 Vijay Mehta
Vijay Mehta Puanani E. Hopson2
Puanani E. Hopson2 Samit B. Patel
Samit B. Patel Devendra I. Mehta
Devendra I. Mehta
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 29 September 2022
Sec. Pediatric Gastroenterology, Hepatology and Nutrition
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.909648
This article is part of the Research TopicInsights in Pediatric Pancreatology 2022View all 12 articles
The pancreas has both endocrine and exocrine function and plays an important role in digestion and glucose control. Understanding the development of the pancreas, grossly and microscopically, and the genetic factors regulating it provides further insight into clinical problems that arise when these processes fail. Animal models of development are known to have inherent issues when understanding human development. Therefore, in this review, we focus on human studies that have reported gross and microscopic development including acinar-, ductal-, and endocrine cells and the neural network. We review the genes and transcription factors involved in organ formation using data from animal models to bridge current understanding where necessary. We describe the development of exocrine function in the fetus and postnatally. A deeper review of the genes involved in pancreatic formation allows us to describe the development of the different groups (proteases, lipids, and amylase) of enzymes during fetal life and postnatally and describe the genetic defects. We discuss the constellation of gross anatomical, as well as microscopic defects that with genetic mutations lead to pancreatic insufficiency and disease states.
The pancreas has both endocrine and exocrine functions. The endocrine system consists of multiple peptide hormones that function to regulate blood glucose but also influence exocrine functions (e.g., somatostatin). Its exocrine function involves the secretion of enzymes, bicarbonate, and water to aid in the digestion of nutrients.
There are many unanswered questions related to embryonic pancreatic development. Due to ethical issues, research in human development of the pancreas has been limited and predominantly conducted via animal studies.
In this review, we describe the current understanding of the development of the human pancreas, including gross and microscopic anatomy with a focus on exocrine function. In addition, we will describe disease states as the consequence of abnormalities in pancreas development and the genetic mutations behind them. We will utilize animal models to highlight the possible development of the human pancreas and its implications in disease states.
The foregut endoderm gives rise to the dorsal and ventral pancreatic buds between days 26 and 31 of embryonic development.
Initially, there are two ventral pancreatic buds but the left side regresses. Figure 1 depicts the rotation of the stomach and duodenum starting at week 5 (Figure 1A) which results in the fusion of the two buds with the ventral bud lying posterior by week 6 (Figure 1B). The majority of the organ is derived from the dorsal bud while the ventral bud will give rise to the uncinate process and part of the head of the pancreas (Figure 1C) (1–3).
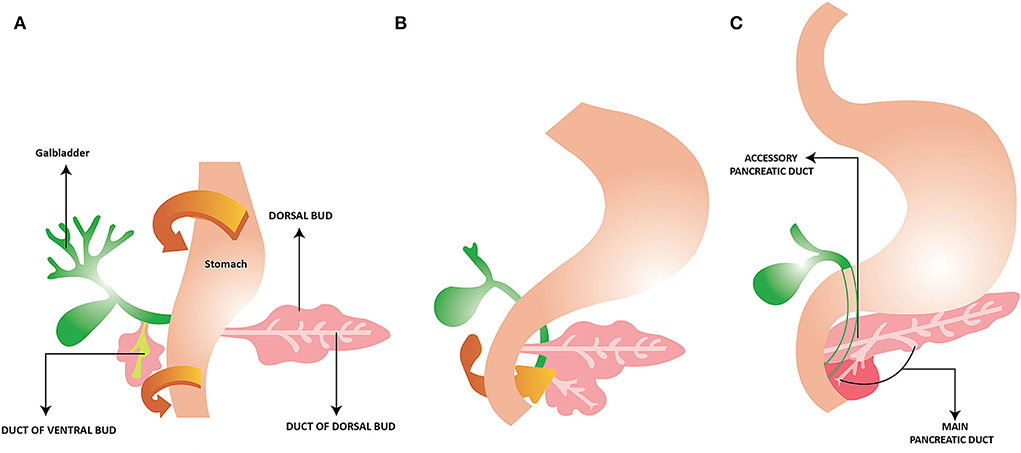
Figure 1. Development of the Pancreas. (A) Rotation of the ventral bud with the stomach and duodenum to the right. (B) The ventral and dorsal bud fuse together. (C) The main pancreatic duct exiting via the ventral bud to the duodenum. Ventral bud gives rise to uncinate process and part of the head of the pancreas. The dorsal bud gives rise to the remainder of the pancreas. Reprinted from “Pancreatic Duct Variations and the Risk of Post-Endoscopic Retrograde Cholangiopancreatography Pancreatitis” Ojo A S. Cureus, 12 (9): e10445. 2020 by Cureus.
The main pain of the pancreatic duct (duct of Wirsung) is formed from the pancreatic duct of the ventral bud and the distal part of the duct of the dorsal bud. The proximal part of the dorsal bud contains an accessory pancreatic duct that opens into the minor duodenal papilla (4, 5). Initially, the ducts of the dorsal and ventral pancreas fuse, followed by partial regression of the dorsal pancreatic duct proximal to the duodenum to form Santorini's canal (6). In some, this will open into the minor papilla, while in others, it will be non-draining or connect with the main duct (6–8). Lack of fusion of the ventral and pancreatic ducts results in pancreatic divisum (9). By the 8th week, the bile tree and main pancreatic duct are joined together at the duodenum (10).
Three variants have been noted in the location of biliary and pancreatic ducts. The most common is when the pancreatic and bile duct joins at variable distances from the duodenum. The second variant includes joining at the duodenal wall and the third variant is when the pancreatic duct and biliary duct open at separate locations in the duodenum. While no particular pathology has been associated, there may be potential for pancreatitis in some individuals with the second and third variants (7, 11). These variations are important during the time of surgery or ERCP (11, 12).
Based on animal models the formation of cells that make up the pancreas starts as the epithelium of the buds begins to fold. It is followed by phases of branching, proliferating, and differentiating. The result is grape-like clusters, in which islet cells form clusters to the periphery of acinar and ductal cells (5).
The pancreas is made predominantly of acinar and duct cells, while the islet cells make up 1 to 2% of the pancreas. By the 9th week, the pancreas exists as tubules and clusters of undifferentiated epithelial cells. The tubules continue to grow, followed by lobule formation by the 14th week. Acinar cells with zymogen granules are noted between the 12th and 15th week, and in significant numbers by the 20th week (13). During this phase, the endoplasmic reticulum and Golgi apparatus undergo significant maturation (6). This is followed by progressive growth with a lumen in the center and lobules to the periphery with numerous acini (6).
Pancreatic stellate cells secrete extracellular matrix proteins and seem to have a significant function in the first trimester of human pancreatic development by enhancing differentiation to exocrine cell lineages (14).
