out as previously described (15).
Patient 1: A 5-year-old white male of European descent presented with asymptomatic proteinuria. He was noted to have nephrotic range proteinuria (urine protein/creatinine ratio 17 mg/mg). He was unresponsive to 6 weeks of corticosteroid therapy and kidney biopsy showed cFSGS (Figure 1A). He had negative testing for HIV and parvovirus B19. He was treated with the calcineurin inhibitor (CNI) cyclosporine for 3 months before progressing to end stage kidney disease (ESKD) at the age of 6 years. He received a living unrelated kidney transplantation at the age of 7 years. He did not develop recurrence of FSGS in his kidney transplant as demonstrated on two kidney allograft biopsies. He continued to have a functioning kidney allograft after more than 10 years of follow-up. The patient’s clinical characteristics are shown in Table 1.
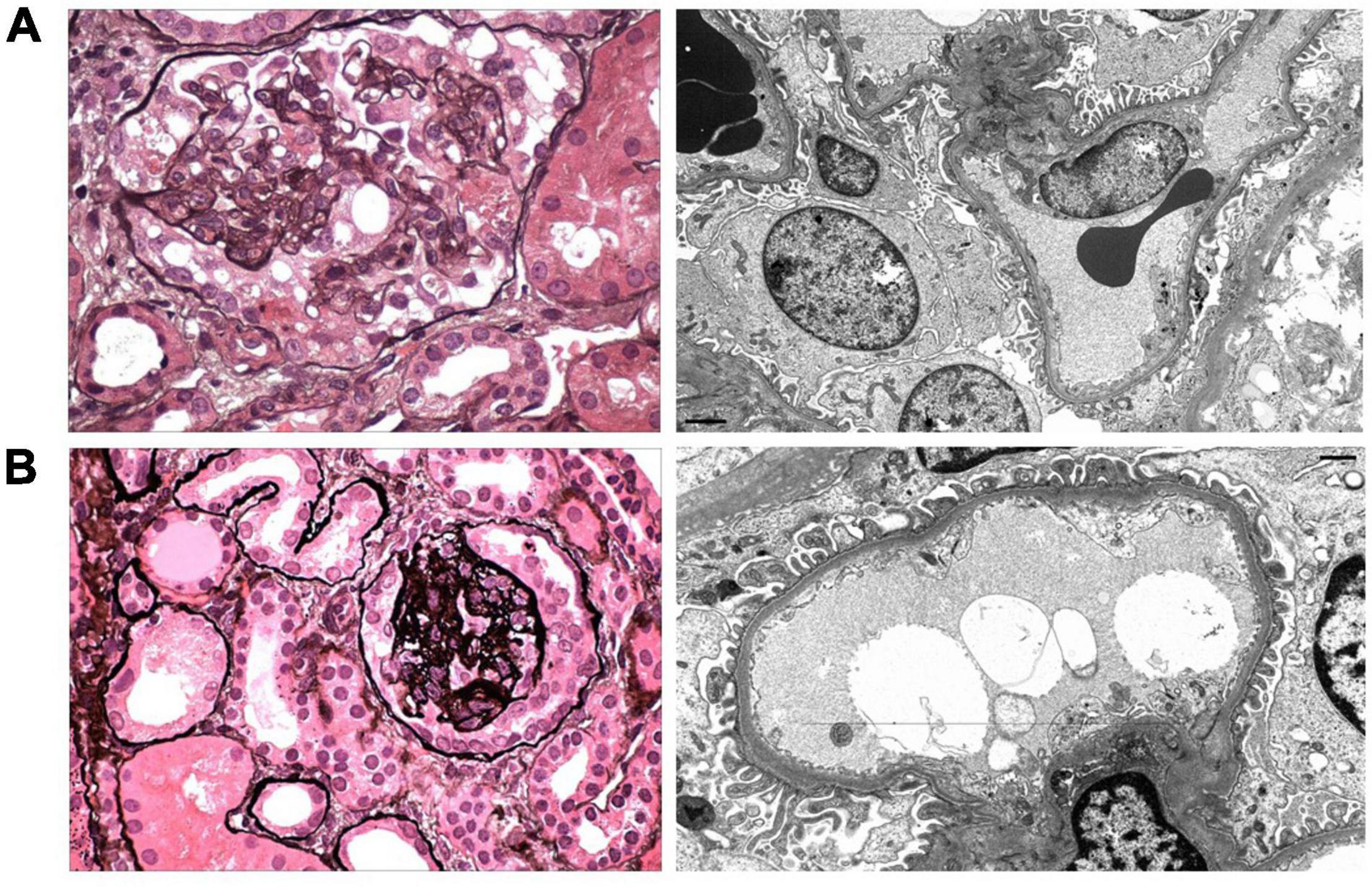
Figure 1. Representative images of the kidney biopsies from Patient 1 (A) and Patient 2 (B) are shown. (A) A representative glomerulus has segmental collapsing sclerosis with implosive wrinkling and collapse of glomerular capillary walls and hypertrophy and hyperplasia of the overlying glomerular epithelial cells (Jones methenamine silver, ×600). Electron microscopy shows irregular (40%) foot process effacement with swollen podocyte cell bodies and open-appearing cytoplasm with reduced organellar content (× 6,000). (B) There is focal global tuft collapse with a corona of hyperplastic glomerular epithelial cells and focal podocyte shedding and apoptosis (Jones methenamine silver, ×600). Electron microscopy reveals mild (25%) foot process effacement (×10,000).
