 Megan Grundy
Megan Grundy Aru Narendran
Aru Narendran- 1Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 2POETIC Laboratory for Preclinical and Drug Discovery Studies, Division of Pediatric Oncology, Alberta Children’s Hospital, University of Calgary, Calgary, AB, Canada
Clinical trials completed in the last two decades have contributed significantly to the improved overall survival of children with cancer. In spite of these advancements, disease relapse still remains a significant cause of death in this patient population. Often, increasing the intensity of current protocols is not feasible because of cumulative toxicity and development of drug resistance. Therefore, the identification and clinical validation of novel targets in high-risk and refractory childhood malignancies are essential to develop effective new generation treatment protocols. A number of recent studies have shown that the hepatocyte growth factor (HGF) and its receptor Mesenchymal epithelial transition factor (c-MET) influence the growth, survival, angiogenesis, and metastasis of cancer cells. Therefore, the c-MET receptor tyrosine kinase and HGF have been identified as potential targets for cancer therapeutics and recent years have seen a race to synthesize molecules to block their expression and function. In this review we aim to summarize the literature that explores the potential and biological rationale for targeting the HGF/c-MET pathway in common and high-risk pediatric solid tumors. We also discuss selected recent and ongoing clinical trials with these agents in relapsed pediatric tumors that may provide applicable future treatments for these patients.
Introduction
Mesenchymal epithelial transition factor (c-MET) is a cell-surface receptor tyrosine kinase that is widely expressed throughout many organ systems. c-MET is predominantly expressed in epithelial cells, while its ligand (hepatocyte growth factor–HGF) is largely expressed by cells of mesenchymal origin (1, 2). Under normal conditions, this signaling pathway plays a crucial role in embryogenesis, wound-healing and regeneration, but has been identified as a driver of cancerous growth, metastasis, and drug resistance (3–5).
The c-MET receptor tyrosine kinase was first discovered as a result of work done to isolate a transforming gene chemically induced in human osteosarcoma cells (6–8). The ligand HGF [or scatter factor (SF) (9)] was later identified as the ligand for the c-MET receptor (10). The HGF/c-MET signaling pathway is complex [reviewed in (11)], and promotes diverse cellular activities including survival, proliferation, motility, and differentiation (12, 13). The HGF/c-MET pathway is crucial during embryogenesis; mice in which either the MET or HGF gene is mutated do not survive in utero. Disruption of the HGF/c-MET pathway results in impaired placentation, liver development, and development of the skeletal muscles of the limbs (14–16). In adult organisms, the HGF/c-MET pathway has been shown to drive wound-healing and regeneration. Increased plasma levels of HGF are seen after various insults including ischemic injury to the myocardium (17, 18), acute renal failure (19), partial hepatectomy (20), and acute lung injury (21). Conditional mutation of the MET gene in mice greatly impairs liver regeneration after injury or partial hepatectomy (20, 22, 23), skin regeneration and wound healing (24), and renal protection following acute kidney injury (25).
Binding of activated HGF to the c-MET cell surface receptor induces receptor dimerization which activates the tyrosine kinase through phosphorylation of two tyrosine residues in the kinase activation loop. This in turn causes autophosphorylation of tyrosine residues in the carboxy-terminal substrate-binding tail of the cytoplasmic MET protein. Here, a wide number of cytoplasmic effector proteins are recruited including phosphoinositide 3 kinase (PI3K), Src homology-2-containing (SHC), Src, Src homology 2 domain-containing phosphatase-2 (SHP2), signal transducer and activator of transcription 3 (STAT3), growth factor receptor-bound protein 2 (GRB2), and GRB2-associated binding protein 1 (GAB1) (26–29). Notably, phosphorylated GAB1 bound to MET (directly or through GRB2) provides additional binding sites for cytoplasmic effector proteins (30, 31). Important signaling cascades activated downstream from c-MET include the RAS-MAPK and PI3K-Akt pathways, which affect transcription ultimately leading to increased cell cycle progression, proliferation, and motility [reviewed in (11)]. Other pathways affected by c-MET signaling–such as RAC-CDC42, p21-activated kinase (P21), and RAP1-FAK–work in the cytoplasm or at the plasma membrane to cause cytoskeleton changes, and reduce cellular adhesion, ultimately promoting cell motility and migration (4).
In addition to the “main” HGF/c-MET signaling pathway, there is evidence for significant crosstalk between c-MET and other cell surface proteins, including integrins, plexins, CD44, and other receptor tyrosine kinases, notably the epidermal growth factor receptor (EGFR) (32, 33). These cross-receptor interactions (often amplified in cancer) have been shown to play a key role in acquired resistance to chemotherapy targeting growth receptors, such as EGFR (34, 35).
Finally, c-MET can be downregulated by degradation after activation. The c-MET receptor is internalized through clathrin-mediated endocytosis and is either ubiquitinated and degraded or recycled back to the cell surface. Ubiquitination is mediated by binding of Cbl (an E3 ubiquitin ligase) to the phosphorylated tail of c-MET, and disruption of this interaction can lead to increased activity of c-MET (36–39). Other mechanisms for regulation of HGF/c-MET signaling include negative feedback by protein kinase C (PKC) (40, 41), degradation of the c-MET receptor by extracellular metalloproteases (42), and dephosphorylation of c-MET tyrosine residues in the activation or docking domains by intracellular phosphatases (43–45).
A more thorough discussion of the molecular signaling underpinning the HGF/c-MET pathway is beyond the scope of this review, and is summarized schematically in (5, 46).
Hepatocyte growth factor/mesenchymal epithelial transition factor axis and cancer
Currently, there is a large body of literature linking the HGF/c-MET pathway to cancer (46). Indeed, the c-MET gene was first identified as a protooncogene transformed by its association with translocated promoter region (TPR), leading to constitutive activation and oncogenic changes (6–8, 47). This same activating TPR-MET oncogene has since been identified in human gastric cancer (48). There are many ways in which the HGF/c-MET pathway–which, as discussed above, is crucial in normal physiology for cell survival, proliferation, motility and migration–can contribute to a progression toward uncontrolled cell growth and invasiveness. Changes that lead to increased levels of active HGF, c-MET expression, c-MET activation, or to decreased c-MET downregulation and degradation generally contribute to a more oncogenic phenotype. While genetic changes such as activating point mutations, or amplification of the MET gene have been identified in a wide range of human cancers, increased activity of the HGF/c-MET pathway is often seen in cancer as a result of physical and chemical changes in the tumor stroma, leading to upregulation of c-MET, or clonal selection of c-MET-expressing cancer cells. The result is often a subset of invasive cancer cells resistant to chemotherapy, and prone to dissemination (46). Figure 1 shows schematically the broad categories of molecular alterations that can contribute to dysregulated HGF/c-MET signaling in cancer.
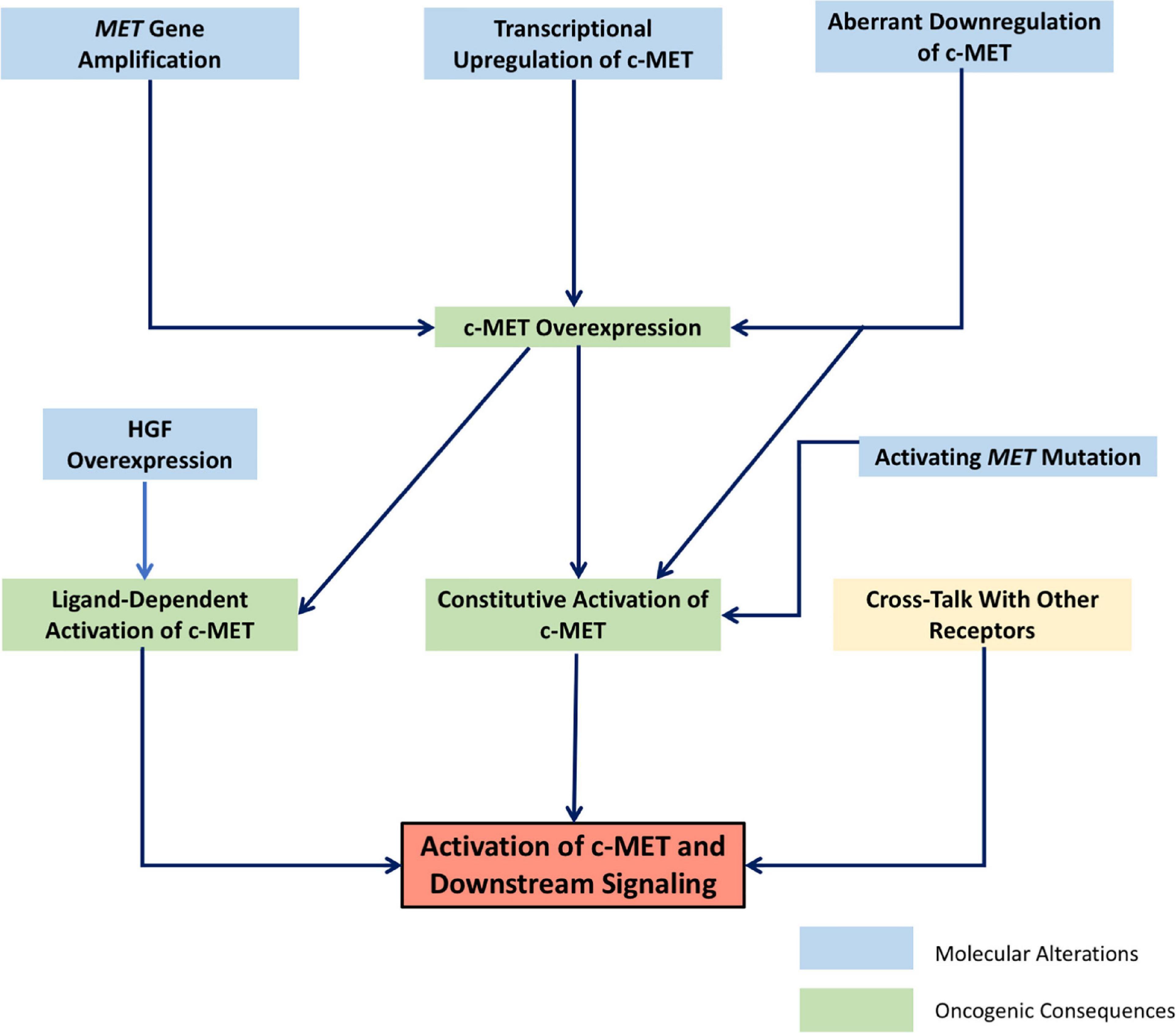
Figure 1. Schematic representation of the molecular mechanisms leading to dysregulated signaling of the HGF/c-MET pathway. Interrelated molecular factors lead to changes in HGF/c-MET expression and activity, resulting in the activation of downstream signaling pathways which promote oncogenic cellular changes.
In the following, the role of the HGF/c-MET pathway in pediatric solid cancers, as is currently understood in the literature, will be reviewed.
Pediatric malignancies
Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is the most common soft-tissue sarcoma in pediatric patients (49). The majority of pediatric RMS can be classified histologically as either “embryonal” (ERMS) or “alveolar” (ARMS) (50). ARMS generally has a worse prognosis than ERMS, is more prone to metastasis, and is more often diagnosed at a later stage (51, 52). In the majority of ARMS tumors, a chromosomal translocation t(2;13) is identified, which leads to the expression of fusion proteins PAX3-FOXO1 or (less frequently) PAX7-FOXO1 [t(1;13)] (50). These fusion proteins combine the PAX3/7 binding domain with the FOXO1 activation domain (aka FOXO1A), resulting in a transcription activator more powerful than PAX3 or PAX7 alone (53). Clinical research indicates that the presence of a fusion protein (“fusion positive”) is a more important prognostic indicator than the histologic classification of ARMS vs. ERMS (54). Early research links the HGF/c-MET pathway to development of RMS; c-MET was identified as a target of the PAX3-FOXO1 fusion protein (55), and a murine model of RMS showed that dysregulation of HGF/c-MET signaling on the background of suppression of INK4a/ARF (locus that encodes two tumor suppressors) was sufficient to lead to the development of RMS tumors with a similar molecular signature to human RMS tumors (56).
In vitro studies of the relationship between c-MET and the PAX3-FOXO1 fusion protein have shown that while expression of c-MET is upregulated by PAX3-FOXO1, PAX3-FOXO1 expression is not necessary for c-MET expression and activity. Ginsberg et al. found that in myogenic cells transfected with PAX3-FOXO1 and fusion-positive ARMS cells, c-MET expression correlated with levels of PAX3-FOXO1. However, high levels of c-MET expression were also found in some samples of ERMS, suggesting that PAX3-FOXO1 is not necessary for c-MET expression (57). The correlation of c-MET upregulation and PAX3-FOXO1 was confirmed by Taulli et al., who also found that while PAX3-FOXO1 could induce oncogenic cell growth, this transformation was dependent on HGF/c-MET signaling. Notably, silencing of the c-MET receptor inhibited cell proliferation, and increased apoptosis in both ARMS- and the fusion-negative ERMS-derived cell lines, which underscores a role for the HGF/c-MET pathway in RMS beyond the effects of the PAX3-FOXO1 transcription activator found largely in ARMS. Indeed, when the c-MET receptor in ERMS xenografts was conditionally silenced with an anti-Met short hairpin RNA (shRNA), tumor growth stopped and subsequently regressed (58).