Comparable with the enteric nervous system, an intra-pancreatic nervous system develops to enable a degree of independence of the pancreas from the central nervous system and the gut. The pancreatic ganglia are the nervous integration centers of the pancreatic exocrine and endocrine secretion. Vagal preganglionic, sympathetic postganglionic, sensory, and enteric fibers innervate the fully developed pancreas. Postganglionic nerve fibers surround almost every acinus, forming a periacinar plexus containing cholinergic, noradrenergic, peptidergic, and nitrergic fibers, which terminate at the acinar cells. The autonomic nervous system of the pancreas interacts with ganglionic structures that are randomly scattered throughout the pancreatic parenchyma and represent the intrinsic neural component of the pancreatic nerve supply (15). Neurons and nerve fibers form complexes with endocrine cells, and epithelial cells located in ducts early in the second trimester and include pain fibers from parasympathetic nerves (16). Interestingly, innervation is found most densely at the head of the pancreas and decreases toward the tail (17). The sympathetic system seems to develop during this early fetal period and may have a role in endocrine pancreas development (18). Understanding sympathetic innervation of the exocrine pancreas is in its infancy. Studies seem to demonstrate sympathetic inhibition of blood flow, resulting in decreased exocrine secretion, and therefore indirect effect (19). On the other hand, parasympathetic innervation has a large role in exocrine function, mediated via vagal nerve activity, especially in the cephalic phase (20). Gastric, intestinal, and absorbed nutrient phases also appear to have both direct and indirect (via vagal nerve) mechanisms of pancreatic enzyme secretion (21). See Neural Control of the Pancreas | Pancreapedia for more information.
The major adult human digestive exocrine pancreatic enzymes include amylase, pancreatic triglyceride lipase (PTL), colipase, trypsinogen, chymotrypsinogen, carboxypeptidase A1 and A2, and elastase. We will discuss the enzymes found in the intrauterine period followed by postnatal development and the maturation process to adult levels of the enzymes.
Most of the human pancreas exocrine development is derived from morphological studies. The exocrine tissue comprises acinar cells that secrete digestive enzymes and a duct system that deliver them to the small intestine.
Pancreatic secretory trypsin inhibitor (PSTI) or serine protease inhibitor kazal type 1 (SPINK1) was first noted by immunohistochemistry at the 8th gestational week (22).
Adult human proteases include trypsinogen, chymotrypsinogen, elastase 1, and carboxypeptidase A1 and A2.
Many enzymes, including trypsinogen, chymotrypsinogen, and elastase-1, appear between 14th and 16th weeks of gestation (22–24).
The trypsinogen and chymotrypsinogen appear to be present from 16th weeks gestation and increase until birth (23). Activation of trypsinogen to trypsin requires enterokinase, while chymotrypsinogen requires trypsin for conversion to chymotrypsin (25, 26). Chymotrypsin was present in the 23-week premature infants with levels similar to term infants (27). While initially the development of enterokinase was thought to be around 26th week gestation, the levels of chymotrypsin found by Kolacek et al. suggest this may be slightly earlier (27, 28).
Neonates from 32 weeks gestation to term appear to have 90% to 100% of trypsin levels of children 2 years of age. On the other hand, levels of chymotrypsin is 50-60% and while carboxypeptidase B is 10-25% the level of 2-year-old children, thus showing the development of protease levels over time (Table 1) (29).

Table 1. Ontogeny of common pancreatic enzymes in humans.
Pancreatic triglyceride lipase (PTL) is the predominant lipase in human adults. However, other enzymes involved in fat breakdown include colipase and phospholipase A and B.
Lipases such as carboxyl ester lipase and pancreatic lipase-related proteins 1 and 2 (PLRP) were present from 14 to 16 weeks gestation (22–24), though PLRP1 has no known activity (38). The mRNA encoding PLRP1 and PLRP2 was present by the 16th week in the human fetal pancreas. In contrast, the mRNA encoding PTL is limited in the fetal pancreas (29), and likely does not start increasing until 41 weeks gestation (24).
Pancreatic amylase (encoded by AMY2) is present in adult humans but has not been detected in the fetus, and in fact is at low levels in humans even until 3 months of age (22, 39, 40).
Digestion of lipid, protein, and carbohydrates in infants relies on the state of the maturation of exocrine pancreatic function. However, diet composition can also affect enzyme production. Infants do not respond to exogenous cholecystokinin (CCK) or secretin well, but the exact maturation time of response of the exocrine pancreas to secretagogues is not well-defined (41, 42). Understanding normal age-based values of pancreatic enzyme activity in duodenal fluid with pancreatic function testing would allow a better understanding of the ontogeny of pancreatic exocrine tissue.
Interestingly, a diet in the form of starch and protein augments the production of α-amylase and trypsin, respectively, but fat does not stimulate the increased lipase levels in infants (36). However, a high protein and low-fat diet stimulates both trypsin and PTL activity (36). After 12 months of age, PTL does appear to be stimulated over baseline activity by meals (29). It may therefore be possible that CCK and secretin more effectively stimulate exocrine function closer to 12 months of age.
Initially, infants have “physiologic” steatorrhea in the first 3 to 6 months postnatally. As previously noted, lipase is at very low levels in the neonatal period. Based on rodent models, PLRP2 may have a role in triglyceride digestion in newborns (38). Along with PLRP2, the presence of colipase has been noted, which based on animal models appears to increase the activity of PLRP2 (43). Overall, PTL output or the coefficient of fat absorption is 5 to 10% of the adult values (29, 33, 44). In a study by Track et al., the PTL levels at 3 weeks were significantly higher than at 3 days of life and approached that of adult levels (34). However, in the study by Lebenthal et al., the PTL levels were low at birth and 1 month, with a substantial rise by 2 years (Table 1) (29, 30).
Amylase in the acinar cells was not detected until 39 weeks gestational age, and the functional amount of amylase does not arise until the 6th week postnatally (22, 29). In premature infants, amylase activity does not increase after a meal, compared to trypsin, and it is presumed that pancreatic amylase needs are met in the form of salivary amylase and amylase in breastmilk (29, 45). Indeed, it is thought that amylase remains at low levels until 6 months, although isolated amylase deficiency was noted to be frequent till 2 years of age (Table 1) (31, 32). Interestingly, in one cohort, isolated amylase deficiency had been noted beyond 2 years of age with a prevalence of up to 3.5% (32).
Elastase on the other hand is found at low levels at birth in meconium but reaches levels at the lower end of normal within 3 to 4 days in term infants and 2 weeks in preterm infants (Table 1) (37).
The immaturity of exocrine pancreas function is a notable factor in infant's vulnerability to metabolic and nutritional stress (33). Thus, additional non-pancreatic sources of digestion are present. Breast milk can be a source of amylase and bile salt-stimulated lipase. Additionally, brush-border glucoamylase is present in newborns with similar concentrations to that of adults, which may help in the digestion of complex carbohydrates.
The process for the development of the pancreas and the fetus is based on specific coordination between genetics and the local environment. Much of our understanding of genes and molecular signaling comes from animal studies with the assumption that human development is similar. Table 2 summarizes genes and roles in pancreas development. We will discuss some of the genes that are involved in pancreas development and more specifically acinar and ductal cells. For further details, please visit Development of the Pancreas | Pancreapedia and work by Jennings et al. (47).

Table 2. Transcription factors and cell signaling involved in human pancreas development.
After the initial formation of pancreatic tissue, activation of Wnt/β-catenin is required for pancreatic tissue growth, particularly in acinar cells (46). Inhibition of another protein, sonic hedgehog (SHH), likely due to the proximity of the notochord, allowed for future expression of pancreas-duodenum homeobox 1 (PDX1), also known as insulin promoter factor 1 (IPF1) (47, 52). PDX1 has been found as early as pancreatic bud formation and continues to be present in all epithelial cells in the embryonic period, and remains in the nuclei of non-endocrine epithelial cells along with the nuclei and cytoplasm of islet cells into adulthood (48).