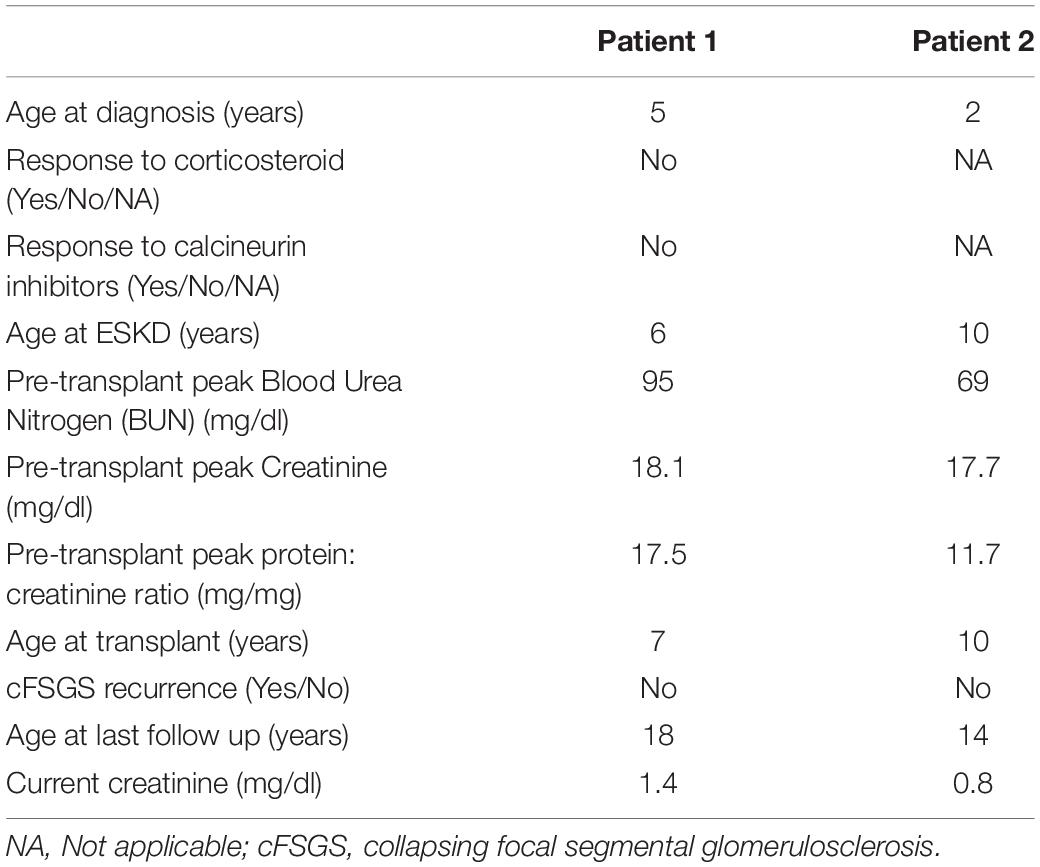
Table 1. Clinical characteristics of siblings with NUP93 mutations.