Particularly in ARMS, the presence of metastases at diagnosis confers a poor prognosis (51, 52). Given the physiological role for the HGF/c-MET pathway in cell motility and migration, particularly in the case of skeletal muscle and limb development (15), this pathway has been keenly studied with regards to its role in promoting metastasis of RMS. Jankowski et al. showed that in response to stimulation with HGF, c-MET-positive (assessed by immunostaining and flow cytometry) RMS cell lines undergo a number of pro-metastatic changes in vitro including increased locomotion, motility-related redistribution of the actin cytoskeleton, and increased cell migration. Notably, while HGF stimulation in vitro did not increase RMS cell proliferation, it did confer increased survival following exposure to radiation or cytotoxic chemotherapy, suggesting that HGF/c-MET might be an important target for therapy-resistant RMS (59). RMS cells were found to migrate toward HGF-rich conditioned medium from bone marrow fibroblasts, and in vivo, RMS cells were attracted into the bone marrow in a manner that was dependent on c-MET signaling. This effect was most pronounced for ARMS cell lines, which showed higher c-MET expression than ERMS cell lines (59). Downregulation of c-MET in RMS cell lines decreased both in vivo tumor growth and bone marrow engraftment (60). A clinical study examining samples from 40 patients with RMS found that in the subset of patients whose cancer had infiltrated the bone marrow (n = 16), the infiltrating cells had greater levels of c-MET expression than did cells from the corresponding primary tumors (61). These findings all implicate the HGF/c-MET pathway as a key driver of RMS invasive and metastatic spread.
RMS arises from dysregulated myogenic progression with loss of terminal cell differentiation. Lower c-MET expression has been correlated with greater levels of differentiation (62). In normal myogenic development, downregulation of c-MET is required for terminal differentiation (63, 64). It has been postulated that the sustained c-MET expression seen in RMS could result from decreased post-transcriptional downregulation by myomiRs (particularly miR-1 and miR-206, microRNA sequences which are physiologically upregulated during myogenesis). Low levels of miR-1 and miR-206 were found in ERMS and ARMS compared to normal muscle, and re-expression of these miRNAs was found to promote differentiation, and inhibit growth of RMS xenografts. c-MET was shown to be downregulated in myogenic precursor cells by miR-206 at the onset of myogenesis, and the expression of miR-206 in RMS cells was found to inhibit c-MET signaling (65, 66).
Histologically, cells from ERMS tumors have a more differentiated “spindle-shaped” morphology compared to the more aggressive small round cells found in ARMS tumors (50). When c-MET signaling was blocked in ARMS cells, they began to resemble ERMS cells morphologically (62). Conversely, when c-MET was constitutively activated (by introduction of the TPR-MET oncogene) ERMS cells did not differentiate. Control ERMS cells developed the typical histology of ERMS tumors with elongated spindle morphology, while TPR-MET-activated ERMS cells retained morphology typical of the more aggressive ARMS tumors. Xenograft tumors derived from the constitutively c-MET-activated ERMS cells were less differentiated, faster growing, had increased vascularization, and more distant metastases compared to controls. Though targeted activation of c-MET promoted these aggressive changes to the typical ERMS phenotype, tumors derived from ARMS cells still grew more aggressively, suggesting that factors other than c-MET activity contribute to the phenotype of ARMS (64).
To date, there are but a few studies linking c-MET activity to clinicopathological parameters and patient outcomes in RMS. Chen et al. examined levels of c-MET expression in primary RMS tumors, and while they identified expression of c-MET in all tumor samples, only a minority of tumors overexpressed c-MET. Their analysis did, however, find a positive correlation between higher expression of c-MET and later staging of cancer, and the presence of a PAX3/7-FOXO1 fusion protein. Higher expression of c-MET was correlated with poorer survival (67). Diomedi-Camassei et al. also found a similar correlation between c-MET expression and advanced disease, however, this study did not identify a relationship between overall survival and c-MET expression (61). A more recent study by Du et al. found a significant effect of c-MET expression on overall survival only in ERMS or “fusion-negative” RMS, and though this study identified greater expression of c-MET in RMS samples compared to normal muscle controls, the difference between c-MET expression in ARMS and ERMS samples was not significant (68). Further analysis of larger cohorts of RMS patients would be beneficial to clarify the role (if any) of c-MET in RMS prognosis.
Osteosarcoma
Osteosarcoma (OS) is the most common primary bone tumor, and occurs in a bimodal age distribution with a peak in the pediatric age range. Survival in OS has plateaued since the 1980’s, and new treatments are needed, especially for those with advanced disease (69). The c-MET gene was first identified in human OS cells that had been treated with a chemical carcinogen (7). A number of studies have since identified overexpression of c-MET in a large portion of OS tissue samples (though the rates of overexpression reported range substantially between studies) (70–73). Human OS-derived cell lines also have been shown to have increased (but variable) c-MET expression (74, 75), and invasive cell behavior (increased motility and invasion) after stimulation with HGF (74).
Patane et al. showed that overexpression of c-MET could transform osteoblasts (with undetectable c-MET levels at baseline) to an OS-like phenotype, capable of anchorage-independent growth, and increased basement membrane and 3-D invasion. Development of this OS-like phenotype was inhibited by blockade of c-MET receptor dimerization or with c-MET targeting shRNA (76). A more recent study from the same group showed that MET-OS “clones”— created from c-MET overexpression in normal bone-derived cells — originated from selective expansion of a subpopulation of committed osteoprogenitor cells at an early stage of differentiation, and that c-MET overexpression inhibited full osteoblast differentiation (77). These studies suggest a role for the HGF/c-MET pathway in the development of OS.
A number of similar reports have suggested a role for c-MET in the development of OS through suppression or downregulation of microRNAs (miRNAs). Specifically, studies have shown that a range of cancer-related miRNAs are decreased in OS, inversely correlated with increased c-MET expression, and directly target the wild-type 3′-UTR of the MET gene. Re-expression of these suppressed miRNAs has been shown to lead to reduced OS growth and invasion (78–84).
Pharmacological c-MET inhibition in OS has been shown to decrease malignant behavior of OS cells in culture, and to suppress in vivo xenograft tumor growth (85, 86). Fioramonti et al. showed that treatment with cabozantinib (an inhibitor of multiple RTKs including c-MET and VEGFR-2), in addition to directly reducing OS proliferation and migration, has an indirect inhibitory effect on RANK-expressing (a poor prognostic marker) OS cell lines by decreasing RANK-L production by osteoblasts (86).
Ewing sarcoma
After OS, Ewing Sarcoma (ES) is the second most common primary pediatric bone tumor. Fleuren et al. found that a majority of samples of ES (including primary tumors, metastases and post-chemotherapy resections) had medium to high levels of cytoplasmic c-MET expression. More specifically, however, they found that expression of c-MET localized to the plasma membrane (and not cytoplasmic c-MET) was associated with worse overall survival. ES cell lines were susceptible to treatment with (non-selective) c-MET inhibitors cabozantinib and crizotinib (87). A recent in vitro study by Charan et al. demonstrated overexpression of HGF in ES xenograft tumors and cell lines, compared to control tissues. The overexpression of HGF was related to an oncogenic p53 isoform, and the authors showed that treatment with a neutralizing anti-HGF antibody could synergize with GD2-targeted CAR-T cell therapy to inhibit ES xenograft growth and increase survival in a murine model (88).
Glioma
Gliomas are the most common pediatric CNS tumors, which after leukemia are responsible for the most cancer deaths in this population (89). Few effective chemotherapeutic options exist for treatment, especially of higher-grade or non-resectable lesions, and outcomes are frequently quite poor. As these cancers are also common in the adult population, much of the literature is comprised of studies of adult tumors.
Early studies of human primary brain tumor samples have shown increased expression of c-MET and/or HGF in higher grade gliomas, such as glioblastoma multiforme (GBM) (90–94). Microscopy analysis of tumor sections, as well as in vitro and in vivo experiments with anti-HGF agents, have suggested that both autocrine and paracrine signaling of HGF occurs in gliomas (90, 91, 93, 95–97). Similar to its effects in sarcomas, stimulation with HGF increased glioma cell proliferation, motility, and invasion (90, 91, 94). Treatment with anti-HGF agents, such as anti-HGF snRNA/ribozymes (95), anti-HGF antibodies (96, 98), or competitive inhibitors of HGF (99) decreased the growth of in vivo xenograft glioma tumors by decreasing cell proliferation and increasing apoptosis (and through effects on angiogenesis which will be addressed separately). Notably, Kim et al. showed in a murine model that glioma xenografts which express c-MET without co-secreting HGF are completely resistant to treatment with an anti-HGF mAb. However, the authors indicated that these results do not necessarily preclude successful therapy in humans, as any native murine HGF in this model (in other words, paracrine HGF) would not have been targeted by the anti-HGF mAb (98). Martens et al. found that an anti-c-MET antibody was effective against GBM xenografts co-expressing HGF and c-MET, while xenografts expressing c-MET but not secreting (human) HGF were resistant to therapy (97).
Studies comparing HGF/c-MET expression in pathological specimens to clinical outcomes have indicated that greater presence of HGF/c-MET is associated with poorer prognosis and outcomes (though this data is largely from adult patients) (100–103). Interestingly, Kong et al. found that a greater percentage of c-MET overexpressing tumors had aggressive radiographic findings on initial MR imaging (invasive or multifocal lesions) compared to c-MET-negative (or low-expressing) tumors (100).
Tumor neovascularization and angiogenesis play a significant role in glial cancers, particularly in aggressive cancers like GBM. The HGF/c-MET pathway influences angiogenesis both directly and indirectly [reviewed in (104)], and treatment with anti-HGF/c-MET agents can reduce tumor microvessel density (95, 97–99). Studies of HGF/c-MET distribution in human primary brain tumors showed that expression was not limited to glial cancer cells, but was also found in supporting tumor microvasculature (91–93). In vitro stimulation with HGF promoted chemotactic migration and increased DNA synthesis in neuro microvascular endothelial cells, suggesting a direct pro-angiogenic role for HGF (94, 105).
Probing the role of HGF as an indirect mediator of angiogenesis, Moriyama et al. demonstrated that treatment of glioma cell lines with HGF increased VEGF mRNA expression and protein secretion in a dose-dependent fashion (106). However, Schmidt et al. quantified the levels of VEGF, HGF, and bFGF in extracts from human gliomas, and found that levels of both VEGF and HGF were increased in higher grade gliomas, and were independent predictors of microvessel density (107). Using both GBM cell lines and primary cultures, Eckerich et al. found that exposure to hypoxia caused an increase in MET transcription and c-MET expression. This effect was linked to hypoxia-inducible-factor 1-alpha (HIF-1a) (the MET promoter is known to have HIF-1 binding sites), and was more pronounced in cell lines that had relatively low basal c-MET expression (108). Martens et al. tested the effects of a one-armed anti-c-MET antibody (OA-5D5) on the in vivo growth of GBM xenografts, and interestingly found that treatment with OA-5D5 decreased intratumoural microvessel density, despite OA-5D5 being unable to bind murine c-MET. The authors suggested that in this model of GBM, anti-c-MET therapy had an indirect anti-angiogenic effect, potentially by decreasing proteolysis within the tumor extracellular matrix, thereby inhibiting neovascular spread (97).
Cancer stem cells are increasingly recognized as key drivers of therapy resistance and tumor progression, particularly in high-grade cancers such as GBM. Studies of GBM-derived cells and neurospheres have identified an association between HGF/c-MET signaling and “stemness” in GBM [reviewed in (109)]. c-MET signaling within neurospheres (which recapitulate the heterogeneity of a GBM tumor more accurately than single cell cultures) was associated with expression of other identifiers of “stemness,” and treatment with HGF sustained clonogenic potential and expression of reprogramming transcription factors (110). However, rather than homogeneously expressing c-MET, subpopulations within c-MET-positive neurospheres of c-MET high- and low-expressing cells were identified, with the high-expressing subpopulations having greater “stem”-like characteristics, including greater clonogenicity and capacity for multipotential differentiation, and enhanced in vivo tumorigenicity (111). Similarly, a study of freshly isolated patient-derived GBM cells showed that within a sorted population of c-MET-positive cells, those with high c-MET expression were more clonogenic, and more tumorigenic in an orthotopic in vivo model, than those with lower c-MET expression (112).
A recent study by Qin et al. found that de novo glioma formation could be instigated by injecting human HGF and c-MET cDNA in combination with siRNA against TRp53 into the lateral ventricle of neonatal mice. These results suggest that increased HGF/c-MET signaling, on a oncogenic background of p53 attenuation, could be sufficient to transform healthy neural stem cells into malignant glioma stem cells (113).
The HGF/c-MET pathway has been shown to contribute to anti-cancer therapy resistance in human gliomas. Indeed, irradiation of glioma stem cells has been shown to induce c-MET upregulation (112), while in vitro studies have shown that pre-treating GBM cells with HGF reduced the cytotoxic effect of DNA damaging agents (114). Furthermore, treatment of GBM xenografts with chimeric U1/ribozymes targeting HGF and c-MET mRNA sensitized tumors to gamma radiation, and greatly improved tumor regression and cure rate (115).
Much of the literature discussing the role of HGF/c-MET in glioma is based upon studies of adult primary tumors, or cell lines derived from adult tumors. However, owing to distinct genetic differences, these findings might not be directly applicable to the pediatric population (116). A recent review of the molecular landscape of pediatric gliomas suggests that in fact pediatric tumors may be a better target for many precision therapies, due to their relative paucity of diverse genetic drivers compared to adult tumors, which tend to be more genetically heterogeneous (117). Wu et al. found amplifications of the MET gene in 6% (7/112) of pediatric high-grade gliomas analyzed (118). Bender et al. analyzed the genetic profiles of pediatric GBMs, and found that up to 10% of tumors harbored gene fusions involving MET–of all the fusion genes identified, MET was the single gene most frequently involved. Notably, all of the GBM samples found to have a MET fusion were also found to have impaired cell cycle regulation. Indeed, the authors found that cell lines transduced with a MET fusion gene were only tumorigenic on the background of a TP53 mutation, or deletion of CDKN2A/CDKN2B, suggesting that while dysregulation of the c-MET pathway plays a role in tumorigenesis, it alone is not sufficient to drive tumor growth (119).
Medulloblastoma
Medulloblastoma (MB) is the most common brain malignancy in pediatrics. Due to the risk of leptomeningeal dissemination, current therapy of MB generally includes surgical resection, chemotherapy and radiation of the neuraxis–an aggressive treatment approach which can result in significant morbidity including endocrinological and neurocognitive deficits. MB develops from neural progenitor cells in the posterior fossa, most commonly in the cerebellum—the HGF/c-MET pathway is crucial for normal cerebellar development (120), and dysregulation of this pathway has been implicated as a driver of MB.