Acinar cells begin initially as several carboxypeptidase A1 pyramidal cells bud from the pancreatic epithelium, likely representing the proacinar population. Additionally, GATA4, basic helix-loop-helix transcription factor (MIST1), and pancreatic secretory trypsin inhibitor are also expressed (1). However, the seminal work by Jennings et al. (56) suggests forkhead box protein A2 (FOXA2), and SRY-related homeobox 9 (SOX9) are other predominant transcription factors that define cell types. In particular, SOX9 and FOXA2 are present in ductal cells, while GATA4 is present in acinar cells from the second trimester onward (56). SOX9 has been noted in dorsal and ventral buds from day 32 with higher levels found in later embryonic stages days 44 to 52 (60). Other cellular markers of ductal cells include cytokeratin 19 (CK19) and CD133. CD133 is found in fetal duct-like cells, while CK19 is widely expressed in fetal pancreatic epithelial cells and continues as a ductal cell marker in adults (48, 65).
Notch signaling interacts with recombination signal binding protein for immunoglobulin kappa J region (RBPJ) to downregulate neurogenin-3 (NEUROG3) expression via hairy and enhancer of split-1 (HES1) (66). NEUROG3 is involved in endocrine commitment. HES1 along with pancreas-associated transcription factor 1a (PTF1a) promotes acinar development (5).
As previously noted, PTF1a is important in the development of acinar cells. This is a class B basic helix-loop-helix (bHLH) transcription factor, which appears to be conserved across species. In animal models, the development of pancreatic buds includes PTF1a along with PDX1 expression (67). However, further development of the pancreas includes an enhancer that binds to the active form of PTF1a and RBPJ to regulate PTF1a expression in acinar cells (68). Over time RBPJ is replaced with RBJPL which in complex with PTF1A and other class A bHLH forms PTF1-L and drives downstream regulators of digestive enzymes (53, 69, 70).
Detailed description of developmental abnormalities that are associated with pancreatitis are outside the scope of this review. For completeness we describe them in brief.
Pancreatic divisum is the most common abnormality related to ductal fusion and drainage. It occurs in up to 10% of individuals (9). The lack of pancreas fusion results in drainage of the dorsal part of the pancreas via the minor papilla, while the ventral portion drains the head of the pancreas, uncinate process, and biliary tree into the major duct (71). It remains unclear if this anomaly increases the risk of recurrent pancreatitis and chronic pancreatitis.
The annular pancreas is a rare cause of pancreatitis and duodenal obstruction, typically with non-bilious emesis (72). It results in a thin band of pancreatic tissue surrounding the second part of the duodenum. It is hypothesized that the two ventral buds remain and fuse with the dorsal bud to create the ring (4). This may be the result of abnormal hedgehog signaling (5). Within the annular pancreas, there are six further classifications of type of annular pancreas depending on ductal drainage, including the main duct, minor duct, and common bile duct (73, 74).
Anomalous pancreaticobiliary junction results from the joining of bile and pancreatic ducts outside the duodenal wall. It is believed that dysplasia and misarrangement of the bile and ventral pancreatic duct around the time of ventral pancreas formation results in pancreaticobiliary malformation (PBM) (75, 76). Typically, the end of the common bile duct is surrounded by the papillary sphincter, regulating bile flow while preventing reflux of pancreatic juices (77). However, with PBM, the sphincter is more distal (more than 15 mm) to the junction of the bile and pancreatic duct. As a result, bile and pancreatic juices may reflux into the other respective ducts, resulting in inflammation, bile duct dilation, pancreatitis, bile duct, and gallbladder cancer (78, 79).
It is further divided into three types, bile duct, pancreatic duct, and complex type, based on each joining at an acute angle. Reflux into the biliary system is thought to result in a higher risk for congenital choledochal cyst, and, in particular, type Ia, Ic, and IV-A are associated with PBM (80). The biliary system is particularly susceptible as the pressure in the pancreatic duct is generally higher (81).
The ansa pancreatica is a rare type of anatomical variation of the pancreatic duct. It is a communication between the main pancreatic duct (of Wirsung) and the accessory pancreatic duct (of Santorini). Ansa pancreatica has been considered a predisposing factor in patients with idiopathic acute pancreatitis (82).
The most common genetic disorder resulting in exocrine pancreatic insufficiency include Cystic fibrosis (>90%), followed by Schwachman-diamond, Johanson-Blizzard, Pearson's bone marrow, pancreatic agenesis/hypoplasia, isolated enzyme deficiencies, and genetic or metabolic causes of pancreatitis (83). These will be discussed here in brief for completeness.
Cystic fibrosis transmembrane conductance regulator (CFTR) is found in ductal epithelial cells and is involved in HCO3 - and Cl− transport between the membranes along with water (84). Impaired movement of the ions, and fluid, results in increased viscosity and obstruction of the lumen (85). It is hypothesized that pancreatic insults begin in utero and progress after birth to affect all ducts resulting in pancreatic insufficiency (86). Pancreatic insufficiency occurs in 85% of this population, with a high prevalence among those with homozygous mutations for Δ508 (83).
Schwachman-Diamond syndrome is an autosomal recessive disorder with bone marrow involvement, skeletal abnormalities, and pancreatic insufficiency with variable penetrance (87). Mutations predominantly on chromosome 7 seem to be responsible, but with a spectrum of homozygous and compound heterozygous mutations (88, 89). It has been identified in 90% of individuals with this syndrome and the protein produced by the gene appears to affect ribosome function and may reduce protein production (90). Biopsies of the pancreas have shown that the duct and islet cell functions are preserved; however, acini are replaced by adipose tissue (91). Interestingly, over time, it seems that patients regain some function, and 40% to 60% have sufficient exocrine function (92).
Johanson-Blizzard syndrome is an autosomal recessive disorder with a defect in the UBR1 gene on Chromosome 15 (15q14–21.1) (93). E3 Ubiquitin ligase UBR1 is involved in the breakdown of intracellular pathways, thus mutations in this affect appropriate protein recycling (94). The typical clinical features are exocrine pancreatic insufficiency, hypoplasia/aplasia of the alae nasi, congenital scalp defects, and growth retardation (95, 96). Zenker et al. reported that individuals with this syndrome did not express UBR1 and had intrauterine-onset destructive pancreatitis with secondary replacement of acinar cells with adipose tissue resulting in pancreatic insufficiency (93, 97, 98).
Pearson syndrome is characterized by the bone marrow with vacuolization of erythroid and myeloid precursors, and sideroblasts, along with pancreatic insufficiency and it was initially reported by Pearson (99). It was later confirmed that this syndrome is the result of mitochondrial DNA deletion, affecting protein-coding and tRNA genes and therefore mitochondrial structure and function (100, 101). However, the prevalence of pancreatic insufficiency varies, likely due to variation in mitochondrial DNA deletions (100, 102–104).
Agenesis of the dorsal pancreas can have non-specific findings, including abdominal pain, and often requires imaging findings such as CT to show a lack of pancreatic tissue (105). Several transcription factors have been implicated in pancreas malformation and agenesis, these include PDX1, Hepatocyte nuclear factor (HNF1B), PTF1A, and SOX9. A case report described a homozygous point deletion mutation in PDX1 resulting in pancreatic agenesis with resulting exocrine and endocrine insufficiency (106). While complete agenesis of the pancreas is incompatible with life, case reports have noted variations in ventral and dorsal agenesis ranging from partial to complete (107, 108).
HNF1B mutations have led to the absence of part of the head, body, and tail of the pancreas, suggesting a role for HNF1B in dorsal pancreas formation (61–63). Another key regulator, particularly of exocrine function, PTF1A, has been implicated in pancreatic and cerebellar agenesis (54). Mutations in downstream enhancers of PTF1A have also been noted with isolated pancreatic agenesis (55). Finally, SOX9 has a role in multiple tissues, and particularly the pancreas through much of its formation. Mutation in one gene results in developmental abnormalities of skeletal, reproductive, and other organs such as pancreas hypoplasia (56, 60).