Patient 2: Patient 2 is the younger brother of Patient 1. He was screened for proteinuria at the age of 2 years because of history of FSGS in his older brother, and he was found to have asymptomatic nephrotic range proteinuria (urine protein/creatinine ratio of 3.2 mg/mg). Kidney biopsy showed collapsing FSGS (Figure 1B). He had negative testing for HIV as part of his transplant evaluation. He was treated with angiotensin converting enzyme inhibitor alone and did not receive immunomodulating agents because of suspicion for monogenic SRNS. He developed ESKD at the age of 10 years and underwent living related kidney transplantation from a maternal uncle shortly after developing ESKD. The donor was not screened for mutations in NUP93 since NUP93 had not been identified at that time as a cause of monogenic SRNS (both brothers had extensive genetic testing, but it did not include NUP93). He did not develop recurrence of FSGS in his kidney transplant as evidenced by four transplant kidney biopsies. He had normal kidney function and no proteinuria after 4 years of follow-up. Clinical characteristics for Patient 2 are shown in Table 1.
Patient 1: (Figure 1A) Kidney biopsy was performed at age 5 years. Among 33 glomeruli present, 13 had global glomerulosclerosis and 9 showed FSGS with predominantly collapsing features. Moderate tubular atrophy and interstitial fibrosis involved approximately 35% of the cortex. Glomerular tuft staining for IgM, C3, and C1q was observed in areas of sclerosis, consistent with non-specific trapping. Foot process effacement affected approximately 40% of the glomerular capillary surface area. The podocyte cell bodies appeared swollen with reduced organellar content.
Patient 2: (Figure 1B) Kidney biopsy was performed at age 2 years. Among 28 glomeruli sampled, 2 displayed global collapsing sclerosis with podocyte capping, hyperplasia, and focal apoptosis. Four glomeruli contained segmental lesions of sclerosis, 2 of which exhibited collapsing features. Podocytes of the remaining glomeruli appeared swollen. Tubular