An early study of the genetic drivers of MB found single copy gains of MET in 38.5% of tumor samples analyzed (121). c-MET and HGF were found to be expressed in tumor specimens and MB cell lines, and levels of c-MET mRNA correlated with poor clinical outcomes. Moreover, it was shown that HGF stimulation of MB cells in vitro induced proliferation, anchorage-independent growth, and protection from chemotherapy-induced cell death. Overexpression of HGF in vivo led to increased growth of xenografts, with unfavorable histologic phenotypes (122). Small molecule inhibition of c-MET in vitro in MB cell lines led to decreased proliferation, motility, and anchorage-independent growth (123), and treatment with an oral c-MET inhibitor (crizotinib) in vivo inhibited growth of subcutaneous MB tumor xenografts (124).
Work done to investigate other molecules and pathways involved in the tumorigenesis of MB found that HGF induced expression of c-Myc (an oncoprotein whose presence can indicate poor prognosis in MB), and inhibition or overexpression of c-Myc in MB modulated the effects of HGF on cell cycle progression and proliferation (125). Additionally, epigenome screening identified serine protease inhibitor kunitz-type 2 (SPINT2–an inhibitor of HGF activation) as a potential tumor suppressor gene in MB. A majority of primary MB tumor samples showed decreased SPINT2 expression, while stable re-expression of SPINT2 in MB cell lines resulted in decreased MB cell proliferation, anchorage-independent growth, and longer overall survival of mice implanted with intercerebellar tumor xenografts (126). Investigation into the relationship between the serine/threonine protein kinase MAP4K4 and c-MET found that in certain MB cell lines, blocking MAP4KR signaling affected c-MET signaling (promoted endocytosis and recycling of c-MET), suggesting a potential combination therapeutic target for a subset of MB (127).
MB can be classified into molecular subgroups: wingless/integrated (WNT), sonic Hedgehog (SHH), and groups 3 and 4. Genetic analysis has revealed an association between increased c-MET expression and the SHH subgroup of MB, though increased c-MET expression is also seen in a subset of tumors classified in groups 3 and 4 (128–130). Higher levels of phosphorylated c-MET were correlated with tumor recurrence and poor prognosis in the SHH subgroup of MB (129). Binning et al. found that activation of SHH in murine models induced formation of MB, while overexpression of HGF on the background of activated SHH resulted in an increased frequency of MB formation. Treatment with a mAb against HGF in this model improved overall survival, though the inhibition of tumor growth was incomplete, leading the authors to hypothesize that dual inhibition of both SHH and HGF would be more effective (131). A further study by the same group interestingly revealed that the combination therapy with SHH and HGF inhibitors did not improve survival (and even reduced survival in one combination therapy) compared to monotherapy (132). Separately, small molecule inhibition of c-MET with foretinib has been shown to be effective against subcutaneous and orthotopic xenografts of disseminated SHH subgroup MB, as well as a transgenic mouse model of metastatic SHH MB (129).
Neuroblastoma
Neuroblastoma (NB) is a malignancy derived from primitive cells of the sympathetic nervous system and is the most common extracranial pediatric solid tumor (133). The clinical behavior and prognosis of NB can range broadly—from tumors which spontaneously regress, to aggressive metastatic tumors causing death.
Hecht et al. provided the first evidence of a role for the HGF/c-MET pathway in NB. They found variable expression of HGF and c-MET across established NB cell lines, with little basal activation of c-MET. However, stimulation of NB cells with HGF promoted invasion and expression of proteolytic extracellular matrix (ECM)-degrading factors. Pre-stimulation of NB cells with HGF in a model of NB in chick embryos (using the chorioallantoic membrane as an epithelial barrier to investigate invasive growth) led to the increased formation of well vascularized tumors (134). A further study by the same group found that expression of the neurotrophin receptor TrkB (a hallmark of invasive and metastatic NB) led to upregulation of HGF and c-MET (and an increase in phosphorylated c-MET, suggesting autocrine stimulation). TrkB-expressing cells were more invasive than the parental cells. A series of stimulation experiments revealed that while anti-BDNF (the stimulating ligand for TrkB) treatment could reduce c-MET phosphorylation, anti-HGF treatment did not similarly affect phosphorylation of TrkB–but did reduce phosphorylation of c-MET (which could be partially rescued by BDNF stimulation). These results suggest that the relationship between TrkB and c-MET is driven at the top by stimulation through TrkB. Analysis of tumor specimens from advanced disease found increased expression of TrkB in a subset of samples, all of which had increased expression of HGF and c-MET (135). Ren et al. investigated the relationship between c-MET and another prognostic marker in NB, macrophage migration inhibitory factor (MIF). When expression of MIF was reduced in an NB cell line, c-MET mRNA levels decreased. Decreased MIF expression led to a less aggressive NB phenotype in vitro (less invasive, less proliferative, and more apoptotic), and decreased tumor growth and metastasis in vivo (136).
Plasma and serum samples from patients with NB were found to have an overall increased concentration of HGF compared to plasma and serum from healthy controls. Notably, HGF concentration was higher in patients with later stage NB and was associated with negative markers of NB prognosis (137). qPCR analysis of c-MET mRNA in NB tumor samples found a correlation between increased c-MET expression and later clinical stage (138). Yan et al. examined the genomic and protein expression of c-MET in NB clinical samples, and across the 54 samples analyzed they identified one example of MET gene amplification, and only one example of increased c-MET expression by IHC. The rest of the samples analyzed did not show any c-MET overexpression, though the authors did identify some alternatively spliced MET isoforms (139). Scorsone et al. looked at a small panel of NB patient samples and found detectable c-MET expression in 38% (3/8) of tumor samples (140). Similarly, established NB cell lines have shown variable (often minimal to no) c-MET expression (134, 140, 141).
In vitro and pre-clinical studies have suggested the potential therapeutic benefit of small molecule inhibition of c-MET in NB (138, 140). Cabozantinib (a small molecule inhibitor of c-MET and VEGFR among other RTKs) was compared to selective anti-VEGFR inhibition in models of NB, and while anti-VEGFR therapy alone led to increased metastasis, cabozantinib decreased metastatic spread to visceral organs (though not to bones). The study was limited, however, by the use of different animal models and cell lines for the study of metastatic spread after VEGFR inhibition or cabozantinib treatment, and thus a direct comparison of the two therapies is not possible (142).
Wilms tumor (nephroblastoma)
Wilms tumor (WT), also known as nephroblastoma, is the most common renal malignancy in children. It is an embryonal malignancy and is thought to derive from aberrant differentiation and development in early nephrogenesis. HGF/c-MET autocrine and paracrine signaling has been shown to play a part in early kidney development (143), and so a role for this pathway in WT tumorigenesis has been posited. The data on a role (if any) for HGF/c-MET in WT is, however, limited. A screen of solid tumors for mutations in the MET gene revealed no mutations in 52 samples of WT (144). In 2002, Alami et al. showed that in a relatively small subset of WT samples, expressions of c-MET and HGF were raised compared to normal kidney tissue, and that in a majority of samples higher levels of c-MET were correlated with a marker of proliferation (145). In contrast, a later study by Vuononvirta et al. reported tissue microarray analysis of a larger cohort of WT and showed that only 8% (15/193) and 14% (25/179) of non-anaplastic tumors expressed HGF or c-MET, respectively, and did not find an association with markers of proliferation. They found greater HGF expression in nephrogenic rests (precursor lesions to WT) compared to WT, suggesting a loss of this signaling pathway with development of malignancy (146). To date the role of HGF/c-MET in WT remains unclear.
Hepatoblastoma
While rare overall, hepatoblastoma (HB) is the most common pediatric liver malignancy (with some geographical differences), particularly in younger children and infants. HB is thought to arise from early hepatic progenitor cells, and owing to their pluripotent nature, histological subtypes of HB include both epithelial and mixed epithelial-mesenchymal variants (147). As previously discussed, HGF/c-MET plays a crucial role in normal liver development and regeneration (14, 20, 22, 23), and so it is perhaps unsurprising that this pathway has been implicated in the pathogenesis of HB.
In pediatric patients, liver regeneration after partial hepatectomy is particularly robust and rapid. In some patients undergoing partial hepatectomy for untreated HB, rapid recurrence of liver tumors and lung metastases is seen post-operatively, coinciding with the time period of maximal liver regeneration. HGF is a crucial growth factor involved in liver regeneration. Von Schweinitz et al. found that sera from HB pediatric patients had significantly elevated HGF concentrations post-hepatectomy compared to healthy controls (with no statistically significant difference in serum HGF concentrations pre-treatment compared to healthy controls). HGF was shown to be produced by stromal cells in co-culture with HB cells, suggesting a paracrine mechanism of HGF stimulation, while analysis of tumor sections revealed high expression of c-MET by epithelial tumor cells. In a subset of patients, HB tumors were compared to normal liver tissue (from the same patient, n = 5) and c-MET mRNA levels were found to be upregulated in HB tumor samples compared to healthy liver tissue (148). The authors of this early paper also showed that in vitro, HGF at physiologic concentrations (matching clinical data of serum HGF concentrations post-hepatectomy) increased proliferation of established HB cell lines, while higher (non-physiologic) concentrations of HGF had the opposite effect (148). However, a more recent study with the same established HB lines and similar concentrations of HGF found that HGF stimulation did not increase viability or proliferation of HB cells, but rather increased HB cell survival only in the face of stressors like serum-starvation, or treatment with chemotherapy, an effect that was mediated through the PI3K/AKT downstream signaling pathway (149).
Aberrant accumulation of β-catenin in the cytoplasm and nucleus can be seen in a large proportion of HB (150), and is likely involved in HB tumorigenesis [reviewed in (147)]. While activation of β-catenin occurs classically through the Wnt pathway, Wnt-independent and HGF-induced nuclear translocation of β-catenin has been shown to occur as a result of a subcellular interaction of β-catenin and c-MET (151). Clinical data suggests significant but variable rates of mutation/deletion in the gene for β-catenin (CTNNB1) affecting β-catenin accumulation in HB. Purcell et al. studied clinical samples of HB, and while 87% (85/98) of tumors showed aberrant β-catenin accumulation, the frequency of CTNNB1 mutations in their patient cohort was substantially lower [at most 20% (20/98) of samples], suggesting an alternative mechanism for β-catenin accumulation. Significant levels of c-MET activated β-catenin [Y654-β-catenin, (152)] were found in the cytoplasm and nucleus of clinical HB samples with wild-type CTNNB1, suggesting that aberrant accumulation β-catenin in these cases was due to HGF/c-MET signaling, a mechanism which was validated in a wild-type CTNNB1 HB cell line after HGF stimulation (153). Recently, Matsumoto et al. found that overexpression of c-MET in addition to overexpression of constitutively active β-catenin and YAP (Yes-associated protein) in vivo led to the formation of murine liver tumors similar to human HB and expressing HB tumor markers (154).
An early study examined the role of various genetic alterations on patient outcomes with HB, and found that clinical outcomes after multimodal therapy were not correlated with MET RNA expression levels, though it should be noted that only 23 patient samples had MET levels quantified (155).
Childhood hepatocellular carcinoma
Hepatocellular carcinoma (HCC) is becoming increasingly prevalent worldwide, although it remains relatively rare in childhood (with geographic differences in incidence). In the vast majority of adult HCC, an etiology related to chronic liver injury/inflammation (alcoholic/non-alcoholic fatty liver disease, viral liver infection etc.) is identified, and patients often present in the context of liver cirrhosis. In children, however, HCC more often develops on the background of a non-cirrhotic liver, and is often considered a separate entity (156, 157).
In 1999, Park et al. sequenced the TK domain of the MET gene in primary liver tumors, from both pediatric and adult patients. Interestingly, they found that 30% (3/10) of childhood HCC samples had missense mutations in the TK domain of the MET gene, while no MET mutations were found across 16 adult HCC tumors. The authors suggest that potentially activating mutations of the MET kinase domain could contribute to the tumorigenesis of childhood HCC, and account for the relatively accelerated timeline compared to adult HCC (which often occurs in the context of longstanding liver inflammation and cirrhosis) (158).
The HGF/c-MET pathway has been explored in adult HCC and is reviewed in (159, 160). Briefly, c-MET has been shown to be overexpressed in a proportion of HCC samples compared to normal liver tissue, and has been linked to advanced disease or poor prognosis (161–165). However, the proportion of tumors overexpressing c-MET varies considerably between studies (160). Pre-clinical studies have shown both pro- and anti-tumorigenic roles for the HGF/c-MET pathway in HCC (166–169), and analysis of clinical samples suggests an overall decreased expression of HGF in adult HCC (160).
Wang et al. showed that transgenic mice with overexpression of c-MET by hepatocytes developed HCC, and that this tumorigenesis was likely due to c-MET activation by cell attachment rather than by HGF stimulation. Furthermore, when expression of c-MET was inhibited, established HCC tumors regressed dramatically, suggesting a role for c-MET in both the development and maintenance of HCC (170). Tao et al. identified a subset of human HCC cases (9–12.5% of analyzed samples) containing both c-MET overexpression and mutation of the β-catenin gene (CTNNB1), and showed that overexpression of c-MET on the background of mutant β-catenin led to the formation of HCC in transgenic mice (171).
Thyroid malignancies
A study by Ramirez et al. examined the implications of c-MET expression with respect to recurrence and metastasis risk in children and young adults with papillary thyroid carcinoma (PTC). Immunohistochemical staining intensities of benign and malignant thyroid lesions were analyzed with respect to clinical outcome. The specimens examined included PTC, follicular thyroid carcinomas (FTC), medullary thyroid carcinomas (MTC), as well as specimens from patients with benign thyroid conditions and normal thyroid tissues. This study found that enhanced expression of c-MET and HGF/SF may be associated with an increased risk for metastasis and tumor recurrence in this patient population (172).
Table 1 broadly summarizes the known effects of HGF/c-MET signaling in the range of pediatric tumor types reviewed here.

Table 1. Effects of the HGF/c-MET pathway in pediatric tumors.