SPINK1 is responsible for inhibiting prematurely activated trypsin in the pancreas. Mutations in the gene have resulted in variations in pathology ranging from increased risk of pancreatitis and exocrine pancreatic insufficiency to inconsistent implications in pancreatic disease (109, 110). Overall, it does appear that the gene is implicated in the earlier cause of pancreatitis and has more pancreatic insufficiency than normal cohorts (111). Indeed, case reports have described exocrine pancreatic insufficiency in infants (112).
Cationic trypsinogen (PRSS1), anionic trypsinogen (PRSS2), and mesotrypsin (PRSS3) are forms of trypsinogen with PRSS1 being the dominant one (104). A hereditary pancreatitis is a rare form of chronic pancreatitis resulting from a mutation in PRSS1, which is autosomal dominant with high penetrance and risk of pancreatic adenocarcinoma (113, 114). Episodes of pancreatitis have been noted to be bimodal with peaks around 6 years and 18 years of age, but with variability in pancreatic exocrine function deficiency, though this was often based on clinical symptoms or stool testing (113, 115, 116).
Carboxypeptidases are metalloproteases that play a role in the digestion of proteins and peptides by hydrolyzing C-terminal peptide bonds (117). Following trypsinogen, carboxypeptidase A1 (CPA1) is the next most common protein excreted in pancreatic fluid (118). Among a cohort of German individuals, CPA1 variants were noted to be a risk factor for chronic pancreatitis. Although the mechanism is uncertain, the authors propose misfolding with subsequent stress in the endoplasmic reticulum as a cause (119).
Chymotrypsinogen C variants (CTRC) is a calcium-dependent serine protease that is important in cationic trypsinogen activation and trypsin degradation (120, 121). Within the pancreas, however, it appears that it has a role in trypsinogen degradation (121, 122). Therefore, mutations result in loss of function and have been associated with early pancreatitis and chronic pancreatitis in pediatrics (123, 124).
Mutations in transcription factors previously noted in the development of the pancreas have been implicated in anatomical variants as well as in endocrine issues, namely, MODY. HNF1B and PDX1 are two that have been implicated (49, 50, 62, 125). Additionally, deficiency in carboxyl ester lipase (CEL) resulted in another form of MODY. CEL, also referred to as bile salt-dependent lipase (BSDL), is one of the four lipases involved in hydrolyses of dietary fat, fat-soluble vitamins, and more specifically cholesterol esters (126). The combined endoscopic pancreatic stimulation test and MRI have shown severely reduced acinar function, along with low pancreas volume with increased lipomatosis in cases of MODY (127, 128).
Knowledge of gene expression and the role of transcription factors allow for further research into stem cell therapy. Current research in both the endocrine and exocrine function was conducted on animal models, though with human pluripotent stem cells (hPSCs). Pluripotent stem cells can be obtained from embryonic cells or can be induced from adult somatic cells, such as fibroblasts or ductal epithelium. Ethical issues are likely to be a big barrier to the use of embryonic cells. Thus, understanding of factors required to convert somatic cells to pluripotent stem cells and then into pancreatic cell lines will hold promise for future. Studies so far seem to show promise in de-differentiating somatic cells-induced pluripotent stem cells (iPSC) and then differentiating into pancreatic progenitor cells (129, 130). In fact, clinical studies are underway in Type 1 diabetes mellitus and the new technologies hold promise in those with exocrine pancreatic dysfunction, particularly in those with chronic pancreatitis requiring islet cell transfer.
There are many answered questions in the human development of the pancreas. How does autonomic innervation, including sympathetic and parasympathetic innervation, develop in the embryo, including when does stimulation such as cephalic input begin? Likewise, a better understanding of innervation by pain fibers may help target therapy.
As current tests for exocrine pancreatic insufficiency include the use of CCK and secretin stimulation, and understanding the maturational process and when the pancreas responds are important.
The use of whole exome sequencing has been increasing with increased access to technology. How certain variants affect exocrine function, and therefore digestion, absorption, and growth will likely provide useful clinical information.
This review shows a framework for the development of the human pancreas including both gross and microanatomy. Pancreas organogenesis is a stepwise process regulated by a complex network of signaling and transcriptional events, that start with the early endoderm toward pancreatic fate. Many crucial players in this process have been identified, including signaling pathways, genes, regulatory elements, and transcription factors. Much of the work is based on the static evaluation of embryonic and fetal specimens that were available due to ethical issues. While gene expressions and transcription factors involved in pancreas formation have been reported, further understanding of cell-to-cell interaction, including those with stellate cells is necessary. It is possible that with a better understanding of iPSC conversion into various pancreatic cell lineages will help understand better the interaction between these cell types along with gene expression and transcription factor production. Molecular understanding of pancreas formation holds exciting promise for future therapies in both the endocrine and exocrine arms.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pan FC, Brissova M. Pancreas development in humans. Curr Opin Endocrinol Diabetes Obes. (2014) 21:77–82. doi: 10.1097/MED.0000000000000047
2. O'Rahilly R, Müller F. Developmental stages in human embryos: revised and new measurements. Cells Tissues Organs. (2010) 192:73–84. doi: 10.1159/000289817
3. Gittes GK. Developmental biology of the pancreas: a comprehensive review. Dev Biol. (2009) 326:4–35. doi: 10.1016/j.ydbio.2008.10.024
4. Moore KL, Persaud TVN, Torchia MG. Alimentary system. In: The Developing Human- Clinically Oriented Embryology. 11th, ed. Amsterdam: Elsevier Publishing Company (2018). p. 193–221.
5. Schoenwolf G, Bleyl S, Brauer P, Francis-West P. Development of the gastrointestinal tract. In: Larsen's Human Embryology. 6th, ed. Amsterdam: Elsevier Publishing Company. (2020).
6. Adda G, Hannoun L, Loygue J. Development of the human pancreas: variations and pathology. A tentative classification. Anat Clin. (1984) 5:275–83. doi: 10.1007/BF01798752
7. Millbourn E. On the excretory ducts of the pancreas in man, with special reference to their relations to each other, to the common bile duct and to the duodenum. Acta Anat. (1950) 9:1–34. doi: 10.1159/000140426
8. Türkvatan A, Erden A, Türkoglu MA, Yener Ö. Congenital variants and anomalies of the pancreas and pancreatic duct: imaging by magnetic resonance cholangiopancreaticography and multidetector computed tomography. Korean J Radiol. (2013) 14:905–13. doi: 10.3348/kjr.2013.14.6.905
9. Agha FP, Williams KD. Pancreas divisum: incidence, detection, and clinical significance. Am J Gastroenterol. (1987) 82:315–20.
10. Wong KC, Lister J. Human fetal development of the hepato-pancreatic duct junction–a possible explanation of congenital dilatation of the biliary tract. J Pediatr Surg. (1981) 16:139–45. doi: 10.1016/S0022-3468(81)80340-5
11. Aljiffry M, Abbas M, Wazzan MAM, Abduljabbar AH, Aloufi S, Aljahdli E. Biliary anatomy and pancreatic duct variations: a cross-sectional study. Saudi J Gastroenterol. (2020) 26:188–93. doi: 10.4103/sjg.SJG_573_19
12. Chavalitdhamrong D, Draganov PV. Unexpected anomaly of the common bile duct and pancreatic duct. World J Clin Cases. (2014) 2:36–8. doi: 10.12998/wjcc.v2.i2.36
13. Laitio M, Lev R, Orlic D. The developing human fetal pancreas: an ultrastructural and histochemical study with special reference to exocrine cells. J Anatomy. (1974) 117(Pt 3):619–34.