atrophy and interstitial fibrosis occupied 10% of the cortex, with prominent proximal tubular protein and lipid resorption droplets and early tubular microcyst formation. Sclerosing glomeruli contained deposits of IgM, C3, and C1q, with podocyte droplet staining for albumin. Foot process effacement involved approximately 25% of the glomerular capillary surface area.
We applied our standard filtering algorithm (Figure 2) (20) and identified segregating compound heterozygous variants (1) c.1772G > T p.G591V, (2) c.2084T > C p.L695S in NUP93 in the two brothers (Figure 3A). Both variants, which occur in evolutionarily conserved amino acid residues, are rare with a minor allele frequency of < 0.00015 in the gnomAD database. Neither variant is present in biallelic (homozygous) state in any public database and both are predicted to be damaging by four in silico analysis tools (Figure 2). In addition we identified a variant in the gene MAP7D2 c.29C > T p.Thr10Met. MAP7D2 encodes for MAP7 domain containing 2 (Figure 2). Variants in MAP7D2 have not been previously reported as a cause of nephrotic syndrome and the function of the gene is unknown.
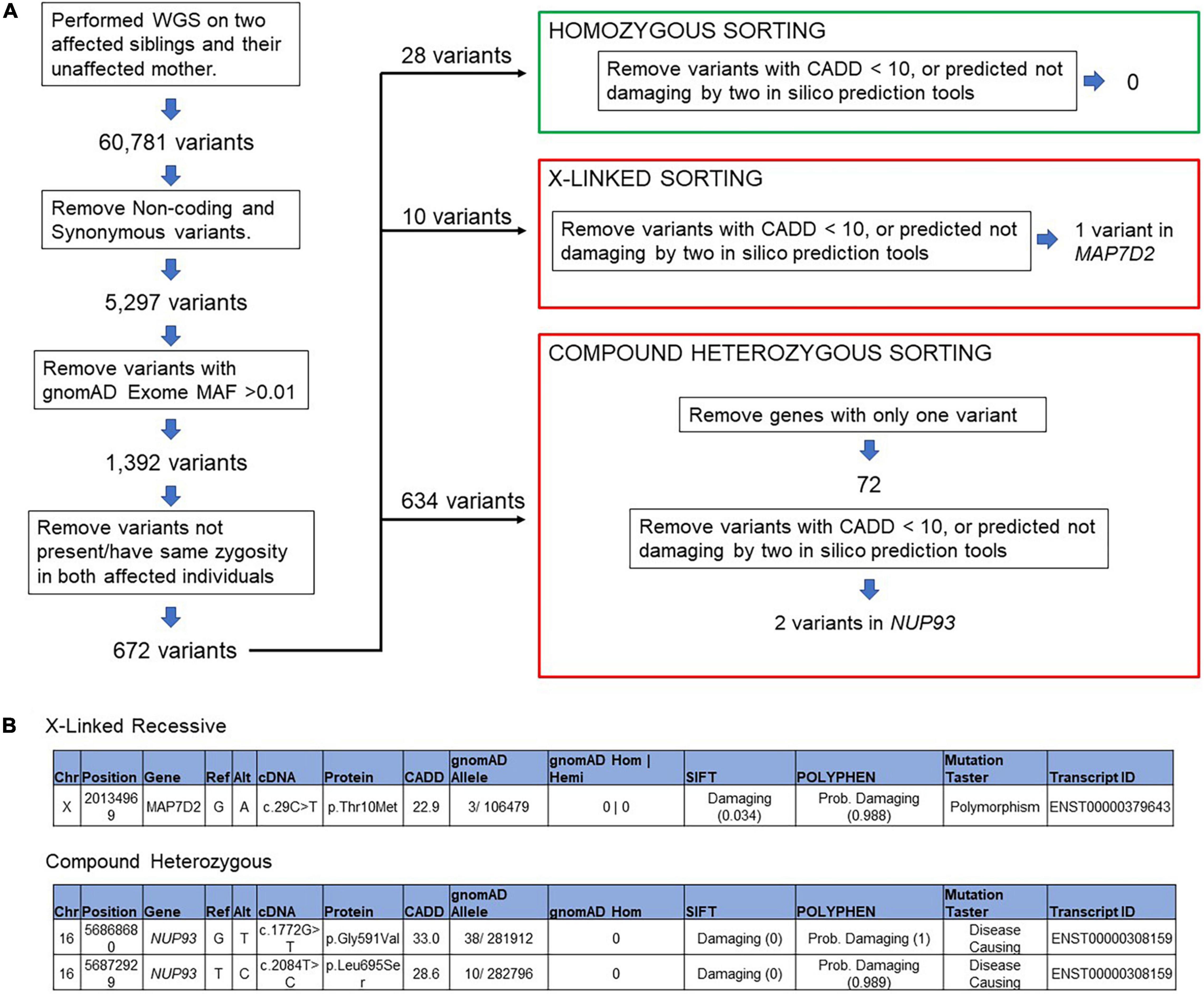
Figure 2. Filtering algorithm (A) variant filtering of the whole genome sequencing data in patient with collapsing FSGS, (B) segregating variants of unknown significant was identified in MAP7D2 and a compound heterozygous variant in NUP93.
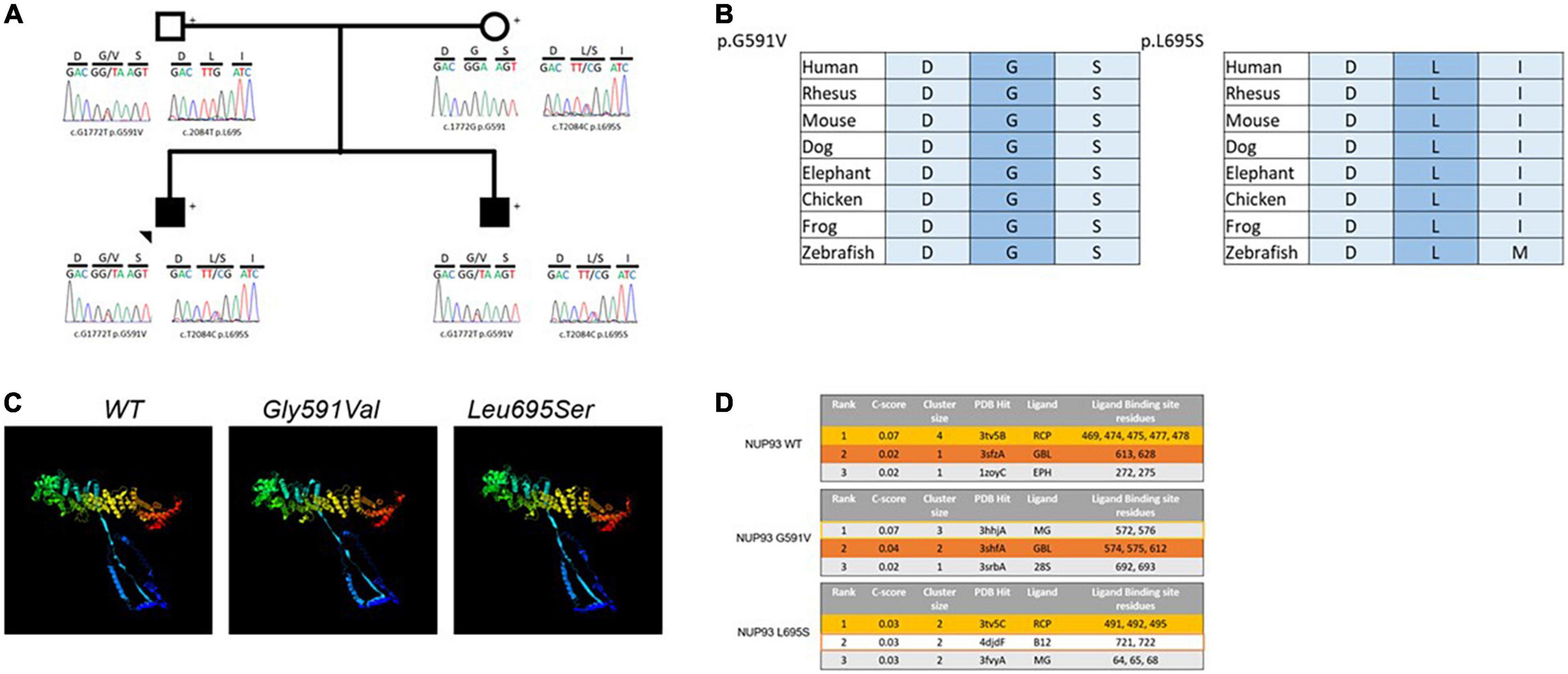
Figure 3. (A) Two rare variants in NUP93 gene segregate with disease in two brothers with cFSGS, (B) the two variants are conserved in evolution down to zebrafish, (C) the 3D structures for the wild type (WT) NUP93 protein (left), the G591V variant (middle) and the L695S variant (right) showing structural changes in the amino terminus (blue) to the carboxyl terminus (red) in the two variants compared with the WT, (D) the top predicted ligands for WT NUP93 (top), G591V (middle) and L695S (bottom). The top two predicted ligands for WT NUP93, Acetyl-coenzyme A carboxylase (RCP, yellow) and Apaf-1 (GBL, orange) are disrupted in the G591V variant and L695AS variant, respectively.