Hepatocyte growth factor/mesenchymal epithelial transition factor pathway as a therapeutic target
Recently, a number of agents that target the HGF/c-MET pathway have been developed and evaluated in preclinical and clinical studies of various cancers. These agents include small molecule inhibitors for c-MET kinase activity, as well as anti-c-MET and anti-HGF antibodies. Currently available small molecule inhibitors include bozitinib, cabozantinib, crizotinib, MSC2156119J, MK-2461, AMG-337, capmatinib, tepotinib, amuvatinib, elzovantinib, savolitinib, glumetinib, and golvatinib (173–176). These molecules have distinct characteristics with respect to their ability to interact with specific residues and regions in the active c-MET molecule. For example, bozitinib (PLB-1001) is a highly selective ATP-competitive c-MET inhibitor with blood-brain barrier permeability allowing for its potential utility in secondary GBM, where a significant proportion of patients carry METex14 alterations which are associated with a poor prognosis (177). Additionally, a number of monoclonal antibodies against c-MET and HGF, as well as competitive analogs of HGF have been developed to target the HGF/c-MET pathway in tumors (178). Compared to small molecule tyrosine kinase inhibitors (TKIs) that often interact with multiple cellular growth regulatory kinases, antibody-based therapeutics inhibit components of the HGF/c-MET pathway with greater specificity, interrupting the ligand binding process. For this reason, these therapeutics are more likely to be effective against tumors with c-MET overexpression, and not restricted to those with oncogenic molecular alterations (175).
Therapeutics targeting the HGF/c-MET pathway tested in clinical trials thus far include c-MET TKIs (selective for only c-MET or targeting additional RTKs), anti-c-MET antibodies, and anti-HGF antibodies. Fu et al. recently published a comprehensive review of clinical trials of HGF/c-MET targeted therapies in adult cancers, which span these three categories of therapeutics (179). Pediatric trials of HGF/c-MET targeted therapies have been more limited, and have thus far only tested selective and non-selective c-MET tyrosine kinase inhibitors (TKIs). Table 2 summarizes the current landscape of completed and recruiting clinical trials of HGF/c-MET targeted therapies with pediatric eligibility and recruitment.
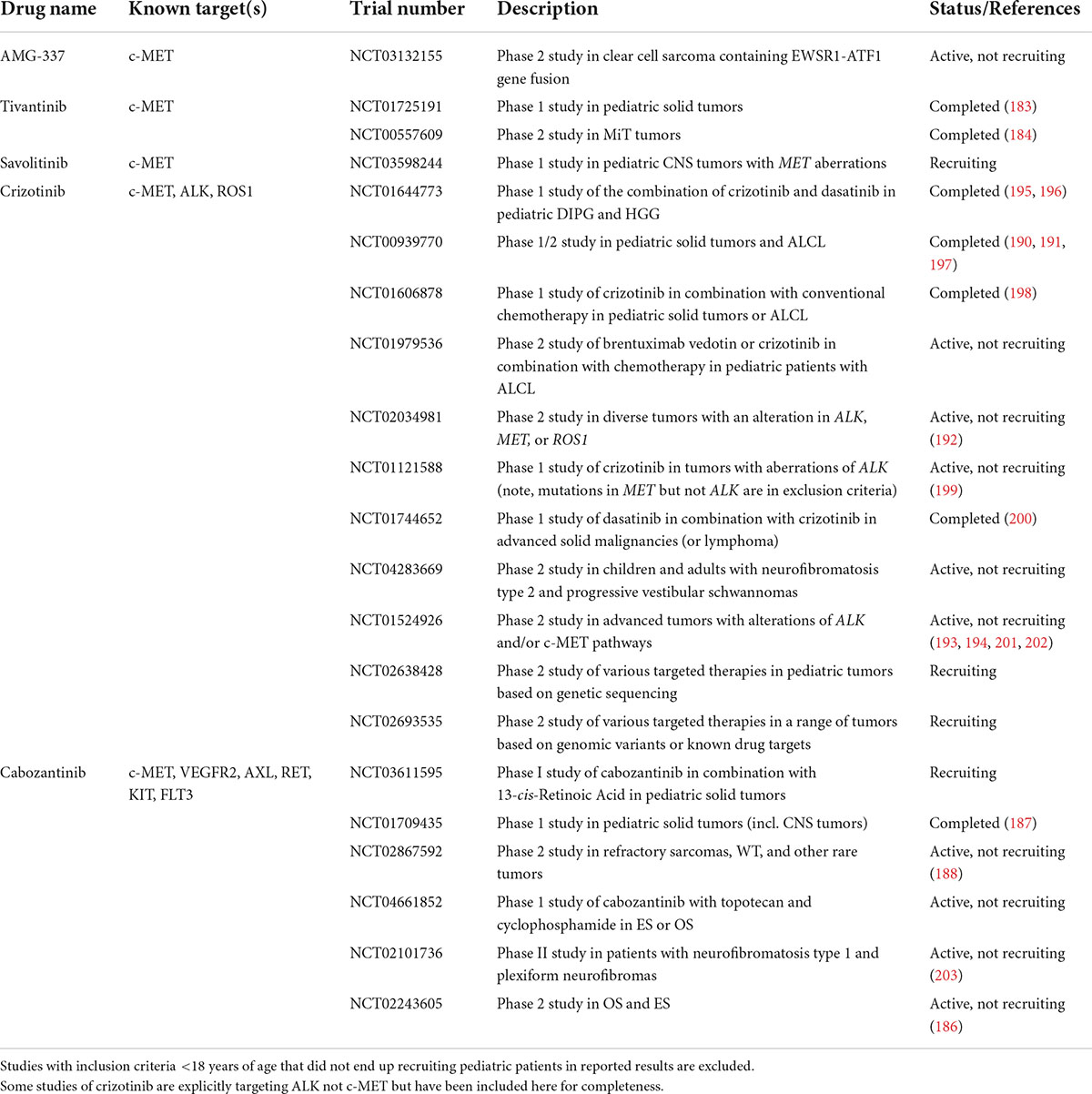
Table 2. Active, recruiting, or completed clinical trials of HGF/c-MET targeting agents that include pediatric patients.
Tivantinib
Tivantinib (ARQ 197), a staurosporine derivative, was identified as a highly selective inhibitor of the c-MET tyrosine kinase, capable of inhibiting c-MET phosphorylation and its downstream effects (180). Tivantinib has also demonstrated significant cytotoxic activity independent of c-MET inhibition, and a mechanism of action involving microtubule dynamics has been posited (181, 182). The uncertainty surrounding tivantinib’s main mechanism of action has important implications for clinical trial design and appropriate biomarker selection.
In the pediatric population, tivantinib has been investigated as a c-MET inhibitor in multiple cancer types. A Children’s Oncology Group phase I trial in pediatric patients with relapsed/refractory solid tumors investigated oral dosing and pharmacokinetics. While oral tivantinib was well tolerated, there was marked pharmacokinetic variability among patients, and no objective responses were seen. Tumor samples (solid tumors including CNS tumors) were analyzed for c-MET expression by IHC, and the majority of samples [81% (26/32)] were found to have undetectable levels of c-MET (183).
Tivantinib has also been evaluated in a phase II trial in adolescent and adult patients (median age of 25) with advanced microphthalmic transcription factor (MITF)-associated tumors (including alveolar soft part sarcoma, translocation-associated renal cell carcinoma, and clear cell sarcoma). The best response in the study was a partial response (PR) in a patient with clear cell sarcoma. Baseline tumor expression of c-MET was evaluated by IHC for 19 patients (out of n = 27). The majority of tumors evaluated were either highly or focally positive for c-MET [74% (14/19)], but no correlation with c-MET staining and best response was observed. Of note, the one patient with clear cell sarcoma who had a PR was negative for c-MET staining (184).
Thus far, the clinical activity of tivantinib in pediatric patients has been limited, and the specificity of this agent as a targeted anti-c-MET therapeutic is uncertain.
Cabozantinib
Cabozantinib is a TKI with multiple targets including c-MET, VEGFR2, RET, KIT, AXL, and FLT3. Cabozantinib has both direct anti-tumor activity and anti-angiogenic effects (185).
A recently reported multi-center phase II clinical trial (the CABONE trial) investigated the effects of cabozantinib in ES and OS. Though the trial was primarily conducted in adult patients (the median age of OS patients was 34), the results showed that in patients with OS, treatment with cabozantinib gave rise to a 12% objective response rate and 33% of patients had 6 months of non-progression (meeting the study’s primary efficacy criterion). Moreover, a biomarker analysis indicated that an increased soluble (plasma) c-MET level at baseline was correlated with improved progression free survival, suggesting that this subpopulation of OS patients with high c-MET expression was responding to the anti-c-MET therapy (186). In a heavily pre-treated ES patient population, an objective response rate of 26% was seen (all PRs), and the primary efficacy criterion was met. However, biomarker analysis did not reveal any associations between serum c-MET and survival outcomes, and as cabozantinib also targets VEGFR2, it is not clear if in these cases the treatment benefit derived primarily from targeting c-MET activity or the anti-angiogenic effects of targeting VEGFR2 (186).
The Children’s Oncology Group has also investigated cabozantinib in relapsed or refractory solid tumors. After a phase I trial found that cabozantinib led to some PRs or stable disease (SD) (187), a phase II trial was conducted to determine activity. The trial was divided into six cohorts: OS (n = 29), ES, RMS, non-RMS soft tissue sarcoma, WT (all n = 13), and other rare tumors (n = 23). Overall, in OS 10/29 patients (34%) had disease control after 4 months (2 PR and 8 SD), while no partial or complete responses were seen in patients with EWS, RMS, or WT (though some PRs were seen in other rare tumors). The study showed potential for cabozantinib in treatment of OS, but limited efficacy in other sarcomas or WT (188).
A recent single-institution case series reported on the use of cabozantinib in four pediatric patients with relapsed NB. All four patients had extended disease control after treatment with cabozantinib for at least 6 months, while two patients had complete responses (>12 months) and remained in clinical remission at the time of publication (189). It is, however, unclear whether this benefit derived from targeting c-MET or rather targeting of VEGFR (or other RTKs sensitive to inhibition by cabozantinib).
Crizotinib
Crizotinib is another non-selective TKI targeting c-MET, though many of the clinical studies of crizotinib have focused primarily on its role targeting anaplastic lymphoma kinase (ALK). In the adult population, crizotinib is approved for treatment of NSCLC with altered ALK.
In pediatric populations, crizotinib has been investigated in a variety of solid tumors and anti-tumor activity has been correlated with the presence of ALK aberrations (190, 191). Crizotinib is approved for treatment of relapsed or refractory anaplastic large cell lymphoma with altered ALK.
From the perspective of targeting c-MET, a number of phase I and II trials which included children and adolescents in their eligibility criteria have examined the role of crizotinib in tumors with MET alterations.
The AcSé program in France supported a multi-tumor phase II trial wherein 107 pediatric tumors were analyzed for alterations in the molecular targets of crizotinib (ALK, MET, and ROS-1). From this cohort, 11 patients with genetic alterations were enrolled and received crizotinib (results from only eight patients were available at the time of abstract publication). The tumor analysis found one MET translocation, and three instances of MET amplification. Notably the single patient with a MET translocation (glioma) had progressive disease with crizotinib therapy (192).
The European Organization for Research and Treatment of Cancer (EORTC) “CREATE” trial was designed as a multi-center multi-tumor trial to evaluate crizotinib in a range of cancer types known to harbor alterations in the targets of crizotinib (ALK, MET, and ROS-1) (193). The trial was open to patients aged 16 and older, and though the patient cohort consisted primarily of adults, the trial contained cohorts of patients with cancers more commonly seen in children, adolescents, and young adults, and so is discussed here.
In advanced ARMS (positive for MET fusion protein), crizotinib (as a single agent) did not have meaningful clinical activity, with just one patient achieving a PR as the best outcome. As only eight patients out of 13 were evaluable for the primary end-point due to the aggressive course of their disease, the authors suggest that targeted therapeutics like crizotinib might be better trialed in aggressive cancers as an earlier treatment (in combination with standard chemotherapy). Given the lack of disease response to anti-c-MET therapy, the authors postulate that c-MET activation is likely not a key driver of disease progression at this advanced stage in ARMS, but may play a role in earlier differentiation (194).
The combination of crizotinib and another TKI, dasatinib, was tested in a Phase I trial in pediatric patients with advanced high-grade glioma or diffuse intrinsic pontine gliomas. Unfortunately, the combination was poorly tolerated, and little anti-tumor activity was appreciated. Notably, only 13% (2/16) of cases available for molecular analysis revealed MET amplification (195).
Discussion
Clearly, the HGF/c-MET pathway contributes to a wide variety of pediatric malignancies. Given its crucial role in development, proliferation, cell motility, and regeneration this is perhaps unsurprising, as aberrations in these cellular functions are commonly identified as drivers of oncogenesis and metastatic spread. There is ample evidence in the literature reviewed above to suggest that targeting the HGF/c-MET pathway could be an effective anti-cancer strategy across a range of tumor types. This promise has not, however, been fully borne out in the limited number of pediatric clinical trials that have been completed to date. Overall, the HGF/c- MET pathway is an exciting target and has shown early promise in cancers such as osteosarcoma. However, in many of the tumor types reviewed here, the principal effects of dysregulated HGF/c-MET signaling have largely been shown to be an increase in invasive/metastatic behavior, resistance to chemo- or radiotherapy, and a promotion of poorly differentiated “stem-like” cells, as opposed to driving cell proliferation and growth. This is likely reflected in the relative paucity of objective clinical responses seen in pediatric clinical trials to date. With the potential exception of tumors containing growth-sustaining genetic alterations in MET, treatment of tumors with anti-HGF/c-MET agents will likely be most impactful in preventing disease progression or relapse after treatment or resection, and when used in combination with cytotoxic therapies. Multiple pre-clinical studies reviewed above suggest that the HGF/c-MET pathway is important for protecting cancer cells from insult, and so it follows that anti-HGF/c-MET therapy could be effective as an adjuvant to other cytotoxic therapies. One of the key hurdles in cancer therapy is overcoming drug resistance developed within heavily pre-treated refractory tumors and targeting the HGF/c-MET pathway could be a promising tool for addressing this.