14. Li J, Chen B, Fellows GF, Goodyer CG, Wang R. Activation of pancreatic stellate cells is beneficial for exocrine but not endocrine cell differentiation in the developing human pancreas. Front Cell Develop Biol. (2021) 9:4276. doi: 10.3389/fcell.2021.694276
15. Kiba T. Relationships between the autonomic nervous system and the pancreas including regulation of regeneration and apoptosis: recent developments. Pancreas. (2004) 29:e51–8. doi: 10.1097/00006676-200408000-00019
16. Krivova Y, Proshchina A, Barabanov V, Leonova O, Saveliev S. Structure of neuro-endocrine and neuro-epithelial interactions in human foetal pancreas. Tissue Cell. (2016) 48:567–76. doi: 10.1016/j.tice.2016.10.005
17. Amella C, Cappello F, Kahl P, Fritsch H, Lozanoff S, Sergi C. Spatial and temporal dynamics of innervation during the development of fetal human pancreas. Neuroscience. (2008) 154:1477–87. doi: 10.1016/j.neuroscience.2008.04.050
18. Krivova YS, Proshchina AE, Otlyga DA, Leonova OG, Saveliev SV. Prenatal development of sympathetic innervation of the human pancreas. Ann Anat. (2022) 240:151880. doi: 10.1016/j.aanat.2021.151880
19. Love JA Yi E, Smith TG. Autonomic pathways regulating pancreatic exocrine secretion. Auton Neurosci. (2007) 133:19–34. doi: 10.1016/j.autneu.2006.10.001
20. DiMagno EP. Regulation of interdigestive gastrointestinal motility and secretion. Digestion. (1997) 58 Suppl 1:53–5. doi: 10.1159/000201527
21. Chandra R, Liddle R. Regulation of Pancreatic Secretion. Pancreapedia: Exocrine Pancreas Knowledge Base (2015). doi: 10.3998/panc.2015.38
22. Fukayama M, Ogawa M, Hayashi Y, Koike M. Development of human pancreas. Immunohistochemical study of fetal pancreatic secretory proteins. Differentiation. (1986) 31:127–33. doi: 10.1111/j.1432-0436.1986.tb00393.x
23. Carrère J, Figarella-Branger D, Senegas-Balas F, Figarella C, Guy-Crotte O. Immunohistochemical study of secretory proteins in the developing human exocrine pancreas. Differentiation. (1992) 51:55–60. doi: 10.1111/j.1432-0436.1992.tb00680.x
24. Yang Y, Sanchez D, Figarella C, Lowe ME. Discoordinate expression of pancreatic lipase and two related proteins in the human fetal pancreas. Pediatr Res. (2000) 47:184–8. doi: 10.1203/00006450-200002000-00006
25. Hermon-Taylor J, Perrin J, Grant DA, Appleyard A, Bubel M, Magee AI. Immunofluorescent localisation of enterokinase in human small intestine. Gut. (1977) 18:259–65. doi: 10.1136/gut.18.4.259
26. McClure WO, Edelman GM. Fluorescent probes for conformational states of proteins. The activation of chymotrypsinogen. Biochemistry. (1967) 6:567–72. doi: 10.1021/bi00854a026
27. Kolacek S, Puntis JW, Lloyd DR, Brown GA, Booth IW. Ontogeny of pancreatic exocrine function. Arch Dis Child. (1990) 65:178–81. doi: 10.1136/adc.65.2.178
28. Antonowicz I, Lebenthal E. Developmental pattern of small intestinal enterokinase and disaccharidase activities in the human fetus. Gastroenterology. (1977) 72:1299–303. doi: 10.1016/S0016-5085(77)80031-0
29. Lebenthal E, Lee PC. Development of functional responses in human exocrine pancreas. Pediatrics. (1980) 66:556–60. doi: 10.1542/peds.66.4.556
30. McClean P, Weaver LT. Ontogeny of human pancreatic exocrine function. Arch Dis Child. (1993) 68(1 Spec No):62–5. doi: 10.1136/adc.68.1_Spec_No.62
31. Hadorn B, Zoppi G, Shmerling DH, Prader A, McIntyre I, Anderson CM. Quantitative assessment of exocrine pancreatic function in infants and children. J Pediatr. (1968) 73:39–50. doi: 10.1016/S0022-3476(68)80037-X
32. Hopson P, Patel S, Bornstein J, Mehta D, Horvath K. Isolated amylase deficiency in children and its clinical implication. J Pediatr Gastroenterol Nutr. (2019) 68:854–60. doi: 10.1097/MPG.0000000000002317
33. Stormon MO, Durie PR. Pathophysiologic basis of exocrine pancreatic dysfunction in childhood. J Pediatr Gastroenterol Nutr. (2002) 35:8–21. doi: 10.1097/00005176-200207000-00004
34. Track NS, Creutzfeldt C, Bokermann M. Enzymatic, functional and ultrastructural development of the exocrine pancreas—II. The human pancreas. Comp Biochem Physiol A Comp Physiol. (1975) 51:95–100. doi: 10.1016/0300-9629(75)90419-3
35. Moriscot C, Renaud W, Carrère J, Figarella-Branger D, Figarella C, Guy-Crotte O. Developmental gene expression of trypsinogen and lipase in human fetal pancreas. J Pediatr Gastroenterol Nutr. (1997) 24:63–7. doi: 10.1097/00005176-199701000-00015
36. Zoppi G, Andreotti G, Pajno-Ferrara F, Njai DM, Gaburro D. Exocrine pancreas function in premature and full term neonates. Pediatr Res. (1972) 6:880–6. doi: 10.1203/00006450-197212000-00005
37. Kori M, Maayan-Metzger A, Shamir R, Sirota L, Dinari G. Faecal elastase 1 levels in premature and full term infants. Arch Dis Child Fetal Neonatal Ed. (2003) 88:F106–8. doi: 10.1136/fn.88.2.F106
38. Lowe ME. The triglyceride lipases of the pancreas. J Lipid Res. (2002) 43:2007–16. doi: 10.1194/jlr.R200012-JLR200
39. Skude G. Sources of the serum isoamylases and their normal range of variation with age. Scand J Gastroenterol. (1975) 10:577–84. doi: 10.1080/00365521.1975.12097015
40. Skude G. Electrophoretic separation, detection, and variation of amylase isoenzymes. Scand J Clin Lab Invest. (1975) 35:41–7. doi: 10.3109/00365517509068003
41. Dumont Y, Larose L, Poirier G, Morisset J. Modulation of pancreatic muscarinic receptors by weaning. Life Sci. (1982) 30:253–7. doi: 10.1016/0024-3205(82)90506-9
42. Leung YK, Lee PC, Lebenthal E. Maturation of cholecystokinin receptors in pancreatic acini of rats. Am J Physiol. (1986) 250:G594–7. doi: 10.1152/ajpgi.1986.250.5.G594
43. D'Agostino D, Lowe ME. Pancreatic lipase-related protein 2 is the major colipase-dependent pancreatic lipase in suckling mice. J Nutr. (2004) 134:132–4. doi: 10.1093/jn/134.1.132
44. Fomon SJ, Ziegler EE, Thomas LN, Jensen RL, Filer LJ Jr. Excretion of fat by normal full-term infants fed various milks and formulas. Am J Clin Nutr. (1970) 23:1299–313. doi: 10.1093/ajcn/23.10.1299
45. Borgstrom B, Lindquist B, Lundh G. Enzyme concentration and absorption of protein and glucose in duodenum of premature infants. AMA J Dis Child. (1960) 99:338–43. doi: 10.1001/archpedi.1960.02070030340010