The variants are located in the carboxyl terminus of the NUP93 protein that is essential for nuclear pore formation. Additionally, in silico 3D modeling predicts that both variants will create structural alterations throughout the protein including the amino and the carboxyl terminal residues that will affect NUP93 ligand binding (Figures 3B–D).
In our cohort of over 200 patients with biopsy proven FSGS, we identified seven children from six families with cFSGS. The clinical characteristics of these seven children are shown in Table 2. To determine whether the variants we found in the index family and NUP93 variants are common cause of monogenic cFSGS, we screened these seven individuals for variants in the coding exons of NUP93 and did not find any patient with biallelic pathogenic variants in NUP93. Since six of the seven individuals are of African ancestry, we screened all seven patients for the high risk APOL1 genotype associated with CKD (21). Five of the seven individuals with cFSGS were found to have APOL1 high-risk genotypes G1/G1, or G1/G2. The other two had the G0/G0 APOL1 genotype. Two of the patients with the high-risk APOL1 genotype also had parvovirus B19 infection at the time of presentation. As expected, the two index patients with the NUP93 pathogenic variants had G0/G0 genotype for APOL1.

Table 2. APOL1 and NUP93 genotype in seven children with cFSGS.
Pathogenic variants in NUP93 have been reported in association with MCD and various morphologic subtypes of FSGS. To the best of our knowledge, cFSGS has not been previously reported in patients with pathogenic NUP93 variants; hence, our report expands the kidney phenotype associated with NUP93 mutations. The reason for our findings is unclear but since other NUP93 pathogenic variants have been associated with other types of FSGS and MCD, our observation is likely due to biallelic heterogeneity but we cannot rule out completely variable expressivity.