It is also important to acknowledge that, while invaluable for pre-clinical research and drug development, in vitro models generally do not reflect the diversity seen across tumors in patients. Many of the studies reviewed above included analysis of HGF/c-MET expression in patient samples, though there existed considerable variability in the methods for quantification, and the “normal” controls used between reports. Biomarker analysis for appropriate patient selection is crucial for any targeted therapy to achieve its predicted potential from pre-clinical data, although in early stages of development it is important to include open Phase I/II studies as to not exclude any groups who might potentially benefit from the therapy for reasons not yet fully elucidated. There are currently multiple ongoing or recruiting trials of anti-c-MET agents including pediatric patients, hence it is expected that additional key data will become available regarding the safety and efficacy of these agents for childhood cancers in the near future.
Author contributions
MG and AN contributed to the review of the literature and writing of the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This study was supported in part by an award from the Alberta Children’s Hospital Foundation and the Kids Cancer Care Foundation of Alberta (Grant #10024168). MG was supported by a Nat Christie Foundation Medical Entrance Award.
Acknowledgments
We thank Andy Tran for his critical review of the manuscript.
Conflict of interest
AN received laboratory research collaboration, funding, and expert panel membership from Apollomics Inc., Provectus Biopharmaceuticals Inc., CorMedix Inc., and Vincerx Pharma.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. (1987) 327:239–42. doi: 10.1038/327239a0
2. Gherardi E, Gray J, Stoker M, Perryman M, Furlong R. Purification of scatter factor, a fibroblast-derived basic protein that modulates epithelial interactions and movement. Proc Natl Acad Sci U.S.A. (1989) 86:5844–8. doi: 10.1073/pnas.86.15.5844
3. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. (2003) 4:915–25. doi: 10.1038/nrm1261
4. Gherardi E, Birchmeier W, Birchmeier C, Woude G. Vande. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. (2012) 12:89–103. doi: 10.1038/nrc3205
5. Koch JP, Aebersold DM, Zimmer Y, Medová M. MET targeting: time for a rematch. Oncogene. (2020) 39:2845–62. doi: 10.1038/s41388-020-1193-8
6. Park M, Dean M, Kaul K, Braun MJ, Gonda MA, Vande Woude G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc Natl Acad Sci U.S.A. (1987) 84:6379–83. doi: 10.1073/pnas.84.18.6379
7. Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. (1984) 311:29–33. doi: 10.1038/311029a0
8. Park M, Dean M, Cooper CS, Schmidt M, O’Brien SJ, Blair DG, et al. Mechanism of met oncogene activation. Cell. (1986) 45:895–904. doi: 10.1016/0092-8674(86)90564-7
9. Weidner KM, Arakaki N, Hartmann G, Vandekerckhove J, Weingart S, Rieder H, et al. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc Natl Acad Sci U.S.A. (1991) 88:7001–5. doi: 10.1073/pnas.88.16.7001
10. Bottaro DP, Rubin JS, Faletto DL, Chan AML, Kmiecik TE, Vande Woude GF, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. (1991) 251:802–4. doi: 10.1126/science.1846706
11. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. (2010) 11:834–48. doi: 10.1038/nrm3012
12. Weidner KM, Behrens J, Vandekerckhove J, Birchmeier W. Scatter factor: molecular characteristics and effect on the invasiveness of epithelial cells. J Cell Biol. (1990) 111:2097–108. doi: 10.1083/jcb.111.5.2097
13. Zhu H, Naujokas MA, Park M. Receptor chimeras indicate that the met tyrosine kinase mediates the motility and morphogenic responses of hepatocyte growth/scatter factor. Cell Growth Dif. (1994) 5:359–66.
14. Schmidt C, Bladt F, Goedecke S, Brlnkmann V, Zschiesche W, Sharpet M, et al. Scatter factor/hepatocyte essential for liver development. Nature. (1995) 373:699–702. doi: 10.1038/373699a0
15. Bladt F, Riethmacher D, Stefan I, Aguzzi A, Birchmeier C. Essential role for the c-mef receptor in the migration of myogenlc precursor cells into the limb bud. Nature. (1995) 376:768–71. doi: 10.1038/376768a0
16. Uehara Y, Minowa O, Mori C, Shiota K, Kuno J, Noda T, et al. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. (1995) 373:702–5. doi: 10.1038/373702a0
17. Matsumori A, Furukawa Y, Hashimoto T, Ono K, Shioi T, Okada M, et al. Increased circulating hepatocyte growth factor in the early stage of acute myocardial infarction. Biochem Biophys Res Commun. (1996) 221:391–5. doi: 10.1006/bbrc.1996.0606
18. Nakamura T, Mizuno S, Matsumoto K, Sawa Y. Myocardial protection from ischemia/reperfusion injury by endogenous and exogenous HGF. J Clin Invest. (2000) 106:1511–9. doi: 10.1172/JCI10226
19. Igawa T, Matsumoto K, Kanda S, Saito Y, Nakamura T. Hepatocyte growth factor may function as a renotropic factor for regeneration in rats with acute renal injury. Am J Physiol Ren Fluid Electrolyte Physiol. (1993) 265:61–9. doi: 10.1152/ajprenal.1993.265.1.F61
20. Borowiak M, Garratt AN, Wüstefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proc Natl Acad Sci U.S.A. (2004) 101:10608–13. doi: 10.1073/pnas.0403412101
21. Yanagita K, Matsumoto K, Sekiguchi K, Ishibashi H, Niho Y, Nakamura T. Hepatocyte growth factor may act as a pulmotrophic factor on lung regeneration after acute lung injury. J Biol Chem. (1993) 268:21212–7. doi: 10.1016/S0021-9258(19)36912-1
22. Huh CG, Factor VM, Sánchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U.S.A. (2004) 101:4477–82. doi: 10.1073/pnas.0306068101
23. Ishikawa T, Factor VM, Marquardt JU, Raggi C, Seo D, Kitade M, et al. Hepatocyte growth factor/c-met signaling is required for stem-cell-mediated liver regeneration in mice. Hepatology. (2012) 55:1215–26. doi: 10.1002/hep.24796
24. Chmielowiec J, Borowiak M, Morkel M, Stradal T, Munz B, Werner S, et al. c-Met is essential for wound healing in the skin. J Cell Biol. (2007) 177:151–62. doi: 10.1083/jcb.200701086
25. Zhou D, Tan RJ, Lin L, Zhou L, Liu Y. Activation of hepatocyte growth factor receptor, c-met, in renal tubules is required for renoprotection after acute kidney injury. Kidney Int. (2013) 84:509–20. doi: 10.1038/ki.2013.102
26. Ponzetto C, Bardelli A, Zhen Z, Maina F, dalla Zonca P, Giordano S, et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. (1994) 77:261–71. doi: 10.1016/0092-8674(94)90318-2
27. Weidner KM, Di Cesare S, Sachs M, Brinkmann V, Behrens J. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature. (1996) 384:173–6. doi: 10.1038/384173a0
28. Fixman, Fournier TM, Kamikura DM, Naujokas MA, Park M. Pathways downstream of Shc and Grb2 are required for cell transformation by the Tpr-Met oncoprotein. J Biol Chem. (1996) 271:13116–22. doi: 10.1074/jbc.271.22.13116
29. Boccaccio C, Andò M, Tamagnone L, Bardelli A, Michieli P, Battistini C, et al. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature. (1998) 391:285–8. doi: 10.1038/34657
30. Maroun CR, Naujokas MA, Holgado-Madruga M, Wong AJ, Park M. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol Cell Biol. (2000) 20:8513–25. doi: 10.1128/MCB.20.22.8513-8525.2000
31. Schaeper U, Gehring NH, Fuchs KP, Sachs M, Kempkes B, Birchmeier W. Coupling of Gab1 to c-Met. Grb2, and Shp2 mediates biological responses. J Cell Biol. (2000) 149:1419–32. doi: 10.1083/jcb.149.7.1419
32. Lai AZ, Abella JV, Park M. Crosstalk in Met receptor oncogenesis. Trends Cell Biol. (2009) 19:542–51. doi: 10.1016/j.tcb.2009.07.002
33. Ishibe S, Karihaloo A, Ma H, Zhang J, Marlier A, Mitobe M, et al. Met and the epidermal growth factor receptor act cooperatively to regulate final nephron number and maintain collecting duct morphology. Development. (2009) 136:337–45. doi: 10.1242/dev.024463
34. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. (2010) 17:77–88. doi: 10.1016/j.ccr.2009.11.022
35. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Joon OP, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. (2007) 316:1039–43. doi: 10.1126/science.1141478
36. Jeffers M, Taylor GA, Weidner KM, Omura S, Vande Woude GF. Degradation of the met tyrosine kinase receptor by the ubiquitin-proteasome pathway. Mol Cell Biol. (1997) 17:799–808. doi: 10.1128/MCB.17.2.799
37. Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, Langdon WY, et al. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol Cell. (2001) 8:995–1004. doi: 10.1016/S1097-2765(01)00378-1
38. Hammond DE, Urbé S, Vande Woude GF, Clague MJ. Down-regulation of MET, the receptor for hepatocyte growth factor. Oncogene. (2001) 20:2761–70. doi: 10.1038/sj.onc.1204475
39. Petrelli A, Gilestro GF, Lanzardo S, Comoglio PM, Migone N, Giordano S. The endophilin-CIN85-Cbl complex mediates ligand-dependent downregulation of c-Met. Nature. (2002) 416:187–90. doi: 10.1038/416187a
40. Gandino L, Longati P, Medico E, Prat M, Comoglio PM. Phosphorylation of serine 985 negatively regulates the hepatocyte growth factor receptor kinase. J Biol Chem. (1994) 269:1815–20. doi: 10.1016/S0021-9258(17)42099-0
41. Hashigasako A, Machide M, Nakamura T, Matsumoto K, Nakamura T. Bi-directional regulation of Ser-985 phosphorylation of c-Met via protein kinase C and protein phosphatase 2A involves c-Met activation and cellular responsiveness to hepatocyte growth factor. J Biol Chem. (2004) 279:26445–52. doi: 10.1074/jbc.M314254200
42. Foveau B, Ancot F, Leroy C, Petrelli A, Reiss K, Vingtdeux V, et al. Down-regulation of the met receptor tyrosine kinase by presenilin-dependent regulated intramembrane proteolysis. Mol Biol Cell. (2009) 20:2495–2250. doi: 10.1091/mbc.e08-09-0969
43. Palka HL, Park M, Tonks NK. Hepatocyte growth factor receptor tyrosine kinase met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J Biol Chem. (2003) 278:5728–35. doi: 10.1074/jbc.M210656200
44. Machide M, Hashigasako A, Matsumoto K, Nakamura T. Contact inhibition of hepatocyte growth regulated by functional association of the c-Met/hepatocyte growth factor receptor and LAR protein-tyrosine phosphatase. J Biol Chem. (2006) 281:8765–72. doi: 10.1074/jbc.M512298200
45. Sangwan V, Paliouras GN, Abella JV, Dubé N, Monast A, Tremblay ML, et al. Regulation of the met receptor-tyrosine kinase by the protein-tyrosine phosphatase 1B and T-cell phosphatase. J Biol Chem. (2008) 283:34374–83. doi: 10.1074/jbc.M805916200
46. Comoglio PM, Trusolino L, Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. (2018) 18:341–58. doi: 10.1038/s41568-018-0002-y
47. Liang TJ, Reid AE, Xavier R, Cardiff RD, Wang TC. Transgenic expression of tpr-met oncogene leads to development of mammary hyperplasia and tumors. J Clin Invest. (1996) 97:2872–7. doi: 10.1172/JCI118744
48. Soman NR, Correa P, Ruiz BA, Wogan GN. The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc Natl Acad Sci U.S.A. (1991) 88:4892–6. doi: 10.1073/pnas.88.11.4892
49. Skapek SX, Ferrari A, Gupta A, Lupo PJ, Butler E, Barr FG, et al. Rhabdomyosarcoma. Nat Rev Dis Prim. (2019) 5:1. doi: 10.1038/s41572-018-0051-2
50. Parham DM, Barr FG. Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol. (2013) 20:387. doi: 10.1097/PAP.0b013e3182a92d0d
51. Breneman JC, Lyden E, Pappo AS, Link MP, Anderson JR, Parham DM, et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma-a report from the intergroup rhabdomyosarcoma study IV. J Clin Oncol. (2003) 21:78–84. doi: 10.1200/JCO.2003.06.129
52. Oberlin O, Rey A, Lyden E, Bisogno G, Stevens MCG, Meyer WH, et al. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J Clin Oncol. (2008) 26:2384–9. doi: 10.1200/JCO.2007.14.7207
53. Bennicelli JL, Advani S, Schäfer BW, Barr FG. PAX3 and PAX7 exhibit conserved cis-acting transcription repression domains and utilize a common gain of function mechanism in alveolar rhabdomyosarcoma. Oncogene. (1999) 18:4348–56. doi: 10.1038/sj.onc.1202812
54. Skapek SX, Anderson J, Barr FG, Bridge JA, Gastier-Foster JM, Parham DM, et al. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children’s oncology group report. Pediatr Blood Cancer. (2013) 60:1411–7. doi: 10.1002/pbc.24532
55. Relaix F, Polimeni M, Rocancourt D, Ponzetto C, Schäfer BW, Buckingham M. The transcriptional activator PAX3-FKHR rescues the defects of Pax3 mutant mice but induces a myogenic gain-of-function phenotype with ligand-independent activation of Met signaling in vivo. Genes Dev. (2003) 17:2950–65. doi: 10.1101/gad.281203
56. Sharp R, Recio JA, Jhappan C, Otsuka T, Liu S, Yu Y, et al. Synergism between INK4a/ARF inactivation and aberrant HGF/SF signaling in rhabdomyosarcomagenesis. Nat Med. (2002) 8:1276–80. doi: 10.1038/nm787
57. Ginsberg JP, Davis RJ, Bennicelli JL, Nauta LE, Barr FG. Up-regulation of MET but not neural cell adhesion molecule expression by the PAX3-FKHR fusion protein in alveolar rhabdomyosarcoma. Cancer Res. (1998) 58:3542–6. doi: 10.1097/00043426-199807000-00084
58. Taulli R, Scuoppo C, Bersani F, Accornero P, Forni PE, Miretti S, et al. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma riccardo. Cancer Res. (2006) 66:4742–9. doi: 10.1016/j.critrevonc.2018.08.007
59. Jankowski K, Kucia M, Wysoczynski M, Reca R, Zhao D, Trzyna E, et al. Both hepatocyte growth factor (HGF) and stromal-derived factor-1 regulate the metastatic behavior of human rhabdomyosarcoma cells, but only HGF enhances their resistance to radiochemotherapy. Cancer Res. (2003) 63:7926–35.