46. Murtaugh LC. The what, where, when and how of Wnt/β-catenin signaling in pancreas development. Organogenesis. (2008) 4:81–6. doi: 10.4161/org.4.2.5853
47. Jennings RE, Berry AA, Strutt JP, Gerrard DT, Hanley NA. Human pancreas development. Development. (2015) 142:3126–37. doi: 10.1242/dev.120063
48. Piper K, Brickwood S, Turnpenny LW, Cameron IT, Ball SG, Wilson DI, et al. Beta cell differentiation during early human pancreas development. J Endocrinol. (2004) 181:11–23. doi: 10.1677/joe.0.1810011
49. Shih DQ, Stoffel M. Molecular etiologies of MODY and other early-onset forms of diabetes. Curr Diab Rep. (2002) 2:125–34. doi: 10.1007/s11892-002-0071-9
50. Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. (1997) 17:138–9. doi: 10.1038/ng1097-138
51. Stoffers DA, Thomas MK, Habener JF. Homeodomain protein IDX-1: a master regulator of pancreas development and insulin gene expression. Trends Endocrinol Metab. (1997) 8:145–51. doi: 10.1016/S1043-2760(97)00008-8
52. Hebrok M, Kim SK, Melton DA. Notochord repression of endodermal Sonic hedgehog permits pancreas development. Genes Dev. (1998) 12:1705–13. doi: 10.1101/gad.12.11.1705
53. Krapp A, Knöfler M, Ledermann B, Bürki K, Berney C, Zoerkler N, et al. The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev. (1998) 12:3752–63. doi: 10.1101/gad.12.23.3752
54. Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, Coleman RJ, Garrett C, et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. (2004) 36:1301–5. doi: 10.1038/ng1475
55. Weedon MN, Cebola I, Patch AM, Flanagan SE, De Franco E, Caswell R, et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet. (2014) 46:61–4. doi: 10.1038/ng.2826
56. Jennings RE, Berry AA, Kirkwood-Wilson R, Roberts NA, Hearn T, Salisbury RJ, et al. Development of the human pancreas from foregut to endocrine commitment. Diabetes. (2013) 62:3514–22. doi: 10.2337/db12-1479
57. Shaw-Smith C, De Franco E, Lango Allen H, Batlle M, Flanagan SE, Borowiec M, et al. GATA4 mutations are a cause of neonatal and childhood-onset diabetes. Diabetes. (2014) 63:2888–94. doi: 10.2337/db14-0061
58. Murtaugh LC. Pancreas and beta-cell development: from the actual to the possible. Development. (2007) 134:427–38. doi: 10.1242/dev.02770
59. Salisbury RJ, Blaylock J, Berry AA, Jennings RE, De Krijger R, Piper Hanley K, et al. The window period of NEUROGENIN3 during human gestation. Islets. (2014) 6:e954436. doi: 10.4161/19382014.2014.954436
60. Piper K, Ball SG, Keeling JW, Mansoor S, Wilson DI, Hanley NA. Novel SOX9 expression during human pancreas development correlates to abnormalities in Campomelic dysplasia. Mech Dev. (2002) 116:223–6. doi: 10.1016/S0925-4773(02)00145-4
61. Haldorsen IS, Vesterhus M, Raeder H, Jensen DK, Søvik O, Molven A, et al. Lack of pancreatic body and tail in HNF1B mutation carriers. Diabet Med. (2008) 25:782–7. doi: 10.1111/j.1464-5491.2008.02460.x
62. Haumaitre C, Fabre M, Cormier S, Baumann C, Delezoide AL, Cereghini S. Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1beta/MODY5 mutations. Hum Mol Genet. (2006) 15:2363–75. doi: 10.1093/hmg/ddl161
63. El-Khairi R, Vallier L. The role of hepatocyte nuclear factor 1β in disease and development. Diabetes Obes Metab. (2016) 18 Suppl 1:23–32. doi: 10.1111/dom.12715
64. Solar M, Cardalda C, Houbracken I, Martín M, Maestro MA, De Medts N, et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell. (2009) 17:849–60. doi: 10.1016/j.devcel.2009.11.003
65. Ramond C, Beydag-Tasöz BS, Azad A, van de Bunt M, Petersen MBK, Beer NL, et al. Understanding human fetal pancreas development using subpopulation sorting, RNA sequencing and single-cell profiling. Development. (2018) 145:5480. doi: 10.1242/dev.165480
66. Puri S, Hebrok M. Cellular plasticity within the pancreas—lessons learned from development. Dev Cell. (2010) 18:342–56. doi: 10.1016/j.devcel.2010.02.005
67. Hald J, Sprinkel AE, Ray M, Serup P, Wright C, Madsen OD. Generation and characterization of Ptf1a antiserum and localization of Ptf1a in relation to Nkx6.1 and Pdx1 during the earliest stages of mouse pancreas development. J Histochem Soc. (2008) 56:587–95. doi: 10.1369/jhc.2008.950675
68. Masui T, Long Q, Beres TM, Magnuson MA, MacDonald RJ. Early pancreatic development requires the vertebrate suppressor of hairless (RBPJ) in the PTF1 bHLH complex. Genes Dev. (2007) 21:2629–43. doi: 10.1101/gad.1575207
69. Masui T, Swift GH, Hale MA, Meredith DM, Johnson JE, Macdonald RJ. Transcriptional autoregulation controls pancreatic Ptf1a expression during development and adulthood. Mol Cell Biol. (2008) 28:5458–68. doi: 10.1128/MCB.00549-08
70. Holmstrom SR, Deering T, Swift GH, Poelwijk FJ, Mangelsdorf DJ, Kliewer SA, et al. LRH-1 and PTF1-L coregulate an exocrine pancreas-specific transcriptional network for digestive function. Genes Dev. (2011) 25:1674–9. doi: 10.1101/gad.16860911
71. Mortelé KJ, Rocha TC, Streeter JL, Taylor AJ. Multimodality imaging of pancreatic and biliary congenital anomalies. Radiographics. (2006) 26:715–31. doi: 10.1148/rg.263055164
72. Ravitch MM, Woods AC Jr. Annular pancreas. Ann Surg. (1950) 132:1116–27. doi: 10.1097/00000658-195012000-00011
73. Cunha JE, de Lima MS, Jukemura J, Penteado S, Jureidini R, Patzina RA, et al. Unusual clinical presentation of annular pancreas in the adult. Pancreatol J Int Assoc Pancreatol (IAP). (2005) 5:81–5. doi: 10.1159/000084493
74. Yogi Y, Shibue T, Hashimoto S. Annular pancreas detected in adults, diagnosed by endoscopic retrograde cholangiopancreatography: report of four cases. Gastroenterol Jpn. (1987) 22:92–9. doi: 10.1007/BF02806340
75. Kamisawa T, Ando H, Suyama M, Shimada M, Morine Y, Shimada H. Japanese clinical practice guidelines for pancreaticobiliary maljunction. J Gastroenterol. (2012) 47:731–59. doi: 10.1007/s00535-012-0611-2
76. Matsumoto Y, Fujii H, Itakura J, Mogaki M, Matsuda M, Morozumi A, et al. Pancreaticobiliary maljunction: etiologic concepts based on radiologic aspects. Gastrointest Endosc. (2001) 53:614–9. doi: 10.1067/mge.2001.113920