Nucleoporins have a critical role in regulating the transfer of signaling molecules between the cell nucleus and cytoplasm. NUP93 encodes a central component of the NUP93 complex of proteins that serves as a major structural backbone of the nuclear pore complex (NPC). The compound heterozygous variants found in this family are located in the carboxyl terminus of the NUP93 protein, a region often associated with both kidney and non-kidney disease-causing variants. This area of the protein is critical for assembly of the NUP93 complex as well as nuclear pore formation (22). Both the G591V and L695S variants are located in binding regions for another component of the NUP93 complex, NUP155, suggesting that formation of this biologically important complex may be disrupted in patients carrying both mutant alleles (23). Additionally, 3D modeling analysis predicts that these variants will result in disruption of NUP93 binding to key nucleoporin-related molecules. The G591V variant is expected to alter binding to acetyl-coenzyme A carboxylase, the most likely predicted ligand of NUP93 and a regulator of long chain fatty acid composition of the nuclear pore-membrane interface (24). The NUP93 ligand with the second highest prediction score, apoptotic protease activating factor 1 (Apaf-1), is expected to be disrupted by L695S. Apaf-1 is known to associate with nucleoporins during translocation between the cytoplasm and nucleus as it serves roles in apoptosis signaling as well as the DNA damage response (25). It is not currently known if these specific altered ligand interactions contribute to the pathogenesis of NUP93-mediated disease, but the use of predictive modeling has uncovered new potential mechanisms that warrant further investigation. Both the G591V and L695S variants have been reported as homozygous variants or as compound heterozygous variants in trans with other variants, but inheritance of both the G591V and L695S variants in trans with each other has not been previously reported (5). All the previous reports of patients with these variants have described non-collapsing FSGS or MCD phenotype on kidney biopsy (4, 26, 27).
Collapsing FSGS has been reported in patients with a variety of infections, including HIV, parvovirus B19, and more recently SARS-CoV-2 (12, 13, 28). It is also associated with use of medications like interferon-alpha, -beta, or -gamma and the bisphosphonate pamidronate (16, 17). Most patients with cFSGS secondary to infections or medication exposure are of African ancestry and carry high-risk APOL1 genotype (29). The index patients in this study did not have clinical evidence of any of these viral infections and they were not exposed to any of the medications associated with cFSGS. Also, they do not carry the high-risk genotype (homozygous G1 or G2 or compound heterozygous G1/G2) for APOL1 genes. To determine if the compound heterozygous variants identified in this family are a common cause of monogenic familial and sporadic cFSGS, we screened other patients with cFSGS in our cohort and did not identify any patients with this combination of variants, suggesting that these compound heterozygous variants are unique to this family. However, we confirmed that APOL1 high risk genotype is the most common risk factor for non-HIV associated cFSGS in our cohort, and our results confirmed previous reports by other Investigators (30). It is interesting to note that some patients with variants in NUP93 have been reported to have recurrence of FSGS following kidney transplantation, suggesting that mutations in NUP93 may cause disease by modulation of the immune cells (26). Our patients have been followed for 4–10 years after transplantation. They have had repeated kidney biopsies during the follow up and neither of them has been diagnosed with disease recurrence. In addition, both patients have functioning grafts and no proteinuria at the last follow-up.
We report a family with cFSGS due to compound heterozygous mutation in NUP93, thereby expanding the spectrum of kidney phenotypes associated with genetic defects in NUP93. As the morphogenesis of cFSGS is postulated to involve rapid rates of podocyte depletion, studies to determine the cellular mechanisms by which these variants cause cFSGS may provide new insights into the pathogenesis of the more common primary and virus-mediated forms of cFSGS.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Duke University IRB. Written informed consent was obtained from the individual(s) next of kin for the publication of any potentially identifiable images or data included in this article.
RG designed the experiments. RG, RC, KB, AW, KH, BL, LG, and VD’A wrote the manuscript. RG, RC, AW, MC-S, KH, GW, and LG performed subjects’ enrollment and sample acquisition. MC-S, BL, KH RC, and RG carried out sequencing, analysis of sequencing data and in silico modeling. VD’A carried out renal histology and electron microscopy reading and interpretation. All authors reviewed and edited the manuscript.
RG was supported by the National Institutes of Health/National Institutes of Diabetes and Digestive and Kidney Diseases (NIH/NIDDK) (grants 5R01DK098135 and 5R01DK094987), NIH/NICHD (1R21HD104176-01), Doris Duke Charitable Foundation Clinical Scientist Development (Award 2009033), Borden Scholars award, and Duke Health Scholars award.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge all the participants in the study. We appreciate the technical support provided by the Duke Molecular Physiology Institute (DMPI) genomics core and the personnel of Duke Center for Genomic and Computational Biology (GCB).
FSGS, Focal segmental glomerulosclerosis; cFSGS, collapsing FSGS; NS, Nephrotic syndrome; SRNS, Steroid Resistant Nephrotic Syndrome; CKD, chronic kidney disease; ESKD, end stage kidney disease; NUPs, Nucleoporins; NPC, Nuclear Pore Complex; MCD, Minimal Change Disease; CNI, calcineurin inhibitor; Apaf-1, apoptotic protease activating factor 1.