60. Lukasiewicz E, Miekus K, Kijowski J, Drabik G, Wilusz M, Bobis-Wozowicz S, et al. Inhibition of rhabdomyosarcoma’s metastatic behavior through downregulation of MET receptor signaling. Folia Histochem Cytobiol. (2009) 47:485–9. doi: 10.2478/v10042-009-0108-x
61. Diomedi-Camassei F, McDowell HP, De loris MA, Uccini S, Altavista P, Raschellà G, et al. Clinical significance of CXC chemokine receptor-4 and c-Met in childhood rhabdomyosarcoma. Clin Cancer Res. (2008) 14:4119–27. doi: 10.1158/1078-0432.CCR-07-4446
62. Miekus K, Lukasiewicz E, Jarocha D, Sekula M, Drabik G, Majka M. The decreased metastatic potential of rhabdomyosarcoma cells obtained through MET receptor downregulation and the induction of differentiation. Cell Death Dis. (2013) 4:1–10. doi: 10.1038/cddis.2012.199
63. Anastasi S, Giordano S, Sthandier O, Gambarotta G, Maione R, Comoglio P, et al. A natural hepatocyte growth factor/scatter factor autocrine loop in myoblast cells and the effect of the constitutive met kinase activation on myogenic differentiation. J Cell Biol. (1997) 137:1057–68. doi: 10.1083/jcb.137.5.1057
64. Skrzypek K, Kusienicka A, Szewczyk B, Adamus T, Lukasiewicz E, Miekus K, et al. Constitutive activation of MET signaling impairs myogenic differentiation of rhabdomyosarcoma and promotes its development and progression. Oncotarget. (2015) 6:31378–98. doi: 10.18632/oncotarget.5145
65. Yan D, Dong Xda E, Chen X, Wang L, Lu C, Wang J, et al. MicroRNA-1/206 targets c-met and inhibits rhabdomyosarcoma development. J Biol Chem. (2009) 284:29596–604. doi: 10.1074/jbc.M109.020511
66. Taulli R, Bersani F, Foglizzo V, Linari A, Vigna E, Ladanyi M, et al. The muscle-specific microRNA miR-206 blocks human rhabdomyosarcoma growth in xenotransplanted mice by promoting myogenic differentiation. J Clin Invest. (2009) 119:2366–78. doi: 10.1172/JCI38075
67. Chen Y, Takita J, Mizuguchi M, Tanaka K, Ida K, Koh K, et al. Mutation and expression analyses of the MET and CDKN2A in rhabdomyosarcoma with emphasis on MET overexpression. Genes Chromosom Cancer. (2007) 46:348–58. doi: 10.1002/gcc.20416
68. Du J, Wang Y, Meng L, Liu Y, Pang Y, Cui W, et al. c-MET expression potentially contributes to the poor prognosis of rhabdomyosarcoma. Int J Clin Exp Pathol. (2018) 11:4083–92.
69. Anderson ME. Update on survival in osteosarcoma. Orthop Clin North Am. (2016) 47:283–92. doi: 10.1016/j.ocl.2015.08.022
70. Ferracini R, Di Renzo MF, Scotlandi K, Baldini N, Olivero M, Lollini P, et al. The MET/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or autocrine circuit. Oncogene. (1995) 10:739–49.
71. Scotlandi K, Baldini N, Oliviero M, Di Renzo MF, Martano M, Serra M, et al. Expression of met/hepatocyte growth factor receptor gene and malignant behavior of musculoskeletal tumors. Am J Pathol. (1996) 149:1209–19.
72. Oda Y, Naka T, Takeshita M, Iwamoto Y, Tsuneyoshi M. Comparison of histological changes and changes in nm23 and c-MET expression between primary and metastatic sites in osteosarcoma: a clinicopathologic and immunohistochemical study. Hum Pathol. (2000) 31:709–16. doi: 10.1053/hupa.2000.8230
73. Naka T, Iwamoto Y, Shinohara N, Ushijima M, Chuman H, Tsuneyoshi M. Expression of c-met proto-oncogene product (c-MET) in benign and malignant bone tumors. Mod Pathol. (1997) 10:832–8.
74. Coltella N, Manara MC, Cerisano V, Trusolino L, Di Renzo MF, Scotlandi K, et al. Role of the MET/HGF receptor in proliferation and invasive behavior of osteosarcoma. FASEB J. (2003) 17:1162–4. doi: 10.1096/fj.02-0576fje
75. Hassan SE, Bekarev M, Kim MY, Lin J, Piperdi S, Gorlick R, et al. Cell surface receptor expression patterns in osteosarcoma. Cancer. (2012) 118:740–9. doi: 10.1002/cncr.26339
76. Patanè S, Avnet S, Coltella N, Costa B, Sponza S, Olivero M, et al. MET overexpression turns human primary osteoblasts into osteosarcomas. Cancer Res. (2006) 66:4750–7. doi: 10.1158/0008-5472.CAN-05-4422
77. Dani N, Olivero M, Mareschi K, Van Duist MM, Miretti S, Cuvertino S, et al. The MET oncogene transforms human primary bone-derived cells into osteosarcomas by targeting committed osteo-progenitors. J Bone Miner Res. (2012) 27:1322–34. doi: 10.1002/jbmr.1578
78. Duan Z, Choy E, Harmon D, Liu X, Susa M, Mankin H, et al. MicroRNA-199a-3p is downregulated in human osteosarcoma and regulates cell proliferation and migration. Mol Cancer Ther. (2011) 10:1337–45. doi: 10.1158/1535-7163.MCT-11-0096
79. Yan K, Gao J, Yang T, Ma Q, Qiu X, Fan Q, et al. MicroRNA-34a inhibits the proliferation and metastasis of osteosarcoma cells both in vitro and in vivo. PLoS One. (2012) 7:e33778. doi: 10.1371/journal.pone.0033778
80. Niu G, Li B, Sun J, Sun L. miR-454 is down-regulated in osteosarcomas and suppresses cell proliferation and invasion by directly targeting c-Met. Cell Prolif. (2015) 48:348–55. doi: 10.1111/cpr.12187
81. Li X, Sun X, Wu J, Li Z. MicroRNA-613 suppresses proliferation, migration and invasion of osteosarcoma by targeting c-MET. Am J Cancer Res. (2016) 6:2869–79.
82. Georges S, Calleja LR, Jacques C, Lavaud M, Moukengue B, Lecanda F, et al. Loss of miR-198 and -206 during primary tumor progression enables metastatic dissemination in human osteosarcoma. Oncotarget. (2018) 9:35726–41. doi: 10.18632/oncotarget.26284
83. Xie W, Xiao J, Wang T, Zhang D, Li Z. MicroRNA-876-5p inhibits cell proliferation, migration and invasion by targeting c-Met in osteosarcoma. J Cell Mol Med. (2019) 23:3293–301. doi: 10.1111/jcmm.14217
84. Li Q, Lu C, Wang J, Gao M, Gao W. MicroRNA-449b-5p suppresses proliferation, migration, and invasian of osteosarcoma by targeting c-Met. Med Sci Monit. (2019) 25:6236–43. doi: 10.12659/MSM.918454
85. Sampson ER, Martin BA, Morris AE, Xie C, Schwarz EM, O’Keefe RJ, et al. The orally bioavailable met inhibitor PF-2341066 inhibits osteosarcoma growth and osteolysis/matrix production in a xenograft model. J Bone Miner Res. (2011) 26:1283–94. doi: 10.1002/jbmr.336
86. Fioramonti M, Fausti V, Pantano F, Iuliani M, Ribelli G, Lotti F, et al. Cabozantinib affects osteosarcoma growth through a direct effect on tumor cells and modifications in bone microenvironment. Sci Rep. (2018) 8:1–11. doi: 10.1038/s41598-018-22469-5
87. Fleuren EDG, Roeffen MHS, Leenders WP, Flucke UE, Vlenterie M, Schreuder HW, et al. Expression and clinical relevance of MET and ALK in Ewing sarcomas. Int J Cancer. (2013) 133:427–36. doi: 10.1002/ijc.28047
88. Charan M, Dravid P, Cam M, Audino A, Gross AC, Arnold MA, et al. GD2-directed CAR-T cells in combination with HGF-targeted neutralizing antibody (AMG102) prevent primary tumor growth and metastasis in Ewing sarcoma. Int J Cancer. (2020) 146:3184–95. doi: 10.1002/ijc.32743
89. Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. (2015) 17:iv1–62. doi: 10.1093/neuonc/nov189
90. Rosen EM, Laterra J, Joseph A, Jin L, Fuchs A, Way D, et al. Scatter factor expression and regulation in human glial tumors. Int J Cancer. (1996) 67:248–55. doi: 10.1002/(SICI)1097-0215(19960717)67:2<248::AID-IJC16>3.0.CO;2-7
91. Koochekpour S, Jeffers M, Rulong S, Taylor G, Klineberg E, Hudson EA, et al. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. (1997) 57:5391–8.
92. Nabeshima K, Shimao Y, Sato S, Kataoka H, Moriyama T, Kawano H, et al. Expression of c-Met correlates with grade of malignancy in human astrocytic tumours: an immunohistochemical study. Histopathology. (1997) 31:436–43. doi: 10.1046/j.1365-2559.1997.3010889.x
93. Kunkel P, Müller S, Schirmacher P, Stavrou D, Fillbrandt R, Westphal M, et al. Expression and localization of scatter factor/hepatocyte growth factor in human astrocytomas. Neuro Oncol. (2001) 3:82–8. doi: 10.1093/neuonc/3.2.82
94. Lamszus K, Schmidt NO, Jin L, Laterra J, Zagzag D, Way D, et al. Scatter factor promotes motility of human glioma and neuromicrovascular endothelial cells. Int J Cancer. (1998) 75:19–28. doi: 10.1002/(SICI)1097-0215(19980105)75:1<19::AID-IJC4>3.0.CO;2-4
95. Abounader R, Lal B, Luddy C, Koe G, Davidson B, Rosen EM, et al. In vivo targeting of SF/HGF and c-met expression via U1snRNA/ribozymes inhibits glioma growth and angiogenesis and promotes apoptosis. FASEB J. (2002) 16:108–10. doi: 10.1096/fj.01-0421fje
96. Cao B, Su Y, Oskarsson M, Zhao P, Kort EJ, Fisher RJ, et al. Neutralizing monoclonal antibodies to hepatocyte growth factor/scatter factor (HGF/SF) display antitumor activity in animal models. Proc Natl Acad Sci U.S.A. (2001) 98:7443–8. doi: 10.1073/pnas.131200498
97. Martens T, Schmidt NO, Eckerich C, Filibrandt R, Merchant M, Schwall R, et al. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res. (2006) 12:6144–52. doi: 10.1158/1078-0432.CCR-05-1418
98. Kim KJ, Wang L, Su YC, Gillespie GY, Salhotra A, Lal B, et al. Systemic anti-hepatocyte growth factor monoclonal antibody therapy induces the regression of intracranial glioma xenografts. Clin Cancer Res. (2006) 12:1292–8. doi: 10.1158/1078-0432.CCR-05-1793
99. Brockmann MA, Papadimitriou A, Brandt M, Fillbrandt R, Westphal M, Lamszus K. Inhibition of intracerebral glioblastoma growth by local treatment with the scatter factor/hepatocyte growth factor-antagonist NK4. Clin Cancer Res. (2003) 9:4578–85.