77. Boyden EA. The anatomy of the choledochoduodenal junction in man. Surg Gynecol Obstet. (1957) 104:641–52.
78. Ishibashi H, Shimada M, Kamisawa T, Fujii H, Hamada Y, Kubota M, et al. Japanese clinical practice guidelines for congenital biliary dilatation. J Hepatobiliary Pancreat Sci. (2017) 24:1–16. doi: 10.1002/jhbp.415
79. Kamisawa T, Takuma K, Anjiki H, Egawa N, Kurata M, Honda G, et al. Pancreaticobiliary maljunction. Clin Gastroenterol Hepatol. (2009) 7:S84–8. doi: 10.1016/j.cgh.2009.08.024
80. Todani T. Congenital choledochal dilatation: classification, clinical features, and long-term results. J Hepatobiliary Pancreat Surg. (1997) 4:276–82. doi: 10.1007/BF02489025
81. Csendes A, Kruse A, Funch-Jensen P, Oster MJ, Ornsholt J, Amdrup E. Pressure measurements in the biliary and pancreatic duct systems in controls and in patients with gallstones, previous cholecystectomy, or common bile duct stones. Gastroenterology. (1979) 77:1203–10. doi: 10.1016/0016-5085(79)90158-6
82. Jarrar MS, Khenissi A, Ghrissi R, Hamila F, Letaief R. Ansa pancreatica: an anatomic variation and a rare cause of acute pancreatitis. Surg Radiol Anat. (2013) 35:745–8. doi: 10.1007/s00276-013-1103-7
83. Durie PR. Inherited causes of exocrine pancreatic dysfunction. Can J Gastroenterol. (1997) 11:145–52. doi: 10.1155/1997/137618
84. Liu F, Zhang Z, Csanády L, Gadsby DC, Chen J. Molecular structure of the human CFTR ion channel. Cell. (2017) 169:85–95. doi: 10.1016/j.cell.2017.02.024
85. Kopelman H, Corey M, Gaskin K, Durie P, Weizman Z, Forstner G. Impaired chloride secretion, as well as bicarbonate secretion, underlies the fluid secretory defect in the cystic fibrosis pancreas. Gastroenterology. (1988) 95:349–55. doi: 10.1016/0016-5085(88)90490-8
86. Wilschanski M, Novak I. The cystic fibrosis of exocrine pancreas. Cold Spring Harb Perspect Med. (2013) 3:a009746. doi: 10.1101/cshperspect.a009746
87. Goobie S, Popovic M, Morrison J, Ellis L, Ginzberg H, Boocock GR, et al. Shwachman-Diamond syndrome with exocrine pancreatic dysfunction and bone marrow failure maps to the centromeric region of chromosome 7. Am J Hum Genet. (2001) 68:1048–54. doi: 10.1086/319505
88. Andolina JR, Morrison CB, Thompson AA, Chaudhury S, Mack AK, Proytcheva M, et al. Shwachman-Diamond syndrome: diarrhea, no longer required? J Pediatr Hematol Oncol. (2013) 35:486–9. doi: 10.1097/MPH.0b013e3182667c13
89. Hashmi SK, Allen C, Klaassen R, Fernandez CV, Yanofsky R, Shereck E, et al. Comparative analysis of Shwachman-Diamond syndrome to other inherited bone marrow failure syndromes and genotype-phenotype correlation. Clin Genet. (2011) 79:448–58. doi: 10.1111/j.1399-0004.2010.01468.x
90. Wong CC, Traynor D, Basse N, Kay RR, Warren AJ. Defective ribosome assembly in Shwachman-Diamond syndrome. Blood. (2011) 118:4305–12. doi: 10.1182/blood-2011-06-353938
91. Rothbaum R, Perrault J, Vlachos A, Cipolli M, Alter BP, Burroughs S, et al. Shwachman-Diamond syndrome: report from an international conference. J Pediatr. (2002) 141:266–70. doi: 10.1067/mpd.2002.125850
92. Cipolli M. Shwachman-Diamond syndrome: clinical phenotypes. Pancreatol J Int Assoc Pancreatol (IAP). (2001) 1:543–8. doi: 10.1159/000055858
93. Zenker M, Mayerle J, Lerch MM, Tagariello A, Zerres K, Durie PR, et al. Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome). Nat Genet. (2005) 37:1345–50. doi: 10.1038/ng1681
94. Sukalo M, Fiedler A, Guzmán C, Spranger S, Addor MC, McHeik JN, et al. Mutations in the human UBR1 gene and the associated phenotypic spectrum. Hum Mutat. (2014) 35:521–31. doi: 10.1002/humu.22538
95. Almashraki N, Abdulnabee MZ, Sukalo M, Alrajoudi A, Sharafadeen I, Zenker M. Johanson-Blizzard syndrome. World J Gastroenterol. (2011) 17:4247–50. doi: 10.3748/wjg.v17.i37.4247
96. Johanson A, Blizzard R, A. syndrome of congenital aplasia of the alae nasi, deafness, hypothyroidism, dwarfism, absent permanent teeth, and malabsorption. J Pediatr. (1971) 79:982–7. doi: 10.1016/S0022-3476(71)80194-4
97. Daentl DL, Frías JL, Gilbert EF, Opitz JM. The Johanson-Blizzard syndrome: case report and autopsy findings. Am J Med Genet. (1979) 3:129–35. doi: 10.1002/ajmg.1320030203
98. Moeschler JB, Polak MJ, Jenkins JJ III, Amato RS. The Johanson-Blizzard syndrome: a second report of full autopsy findings. Am J Med Gen. (1987) 26:133–8. doi: 10.1002/ajmg.1320260120
99. Pearson HA, Lobel JS, Kocoshis SA, Naiman JL, Windmiller J, Lammi AT, et al. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr. (1979) 95:976–84. doi: 10.1016/S0022-3476(79)80286-3
100. Rotig A, Colonna M, Bonnefont JP, Blanche S, Fischer A, Saudubray JM, et al. Mitochondrial DNA deletion in Pearson's marrow/pancreas syndrome. Lancet. (1989) 1:902–3. doi: 10.1016/S0140-6736(89)92897-3
101. Khasawneh R, Alsokhni H, Alzghoul B, Momani A, Abualsheikh N, Kamal N, et al. A novel mitochondrial DNA deletion in patient with Pearson syndrome. Med Arch. (2018) 72:148–50. doi: 10.5455/medarh.2018.72.148-150
102. Broomfield A, Sweeney MG, Woodward CE, Fratter C, Morris AM, Leonard JV, et al. Paediatric single mitochondrial DNA deletion disorders: an overlapping spectrum of disease. J Inherit Metab Dis. (2015) 38:445–57. doi: 10.1007/s10545-014-9778-4
103. Farruggia P, Di Cataldo A, Pinto RM, Palmisani E, Macaluso A, Valvo LL, et al. Pearson syndrome: a retrospective cohort study from the marrow failure study group of a IEOP (Associazione Italiana Emato-Oncologia Pediatrica). JIMD Rep. (2016) 26:37–43. doi: 10.1007/8904_2015_470
104. Whitcomb DC, Lowe ME. Genetic disorders of the pancreas and pancreatic disorders of childhood. In:Feldman M, Friedman LS, Brandt LJ, , editors. Sleisenger and Fordtran's Gastrointestinal and Liver Disease - 2 Volume Set: Pathophysiology, Diagnosis, Management. 11th ed. Amsterdam: Elsevier (2020). p. 862–92.