1. Veltkamp F, Rensma LR, Bouts A, Learns consortium. Incidence and relapse of idiopathic nephrotic syndrome: meta-analysis. Pediatrics. (2021) 148:e2020029249. doi: 10.1542/peds.2020-029249
2. No Author. The primary nephrotic syndrome in children. identification of patients with minimal change nephrotic syndrome from initial response to prednisone. A report of the international study of kidney disease in children. J Pediatr. (1981) 98:561–4. doi: 10.1016/s0022-3476(81)80760-3
3. Bensimhon AR, Williams AE, Gbadegesin RA. Treatment of steroid-resistant nephrotic syndrome in the genomic era. Pediatr Nephrol. (2019) 34:2279–93. doi: 10.1007/s00467-018-4093-1
4. Braun DA, Sadowski CE, Kohl S, Lovric S, Astrinidis SA, Pabst WL, et al. Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet. (2016) 48:457–65. doi: 10.1038/ng.3512
5. Pei Y, Rong L, Jiang M, Lin Z, Cheng C, Chen L, et al. The New Compound Heterozygous Mutation of NUP Nephropathy: Report of Two Cases and Literature Review. (2022). Available online at: https://www.researchsquare.com/article/rs-238609/v1 (accessed January 06, 2022)
6. Feng D, Steinke JM, Krishnan R, Birrane G, Pollak MR. Functional validation of an alpha-actinin-4 mutation as a potential cause of an aggressive presentation of adolescent focal segmental glomerulosclerosis: implications for genetic testing. PLoS One. (2016) 11:e0167467. doi: 10.1371/journal.pone.0167467
7. Barisoni L, Diomedi-Camassei F, Santorelli FM, Caridi G, Thomas DB, Emma F, et al. Collapsing glomerulopathy associated with inherited mitochondrial injury. Kidney Int. (2008) 74:237–43. doi: 10.1038/sj.ki.5002767
8. Berkovic SF, Dibbens LM, Oshlack A, Silver JD, Katerelos M, Vears DF, et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet. (2008) 82:673–84. doi: 10.1016/j.ajhg.2007.12.019
9. Gigante M, Caridi G, Montemurno E, Soccio M, d’Apolito M, Cerullo G, et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. (2011) 6:1626–34. doi: 10.2215/CJN.07830910
10. D’Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. (2011) 365:2398–411. doi: 10.1056/NEJMra1106556
11. Thomas DB, Franceschini N, Hogan SL, Ten Holder S, Jennette CE, Falk RJ, et al. Clinical and pathologic characteristics of focal segmental glomerulosclerosis pathologic variants. Kidney Int. (2006) 69:920–6. doi: 10.1038/sj.ki.5000160
12. Kudose S, Santoriello D, Bomback AS, Sekulic M, Batal I, Stokes MB, et al. Longitudinal outcomes of COVID-19-associated collapsing glomerulopathy and other podocytopathies. J Am Soc Nephrol. (2021) 32:2958–69. doi: 10.1681/ASN.2021070931
13. Moudgil A, Nast CC, Bagga A, Wei L, Nurmamet A, Cohen AH, et al. Association of parvovirus B19 infection with idiopathic collapsing glomerulopathy. Kidney Int. (2001) 59:2126–33. doi: 10.1046/j.1523-1755.2001.00727.x
14. Tomlinson L, Boriskin Y, McPhee I, Holwill S, Rice P. Acute cytomegalovirus infection complicated by collapsing glomerulopathy. Nephrol Dia Transplant. (2003) 18:187–9. doi: 10.1093/ndt/18.1.187
15. D’Agati V, Suh JI, Carbone L, Cheng JT, Appel G. Pathology of HIV-associated nephropathy: a detailed morphologic and comparative study. Kidney Int. (1989) 35:1358–70. doi: 10.1038/ki.1989.135
16. Markowitz GS, Appel GB, Fine PL, Fenves AZ, Loon NR, Jagannath S, et al. Collapsing focal segmental glomerulosclerosis following treatment with high-dose pamidronate. J Am Soc Nephrol. (2001) 12:1164–72. doi: 10.1681/ASN.V1261164
17. Markowitz GS, Nasr SH, Stokes MB, D’Agati VD. Treatment with IFN-{alpha}, -{beta}, or -{gamma} is associated with collapsing focal segmental glomerulosclerosis. Erratum Clin J Am Soc Nephrol. (2010) 5:607–15. doi: 10.2215/CJN.07311009