100. Kong DS, Song SY, Kim DH, Kyeung MJ, Yoo JS, Jong SK, et al. Prognostic significance of c-Met expression in glioblastomas. Cancer. (2009) 115:140–8. doi: 10.1002/cncr.23972
101. Pierscianek D, Kim YH, Motomura K, Mittelbronn M, Paulus W, Brokinkel B, et al. MET gain in diffuse astrocytomas is associated with poorer outcome. Brain Pathol. (2013) 23:13–8. doi: 10.1111/j.1750-3639.2012.00609.x
102. Olmez OF, Cubukcu E, Evrensel T, Kurt M, Avci N, Tolunay S, et al. The immunohistochemical expression of c-Met is an independent predictor of survival in patients with glioblastoma multiforme. Clin Transl Oncol. (2014) 16:173–7. doi: 10.1007/s12094-013-1059-4
103. Petterson SA, Dahlrot RH, Hermansen SK, K A. Munthe S, Gundesen MT, Wohlleben H, et al. High levels of c-Met is associated with poor prognosis in glioblastoma. J Neurooncol. (2015) 122:517–27. doi: 10.1007/s11060-015-1723-3
104. Abounader R, Laterra J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol. (2005) 7:436–51. doi: 10.1215/S1152851705000050
105. Brockmann MA, Ulbricht U, Grüner K, Fillbrandt R, Westphal M, Lamszus K, et al. Glioblastoma and cerebral microvascular endothelial cell migration in response to tumor-associated growth factors. Neurosurgery. (2003) 52:1391–9. doi: 10.1227/01.NEU.0000064806.87785.AB
106. Moriyama T, Kataoka H, Hamasuna R, Yokogami K, Uehara H, Kawano H, et al. Up-regulation of vascular endothelial growth factor induced by hepatocyte growth factor/scatter factor stimulation in human glioma cells. Biochem Biophys Res Commun. (1998) 249:73–7. doi: 10.1006/bbrc.1998.9078
107. Schmidt NO, Westphal M, Hagel C, Ergün S, Stavrou D, Rosen EM, et al. Levels of vascular endothelial growth factor, hepatocyte growth factor/scatter factor and basic fibroblast growth factor in human gliomas and their relation to angiogenesis. Int J Cancer. (1999) 84:10–8. doi: 10.1002/(SICI)1097-0215(19990219)84:1<10::AID-IJC3>3.0.CO;2-L
108. Eckerich C, Zapf S, Fillbrandt R, Loges S, Westphal M, Lamszus K. Hypoxia can induce c-Met expression in glioma cells and enhance SF/HGF-induced cell migration. Int J Cancer. (2007) 121:276–83. doi: 10.1002/ijc.22679
109. Boccaccio C, Comoglio PM. The MET oncogene in glioblastoma stem cells: implications as a diagnostic marker and a therapeutic target. Cancer Res. (2013) 73:3193–9. doi: 10.1158/0008-5472.CAN-12-4039
110. Li Y, Li A, Glas M, Lal B, Ying M, Sang Y, et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci U.S.A. (2011) 108:9951–6. doi: 10.1073/pnas.1016912108
111. De Bacco F, Casanova E, Medico E, Pellegatta S, Orzan F, Albano R, et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. (2012) 72:4537–50. doi: 10.1158/0008-5472.CAN-11-3490
112. Joo KM, Jin J, Kim E, Kim KH, Kim Y, Kang BG, et al. MET signaling regulates glioblastoma stem cells. Cancer Res. (2012) 72:3828–38. doi: 10.1158/0008-5472.CAN-11-3760
113. Qin Y, Musket A, Kou J, Preiszner J, Tschida BR, Qin A, et al. Overexpression of HGF/MET axis along with p53 inhibition induces de novo glioma formation in mice. Neuro-Oncology Adv. (2020) 2:1–12. doi: 10.1093/noajnl/vdaa067
114. Bowers DC, Fan S, Walter KA, Abounader R, Williams JA, Rosen EM, et al. Scatter factor/hepatocyte growth factor protects against cytotoxic death in human glioblastoma via phosphatidylinositol 3-kinase- and AKT-dependent pathways. Cancer Res. (2000) 60:4277–83.
115. Lal B, Xia S, Abounader R, Laterra J. Targeting the c-Met pathway potentiates glioblastoma responses to γ-radiation. Clin Cancer Res. (2005) 11:4479–86. doi: 10.1158/1078-0432.CCR-05-0166
116. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell. (2017) 32:520.e–37.e. doi: 10.1016/j.ccell.2017.08.017
117. Miklja Z, Pasternak A, Stallard S, Nicolaides T, Kline-Nunnally C, Cole B, et al. Molecular profiling and targeted therapy in pediatric gliomas: review and consensus recommendations. Neuro Oncol. (2019) 21:968–80. doi: 10.1093/neuonc/noz022
118. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. (2014) 46:444–50. doi: 10.1038/ng.2938
119. Bender S, Gronych J, Warnatz HJ, Hutter B, Gröbner S, Ryzhova M, et al. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat Med. (2016) 22:1314–20. doi: 10.1038/nm.4204
120. Ieraci A, Forni PE, Ponzetto C. Viable hypomorphic signaling mutant of the met receptor reveals a role for hepatocyte growth factor in postnatal cerebellar development. Proc Natl Acad Sci U.S.A. (2002) 99:15200–5. doi: 10.1073/pnas.222362099
121. Tong CYK, Hui ABY, Yin XL, Pang JCS, Zhu XL, Poon WS, et al. Detection of oncogene amplifications in medulloblastomas by comparative genomic hybridization and array-based comparative genomic hybridization. J Neurosurg. (2004) 100:187–93. doi: 10.3171/ped.2004.100.2.0187
122. Li Y, Lal B, Kwon S, Fan X, Saldanha U, Reznik TE, et al. The scatter factor/hepatocyte growth factor: c-Met pathway in human embryonal central nervous system tumor malignancy. Cancer Res. (2005) 65:9355–62. doi: 10.1158/0008-5472.CAN-05-1946
123. Kongkham PN, Onvani S, Smith CA, Rutka JT. Inhibition of the met receptor tyrosine kinase as a novel therapeutic strategy in medulloblastoma. Transl Oncol. (2010) 3:336–43. doi: 10.1593/tlo.10121
124. Guessous F, Yang Y, Johnson E, Marcinkiewicz L, Smith M, Zhang Y, et al. Cooperation between c-Met and focal adhesion kinase family members in medulloblastoma and implications for therapy. Mol Cancer Ther. (2012) 11:288–97. doi: 10.1158/1535-7163.MCT-11-0490
125. Li Y, Guessous F, Johnson EB, Eberhart CG, Li XN, Shu Q, et al. Functional and molecular interactions between the HGF/c-Met pathway and c-Myc in large-cell medulloblastoma. Lab Investig. (2008) 88:98–111. doi: 10.1038/labinvest.3700702
126. Kongkham PN, Northcott PA, Ra YS, Nakahara Y, Mainprize TG, Croul SE, et al. An epigenetic genome-wide screen identifies SPINT2 as a novel tumor suppressor gene in pediatric medulloblastoma. Cancer Res. (2008) 68:9945–53. doi: 10.1158/0008-5472.CAN-08-2169
127. Tripolitsioti D, Kumar KS, Neve A, Migliavacca J, Capdeville C, Rushing EJ, et al. MAP4K4 controlled integrin β1 activation and c-Met endocytosis are associated with invasive behavior of medulloblastoma cells. Oncotarget. (2018) 9:23220–36. doi: 10.18632/oncotarget.25294
128. Onvani S, Terakawa Y, Smith C, Northcott P, Taylor M, Rutka J. Molecular genetic analysis of the hepatocyte growth factor/MET signaling pathway in pediatric medulloblastoma. Genes Chromosom Cancer. (2012) 57:675–88. doi: 10.1002/gcc.21954
129. Faria CC, Golbourn BJ, Dubuc AM, Remke M, Diaz RJ, Agnihotri S, et al. Foretinib is effective therapy for metastatic sonic hedgehog medulloblastoma. Cancer Res. (2015) 75:134–46. doi: 10.1158/0008-5472.CAN-13-3629
130. Santhana Kumar K, Tripolitsioti D, Ma M, Grählert J, Egli KB, Fiaschetti G, et al. The Ser/Thr kinase MAP4K4 drives c-Met-induced motility and invasiveness in a cell-based model of SHH medulloblastoma. Springerplus. (2015) 4:19. doi: 10.1186/s40064-015-0784-2
131. Binning MJ, Niazi T, Pedone CA, Lal B, Eberhart CG, Kim KJ, et al. Hepatocyte growth factor and Sonic hedgehog expression in cerebellar neural progenitor cells costimulate medulloblastoma initiation and growth. Cancer Res. (2008) 68:7838–45. doi: 10.1158/0008-5472.CAN-08-1899
132. Coon V, Laukert T, Pedone CA, Laterra J, Kim KJ, Fults DW. Molecular therapy targeting sonic hedgehog and hepatocyte growth factor signaling in a mouse model of medulloblastoma. Mol Cancer Ther. (2010) 9:2627–36. doi: 10.1158/1535-7163.MCT-10-0486
133. Van Arendonk KJ, Chung DH. Neuroblastoma: tumor biology and its implications for staging and treatment. Children. (2019) 6:12. doi: 10.3390/children6010012
134. Hecht M, Papoutsi M, Tran HD, Wilting J, Schweigerer L. Hepatocyte growth factor/c-Met signaling promotes the progression of experimental human neuroblastomas. Cancer Res. (2004) 64:6109–18. doi: 10.1158/0008-5472.CAN-04-1014
135. Hecht M, Schulte JH, Eggert A, Wilting J, Schweigerer L. The neurotrophin receptor TrkB cooperates with c-Met in enhancing neuroblastoma invasiveness. Carcinogenesis. (2005) 26:2105–15. doi: 10.1093/carcin/bgi192
136. Ren Y, Chan HM, Fan J, Xie Y, Chen YX, Li W, et al. Inhibition of tumor growth and metastasis in vitro and in vivo by targeting macrophage migration inhibitory factor in human neuroblastoma. Oncogene. (2006) 25:3501–8. doi: 10.1038/sj.onc.1209395
137. Sköldenberg EG, Larsson A, Jakobson Å, Hedborg F, Kogner P, Christofferson RH, et al. The angiogenic growth factors HGF and VEGF in serum and plasma from neuroblastoma patients. Anticancer Res. (2009) 29:3311–9.
138. Crosswell HE, Dasgupta A, Alvarado CS, Watt T, Christensen JG, De P, et al. PHA665752, a small-molecule inhibitor of c-Met, inhibits hepatocyte growth factor-stimulated migration and proliferation of c-Met-positive neuroblastoma cells. BMC Cancer. (2009) 9:1–10. doi: 10.1186/1471-2407-9-411
139. Yan B, Lim M, Zhou L, Kuick CH, Leong MY, Yong KJ, et al. Identification of MET genomic amplification, protein expression and alternative splice isoforms in neuroblastomas. J Clin Pathol. (2013) 66:985–91. doi: 10.1136/jclinpath-2012-201375
140. Scorsone K, Zhang L, Woodfield SE, Hicks J, Zage PE. The novel kinase inhibitor EMD1214063 is effective against neuroblastoma. Invest New Drugs. (2014) 32:815–24. doi: 10.1007/s10637-014-0107-4
141. Zhang L, Scorsone K, Woodfield SE, Zage PE. Sensitivity of neuroblastoma to the novel kinase inhibitor cabozantinib is mediated by ERK inhibition. Cancer Chemother Pharmacol. (2015) 76:977–87. doi: 10.1007/s00280-015-2871-z
142. Daudigeos-Dubus E, Le Dret L, Bawa O, Opolon P, Vievard A, Villa I, et al. Dual inhibition using cabozantinib overcomes HGF/MET signaling mediated resistance to pan-VEGFR inhibition in orthotopic and metastatic neuroblastoma tumors. Int J Oncol. (2017) 50:203–11. doi: 10.3892/ijo.2016.3792
143. Woolf AS, Kolatsi-Joannou M, Hardman P, Andermarcher E, Moorby C, Fine LG, et al. Roles of hepatocyte growth factor/scatter factor and the met receptor in the early development of the metanephros. J Cell Biol. (1995) 128:171–84. doi: 10.1083/jcb.128.1.171
144. Schmidt L, Junker K, Nakaigawa N, Kinjerski T, Weirich G, Miller M, et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. (1999) 18:2343–50. doi: 10.1038/sj.onc.1202547
145. Alami J, Williams BRG, Yeger H. Expression and localization of HGF and met in Wilms’ tumours. J Pathol. (2002) 196:76–84. doi: 10.1002/path.997
146. Vuononvirta R, Sebire NJ, Messahel B, Perusinghe N, Reis-Filho JS, Pritchard-Jones K, et al. Expression of hepatocyte growth factor and its receptor met in wilms’ tumors and nephrogenic rests reflects their roles in kidney development. Clin Cancer Res. (2009) 15:2723–30. doi: 10.1158/1078-0432.CCR-08-1898
147. Bell D, Ranganathan S, Tao J, Monga SPS. Novel advances in understanding of molecular pathogenesis of hepatoblastoma: a Wnt/β-Catenin perspective. Gene Expr. (2017) 17:141–54. doi: 10.3727/105221616X693639
148. Von Schweinitz D, Faundez A, Teichmann B, Birnbaum T, Koch A, Hecker H, et al. Hepatocyte growth-factor-scatter factor can stimulate post-operative tumor-cell proliferation in childhood hepatoblastoma. Int J Cancer. (2000) 85:151–9. doi: 10.1002/(SICI)1097-0215(20000115)85:2<151::AID-IJC1>3.0.CO;2-6
149. Grotegut S, Kappler R, Tarimoradi S, Lehembre F, Christofori G, Von Schweinitz D. Hepatocyte growth factor protects hepatoblastoma cells from chemotherapy-induced apoptosis by AKT activation. Int J Oncol (2010) 36:1261–7. doi: 10.3892/ijo_00000610
150. Ranganathan S, Tan X, Monga SPS. B -Catenin and met deregulation in childhood hepatoblastomas. Pediatr Dev Pathol. (2005) 8:435–47. doi: 10.1007/s10024-005-0028-5
151. Monga SPS, Mars WM, Pediaditakis P, Bell A, Mulé K, Bowen WC, et al. Hepatocyte growth factor induces Wnt-independent nuclear translocation of β-catenin after Met-β-catenin dissociation in hepatocytes. Cancer Res. (2002) 62:2064–71.
152. Zeng G, Apte U, Micsenyi A, Bell A, Monga SPS. Tyrosine residues 654 and 670 in beta-catenin are crucial in regulation of Met-β-catenin interactions. Cell. (2006) 312:3620–30. doi: 10.1016/j.yexcr.2006.08.003
153. Purcell R, Childs M, Maibach R, Miles C, Turner C, Zimmermann A, et al. HGF/c-Met related activation of beta-catenin in hepatoblastoma. J Exp Clin Cancer Res. (2011) 30:17–9. doi: 10.1186/1756-9966-30-96
154. Matsumoto S, Yamamichi T, Shinzawa K, Kasahara Y, Nojima S, Kodama T, et al. GREB1 induced by Wnt signaling promotes development of hepatoblastoma by suppressing TGFβ signaling. Nat Commun. (2019) 10:3882. doi: 10.1038/s41467-019-11533-x
155. von Schweinitz D, Kraus JA, Albrecht S, Koch A, Fuchs J, Pietsch T. Prognostic impact of molecular genetic alterations in hepatoblastoma. Med Pediatr Oncol. (2002) 38:104–8. doi: 10.1002/mpo.1280
156. Schmid I, von Schweinitz D. Pediatric hepatocellular carcinoma: challenges and solutions. J Hepatocell Carcinoma. (2017) 4:15–21. doi: 10.2147/JHC.S94008
157. Weeda VB, Aronson DC, Verheij J, Lamers WH. Is hepatocellular carcinoma the same disease in children and adults? Comparison of histology, molecular background, and treatment in pediatric and adult patients. Pediatr Blood Cancer. (2019) 66:1–9. doi: 10.1002/pbc.27475
158. Park WS, Dong SM, Kim SY, Na EY, Shin MS, Pi JH, et al. Somatic mutations in the kinase domain of the MET/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res. (1999) 59:307–10.