105. Güçlü M, Serin E, Ulucan S, Kul K, Ozer B, Gümürdülü Y, et al. Agenesis of the dorsal pancreas in a patient with recurrent acute pancreatitis: case report and review. Gastrointest Endosc. (2004) 60:472–5. doi: 10.1016/S0016-5107(04)01733-X
106. Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. (1997) 15:106–10. doi: 10.1038/ng0197-106
107. Mei W, Cao F, Li F. Two cases of agenesis of the dorsal pancreas and a review of the literature. BMC Gastroenterol. (2020) 20:94. doi: 10.1186/s12876-020-01245-8
108. Valiyeva S, Romano L, Schietroma M, Carlei F, Giuliani A. Partial agenesis of dorsal pancreas. Report of two cases. Int J Surg Case Rep. (2020) 77s(Suppl):S17–20. doi: 10.1016/j.ijscr.2020.10.029
109. Drenth JP, te Morsche R, Jansen JB. Mutations in serine protease inhibitor Kazal type 1 are strongly associated with chronic pancreatitis. Gut. (2002) 50:687–92. doi: 10.1136/gut.50.5.687
110. Midha S, Khajuria R, Shastri S, Kabra M, Garg PK. Idiopathic chronic pancreatitis in India: phenotypic characterisation and strong genetic susceptibility due to SPINK1 and CFTR gene mutations. Gut. (2010) 59:800–7. doi: 10.1136/gut.2009.191239
111. Muller N, Sarantitis I, Rouanet M, de Mestier L, Halloran C, Greenhalf W, et al. Natural history of SPINK1 germline mutation related-pancreatitis. EBioMed. (2019) 48:581–91. doi: 10.1016/j.ebiom.2019.09.032
112. Venet T, Masson E, Talbotec C, Billiemaz K, Touraine R, Gay C, et al. Severe infantile isolated exocrine pancreatic insufficiency caused by the complete functional loss of the SPINK1 gene. Hum Mutat. (2017) 38:1660–5. doi: 10.1002/humu.23343
113. Rebours V, Boutron-Ruault MC, Schnee M, Férec C, Le Maréchal C, Hentic O, et al. The natural history of hereditary pancreatitis: a national series. Gut. (2009) 58:97–103. doi: 10.1136/gut.2008.149179
114. Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. (1996) 14:141–5. doi: 10.1038/ng1096-141
115. Keim V, Bauer N, Teich N, Simon P, Lerch MM, Mössner J. Clinical characterization of patients with hereditary pancreatitis and mutations in the cationic trypsinogen gene. Am J Med. (2001) 111:622–6. doi: 10.1016/S0002-9343(01)00958-5
116. Sossenheimer MJ, Aston CE, Preston RA, Gates LK Jr, Ulrich CD, Martin SP, et al. Clinical characteristics of hereditary pancreatitis in a large family, based on high-risk haplotype the midwest multicenter pancreatic study group (MMPSG). Am J Gastroenterol. (1997) 92:1113–6.
117. Vendrell J, Querol E, Avilés FX. Metallocarboxypeptidases and their protein inhibitors. Structure, function and biomedical properties. Biochim Biophys Acta. (2000) 1477:284–98. doi: 10.1016/S0167-4838(99)00280-0
118. Scheele G, Bartelt D, Bieger W. Characterization of human exocrine pancreatic proteins by two-dimensional isoelectric focusing/sodium dodecyl sulfate gel electrophoresis. Gastroenterology. (1981) 80:461–73. doi: 10.1016/0016-5085(81)90007-X
119. Witt H, Beer S, Rosendahl J, Chen JM, Chandak GR, Masamune A, et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat Genet. (2013) 45:1216–20. doi: 10.1038/ng.2730
120. Nemoda Z, Sahin-Tóth M. Chymotrypsin C (caldecrin) stimulates autoactivation of human cationic trypsinogen. J Biol Chem. (2006) 281:11879–86. doi: 10.1074/jbc.M600124200
121. Zhou J, Sahin-Tóth M. Chymotrypsin C mutations in chronic pancreatitis. J Gastroenterol Hepatol. (2011) 26:1238–46. doi: 10.1111/j.1440-1746.2011.06791.x
122. Rinderknecht H, Adham NF, Renner IG, Carmack C. A possible zymogen self-destruct mechanism preventing pancreatic autodigestion. Int J Pancreatol. (1988) 3:33–44. doi: 10.1007/BF02788221
123. Giefer MJ, Lowe ME, Werlin SL, Zimmerman B, Wilschanski M, Troendle D, et al. Early-onset acute recurrent and chronic pancreatitis is associated with PRSS1 or CTRC gene mutations. J Pediatr. (2017) 186:95–100. doi: 10.1016/j.jpeds.2017.03.063
124. Grabarczyk AM, Oracz G, Wertheim-Tysarowska K, Kujko AA, Wejnarska K, Kolodziejczyk E, et al. Chymotrypsinogen C genetic variants, including c.180TT, are strongly associated with chronic pancreatitis in pediatric patients. J Pediatr Gastroenterol Nutr. (2017) 65:652–7. doi: 10.1097/MPG.0000000000001767
125. Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. (1997) 17:384–5. doi: 10.1038/ng1297-384
126. Johansson BB, Fjeld K, El Jellas K, Gravdal A, Dalva M, Tjora E, et al. The role of the carboxyl ester lipase (CEL) gene in pancreatic disease. Pancreatol J Int Assoc Pancreatol (IAP). (2018) 18:12–9. doi: 10.1016/j.pan.2017.12.001
127. Raeder H, Haldorsen IS, Ersland L, Grüner R, Taxt T, Søvik O, et al. Pancreatic lipomatosis is a structural marker in nondiabetic children with mutations in carboxyl-ester lipase. Diabetes. (2007) 56:444–9. doi: 10.2337/db06-0859
128. Tjora E, Wathle G, Engjom T, Erchinger F, Molven A, Aksnes L, et al. Severe pancreatic dysfunction but compensated nutritional status in monogenic pancreatic disease caused by carboxyl-ester lipase mutations. Pancreas. (2013) 42:1078–84. doi: 10.1097/MPA.0b013e3182920e9c
129. Ito K, Matsuura K, Mihara Y, Sakamoto Y, Hasegawa K, Kokudo N, et al. Delivery of pancreatic digestive enzymes into the gastrointestinal tract by pancreatic exocrine tissue transplant. Sci Rep. (2019) 9:5922. doi: 10.1038/s41598-019-42362-z
Keywords: pancreas, ontogeny, genes, development, prenatal and postnatal of enzyme secretion
Citation: Mehta V, Hopson PE, Smadi Y, Patel SB, Horvath K and Mehta DI (2022) Development of the human pancreas and its exocrine function. Front. Pediatr. 10:909648. doi: 10.3389/fped.2022.909648
Received: 31 March 2022; Accepted: 11 August 2022;
Published: 29 September 2022.
Edited by:
Cheryl Gariepy, Nationwide Children's Hospital, United StatesReviewed by:
Atchariya Chanpong, University College London, United KingdomCopyright © 2022 Mehta, Hopson, Smadi, Patel, Horvath and Mehta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Devendra I. Mehta, RGV2ZW5kcmEubWVodGFAb3JsYW5kb2hlYWx0aC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.