18. Larsen CP, Beggs ML, Saeed M, Walker PD. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J Am Soc Nephrol. (2013) 24:722–5. doi: 10.1681/ASN.2012121180
19. Buob D, Decambron M, Gnemmi V, Frimat M, Hoffmann M, Azar R, et al. Collapsing glomerulopathy is common in the setting of thrombotic microangiopathy of the native kidney. Kidney Int. (2016) 90:1321–31. doi: 10.1016/j.kint.2016.07.021
20. Lane BM, Chryst-Stangl M, Wu G, Shalaby M, El Desoky S, Middleton CC, et al. Steroid-sensitive nephrotic syndrome candidate gene CLVS1 regulates podocyte oxidative stress and endocytosis. JCI Insight. (2022) 7:e152102. doi: 10.1172/jci.insight.152102
21. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. (2010) 329:841–5. doi: 10.1126/science.1193032
22. Sachdev R, Sieverding C, Flötenmeyer M, Antonin W. The C-terminal domain of Nup93 is essential for assembly of the structural backbone of nuclear pore complexes. Mol Biol Cell. (2012) 23:740–9. doi: 10.1091/mbc.E11-09-0761
23. Zanni G, De Magistris P, Nardella M, Bellacchio E, Barresi S, Sferra A, et al. Biallelic variants in the nuclear pore complex protein NUP93 are associated with non-progressive congenital ataxia. Cerebellum. (2019) 18:422–32. doi: 10.1007/s12311-019-1010-5
24. Schneiter R, Hitomi M, Ivessa AS, Fasch EV, Kohlwein SD, Tartakoff AM. A yeast acetyl coenzyme A carboxylase mutant links very-long-chain fatty acid synthesis to the structure and function of the nuclear membrane-pore complex. Mol Cell Biol. (1996) 16:7161–72. doi: 10.1128/MCB.16.12.7161
25. Jagot-Lacoussiere L, Faye A, Bruzzoni-Giovanelli H, Villoutreix BO, Rain JC, Poyet JL. DNA damage-induced nuclear translocation of Apaf-1 is mediated by nucleoporin Nup107. Cell Cycle. (2015) 14:1242–51. doi: 10.1080/15384101.2015.1014148
26. Bezdíčka M, Štolbová Š, Seeman T, Cinek O, Malina M, Šimánková N, et al. Genetic diagnosis of steroid-resistant nephrotic syndrome in a longitudinal collection of Czech and Slovak patients: a high proportion of causative variants in NUP93. Pediatri Nephrol. (2018) 33:1347–63. doi: 10.1007/s00467-018-3950-2
27. Bierzynska A, McCarthy HJ, Soderquest K, Sen ES, Colby E, Ding WY, et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. (2017) 91:937–47. doi: 10.1016/j.kint.2016.10.013
28. Kudose S, Santoriello D, Bomback AS, Stokes MB, Batal I, Markowitz GS, et al. The spectrum of kidney biopsy findings in HIV-infected patients in the modern era. Kidney Int. (2020) 97:1006–16. doi: 10.1016/j.kint.2020.01.018
29. Kasembeli AN, Duarte R, Ramsay M, Mosiane P, Dickens C, Dix-Peek T, et al. APOL1 risk variants are strongly associated with HIV-associated nephropathy in black South Africans. J Am Soc Nephrol. (2015) 26:2882–90. doi: 10.1681/ASN.2014050469
Keywords: nephrotic syndrome, focal segmental glomerulosclerosis, collapsing FSGS, nucleoporin genes, APOL1
Citation: Cason RK, Williams A, Chryst-Stangl M, Wu G, Huggins K, Brathwaite KE, Lane BM, Greenbaum LA, D’Agati VD and Gbadegesin RA (2022) Collapsing Focal Segmental Glomerulosclerosis in Siblings With Compound Heterozygous Variants in NUP93 Expand the Spectrum of Kidney Phenotypes Associated With Nucleoporin Gene Mutations. Front. Pediatr. 10:915174. doi: 10.3389/fped.2022.915174
Received: 07 April 2022; Accepted: 07 June 2022;
Published: 07 July 2022.
Edited by:
Marina Vivarelli, Bambino Gesú Ospedale Pediatrico, ItalyReviewed by:
Fatih Ozaltin, Hacettepe University, TurkeyCopyright © 2022 Cason, Williams, Chryst-Stangl, Wu, Huggins, Brathwaite, Lane, Greenbaum, D’Agati and Gbadegesin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rasheed A. Gbadegesin, cmFzaGVlZC5nYmFkZWdlc2luQGR1a2UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.