159. Goyal L, Muzumdar MD, Zhu AX. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res. (2013) 19:2310–8. doi: 10.1158/1078-0432.CCR-12-2791
160. Giordano S, Columbano A. Met as a therapeutic target in HCC: facts and hopes. J Hepatol. (2014) 60:442–52. doi: 10.1016/j.jhep.2013.09.009
161. Suzuki K, Hayashi N, Yamada Y, Yoshihara H, Miyamoto Y, Ito Y, et al. Expression of the c-met protooncogene in human hepatocellular carcinoma. Hepatology. (1994) 20:1231–6. doi: 10.1002/hep.1840200520
162. Ueki T, Fujimoto J, Suzuki T, Yamamoto H, Okamoto E. Expression of hepatocyte growth factor and its receptor, the c-met proto-oncogene, in hepatocellular carcinoma. Hepatology. (1997) 25:619–23. doi: 10.1002/hep.510250321
163. D’Errico A, Fiorentino M, Ponzetto A, Daikuhara Y, Tsubouchi H, Brechot C, et al. Liver hepatocyte growth factor does not always correlate with hepatocellular proliferation in human liver lesions: its specific receptor c- met does. Hepatology. (1996) 24:60–4. doi: 10.1002/hep.510240112
164. Wang ZL, Liang P, Dong BW, Yu XL, Yu DJ. Prognostic factors and recurrence of small hepatocellular carcinoma after hepatic resection or microwave ablation: a retrospective study. J Gastrointest Surg. (2008) 12:327–37. doi: 10.1007/s11605-007-0310-0
165. Kondo S, Ojima H, Tsuda H, Hashimoto J, Morizane C, Ikeda M, et al. Clinical impact of c-Met expression and its gene amplification in hepatocellular carcinoma. Int J Clin Oncol. (2013) 18:207–13. doi: 10.1007/s10147-011-0361-9
166. Shiota G, Rhoads DB, Wang TC, Nakamura T, Schmidt EV. Hepatocyte growth factor inhibits growth of hepatocellular carcinoma cells. Proc Natl Acad Sci U.S.A. (1992) 89:373–7. doi: 10.1073/pnas.89.1.373
167. Horiguchi N, Takayama H, Toyoda M, Otsuka T, Fukusato T, Merlino G, et al. Hepatocyte growth factor promotes hepatocarcinogenesis through c-Met autocrine activation and enhanced angiogenesis in transgenic mice treated with diethylnitrosamine. Oncogene. (2002) 21:1791–9. doi: 10.1038/sj.onc.1205248
168. Liu ML, Mars WM, Michalopoulos GK. Hepatocyte growth factor inhibits cell proliferation in vivo of rat hepatocellular carcinomas induced by diethylnitrosamine. Carcinogenesis. (1995) 16:841–3. doi: 10.1093/carcin/16.4.841
169. Takami T, Kaposi-Novak P, Uchida K, Gomez-Quiroz LE, Conner EA, Factor VM, et al. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine-induced hepatocarcinogenesis. Cancer Res. (2007) 67:9844–51. doi: 10.1158/0008-5472.CAN-07-1905
170. Wang R, Ferrell LD, Faouzi S, Maher JJ, Michael Bishop J. Activation of the met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J Cell Biol. (2001) 153:1023–33. doi: 10.1083/jcb.153.5.1023
171. Tao J, Xu E, Zhao Y, Singh S, Xiaolei L, Couchy G, et al. Modeling a human hcc subset in mice through co- expression of met and point-mutant β-catenin. Hepatology. (2016) 64:1587–605. doi: 10.1002/hep.28601
172. Ramirez R, Hsu D, Patel A, Fenton C, Dinauer C, Tuttle RM, et al. Over-expression of hepatocyte growth factor/scatter factor (HGF/SF) and the HGF/SF receptor (cMET) are associated with a high risk of metastasis and recurrence for children and young adults with papillary thyroid carcinoma. Clin Endocrinol. (2000) 53:635–44. doi: 10.1046/j.1365-2265.2000.01124.x
173. Puccini A, Marin-Ramos NI, Bergamo F, Schirripa M, Lonardi S, Lenz H-J, et al. Safety and tolerability of c-MET inhibitors in cancer. Drug Saf. (2019) 42:211–33. doi: 10.1007/s40264-018-0780-x
174. Wang H, Rao B, Lou J, Li J, Liu Z, Li A, et al. The function of the HGF/c-Met axis in hepatocellular carcinoma. Front Cell Dev Biol. (2020) 8:1–15. doi: 10.3389/fcell.2020.00055
175. Hong L, Zhang J, Heymach JV, Le X. Current and future treatment options for MET exon 14 skipping alterations in non-small cell lung cancer. Ther Adv Med Oncol. (2021) 13:1–16. doi: 10.1177/1758835921992976
176. Goździk-Spychalska J, Szyszka-Barth K, Spychalski Ł, Ramlau K, Wójtowicz J, Batura-Gabryel H, et al. c-MET inhibitors in the treatment of lung cancer. Curr Treat Options Oncol. (2014) 15:670–82. doi: 10.1007/s11864-014-0313-5
177. Hu H, Mu Q, Bao Z, Chen Y, Liu Y, Chen J, et al. Mutational landscape of secondary glioblastoma guides MET-targeted trial in brain tumor. Cell. (2018) 175:1665.e–78.e. doi: 10.1016/j.cell.2018.09.038
178. Kim KH, Kim H. Progress of antibody-based inhibitors of the HGF-cMET axis in cancer therapy. Exp Mol Med. (2017) 49:e307–307. doi: 10.1038/emm.2017.17
179. Fu J, Su X, Li Z, Deng L, Liu X, Feng X, et al. HGF/c-MET pathway in cancer: from molecular characterization to clinical evidence. Oncogene. (2021) 40:4625–51. doi: 10.1038/s41388-021-01863-w
180. Munshi N, Jeay S, Li Y, Chen CR, France DS, Ashwell MA, et al. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. (2010) 9:1544–53. doi: 10.1158/1535-7163.MCT-09-1173
181. Katayama R, Aoyama A, Yamori T, Qi J, Oh-hara T, Song Y, et al. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. (2013) 73:3087–96. doi: 10.1158/0008-5472.CAN-12-3256
182. Basilico C, Pennacchietti S, Vigna E, Chiriaco C, Arena S, Bardelli A, et al. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin Cancer Res. (2013) 19:2381–92. doi: 10.1158/1078-0432.CCR-12-3459
183. Geller JI, Perentesis JP, Liu X, Minard CG, Kudgus RA, Reid JM, et al. A phase 1 study of the c-Met inhibitor, tivantinib (ARQ197) in children with relapsed or refractory solid tumors: a children’s oncology group study phase 1 and pilot consortium trial (ADVL1111). Pediatr Blood Cancer. (2017) 64:1–21. doi: 10.1002/pbc.26565
184. Wagner AJ, Goldberg JM, Dubois SG, Choy E, Lee R, Pappo A, et al. Tivantinib (ARQ 197), a selective inhibitor of MET, in patients with microphthalmia transcription factor-associated tumors: results of a multicenter phase 2 trial. Cancer. (2012) 118:5894–902. doi: 10.1002/cncr.27582
185. Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. (2011) 10:2298–308. doi: 10.1158/1535-7163.MCT-11-0264
186. Italiano A, Mir O, Mathoulin-Pelissier S, Penel N, Piperno-Neumann S, Bompas E, et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol. (2020) 21:446–55. doi: 10.1016/S1470-2045(19)30825-3
187. Chuk MK, Widemann BC, Minard CG, Liu X, Kim AR, Bernhardt MB, et al. A phase 1 study of cabozantinib in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: trial ADVL1211, a report from the children’s oncology group. Pediatr Blood Cancer. (2018) 65:1–7. doi: 10.1002/pbc.27077
188. Akshintala S, Widemann BC, Barkauskas DA, Hall D, Reid JM, Voss SD, et al. Phase 2 trial of cabozantinib in children and young adults with refractory sarcomas, Wilms tumor, and rare tumors: children’s oncology group study (ADVL1622). J Clin Oncol. (2021) 39:10010. doi: 10.1200/JCO.2021.39.15_suppl.10010
189. Perisa MP, Storey M, Streby KA, Ranalli MA, Skeens M, Shah N. Cabozantinib for relapsed neuroblastoma: single institution case series. Pediatr Blood Cancer. (2020) 67:1–4. doi: 10.1002/pbc.28317
190. Mossé YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a children’s oncology group phase 1 consortium study. Lancet Oncol. (2013) 14:472–80. doi: 10.1016/S1470-2045(13)70095-0
191. Mossé YP, Voss SD, Lim MS, Rolland D, Minard CG, Fox E, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a Children’s Oncology Group study. J Clin Oncol. (2017) 35:3215–21. doi: 10.1200/JCO.2017.73.4830
192. Vassal G, Faivre L, Geoerger B, Plantaz D, Auvrignon A, Coze C, et al. Crizotinib in children and adolescents with advanced ROS1, MET, or ALK-rearranged cancer: results of the AcSé phase II trial. J Clin Oncol. (2016) 34:11509. doi: 10.1200/JCO.2016.34.15_suppl.11509
193. Péron J, Marreaud S, Staelens D, Raveloarivahy T, Nzokirantevye A, Flament J, et al. A multinational, multi-tumour basket study in very rare cancer types: the European organization for research and treatment of cancer phase II 90101 ‘CREATE’ trial. Eur J Cancer. (2019) 109:192–5. doi: 10.1016/j.ejca.2018.12.013
194. Schöffski P, Wozniak A, Leahy MG, Aamdal S, Rutkowski P, Bauer S, et al. The tyrosine kinase inhibitor crizotinib does not have clinically meaningful activity in heavily pre-treated patients with advanced alveolar rhabdomyosarcoma with FOXO rearrangement: European organisation for research and treatment of cancer phase 2 trial. Eur J Cancer. (2018) 94:156–67. doi: 10.1016/j.ejca.2018.02.011
195. Broniscer A, Jia S, Mandrell B, Hamideh D, Huang J, Onar-Thomas A, et al. Phase 1 trial, pharmacokinetics, and pharmacodynamics of dasatinib combined with crizotinib in children with recurrent or progressive high-grade and diffuse intrinsic pontine glioma. Pediatr Blood Cancer. (2018) 65:1–8. doi: 10.1002/pbc.27035
196. Gibson EG, Selvo NS, Campagne O, Gajjar A, Stewart CF. Abstract 1357: population pharmacokinetic analysis of crizotinib in children with progressive/recurrent high-grade and diffuse intrinsic pontine gliomas. Cancer Res (2021) 81:1357L–1357. doi: 10.1158/1538-7445.AM2021-1357
197. Balis FM, Thompson PA, Mosse YP, Blaney SM, Minard CG, Weigel BJ, et al. First-dose and steady-state pharmacokinetics of orally administered crizotinib in children with solid tumors: a report on ADVL0912 from the children’s oncology group phase 1/Pilot consortium. Cancer Chemother Pharmacol. (2017) 79:181–7. doi: 10.1007/s00280-016-3220-6
198. Greengard E, Mosse YP, Liu X, Minard CG, Reid JM, Voss S, et al. Safety, tolerability and pharmacokinetics of crizotinib in combination with cytotoxic chemotherapy for pediatric patients with refractory solid tumors or anaplastic large cell lymphoma (ALCL): a children’s oncology group phase 1 consortium study (ADVL1212. Cancer Chemother Pharmacol. (2020) 86:829–40. doi: 10.1007/s00280-020-04171-4
199. Gambacorti-Passerini C, Orlov S, Zhang L, Braiteh F, Huang H, Esaki T, et al. Long-term effects of crizotinib in ALK-positive tumors (excluding NSCLC): a phase 1b open-label study. Am J Hematol. (2018) 93:607–14. doi: 10.1002/ajh.25043
200. Kato S, Jardim DL, Johnson FM, Subbiah V, Piha-Paul S, Tsimberidou AM, et al. Phase I study of the combination of crizotinib (as a MET inhibitor) and dasatinib (as a c-SRC inhibitor) in patients with advanced cancer. Invest New Drugs. (2018) 36:416–23. doi: 10.1007/s10637-017-0513-5
201. Schöffski P, Wozniak A, Stacchiotti S, Rutkowski P, Blay JY, Lindner LH, et al. Activity and safety of crizotinib in patients with advanced clear-cell sarcoma with MET alterations: European organization for research and treatment of cancer phase II trial 90101 “CREATE.”. Ann Oncol. (2017) 28:3000–8. doi: 10.1093/annonc/mdx527
202. Schöffski P, Wozniak A, Kasper B, Aamdal S, Leahy MG, Rutkowski P, et al. Activity and safety of crizotinib in patients with alveolar soft part sarcoma with rearrangement of TFE3: European organization for research and treatment of cancer (EORTC) phase II trial 90101 “CREATE.”. Ann Oncol. (2018) 29:758–65. doi: 10.1093/annonc/mdx774
Keywords: mesenchymal epithelial transition factor (MET), hepatocyte growth factor (HGF), scatter factor, pediatric solid tumor, metastatic, targeted therapy
Citation: Grundy M and Narendran A (2022) The hepatocyte growth factor/mesenchymal epithelial transition factor axis in high-risk pediatric solid tumors and the anti-tumor activity of targeted therapeutic agents. Front. Pediatr. 10:910268. doi: 10.3389/fped.2022.910268
Received: 01 April 2022; Accepted: 15 July 2022;
Published: 10 August 2022.
Edited by:
Yong-mi Kim, Children’s Hospital of Los Angeles, United StatesReviewed by:
Joanna Kitlinska, Georgetown University, United StatesTakashi Kato, Yasuda Women’s University, Japan
Copyright © 2022 Grundy and Narendran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aru Narendran, YS5uYXJlbmRyYW5AdWNhbGdhcnkuY2E=