 Carmen Muntean
Carmen Muntean Iuliana Magdalena Starcea
Iuliana Magdalena Starcea Cristina Stoica
Cristina Stoica Claudia Banescu
Claudia Banescu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 01 June 2022
Sec. Pediatric Nephrology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.908657
This article is part of the Research TopicRare Diseases with Renal Implications in ChildrenView all 4 articles
Inherited renal diseases represent 20% of the causes of end-stage renal diseases. Fabry disease, an X-linked lysosomal storage disorder, results from α-galactosidase A deficient or absent activity followed by globotriaosylceramide (Gb3) accumulation and multiorgan involvement. In Fabry disease, kidney involvement starts early, during intrauterine life by the Gb3 deposition. Even if chronic kidney disease (CKD) is discovered later in adult life in Fabry disease patients, a decline in glomerular filtration rate (GFR) can occur during adolescence. The first clinical sign of kidney involvement is represented by albuminuria. So, early and close monitoring of kidneys function is required: albuminuria and proteinuria, urinary albumin-to-creatinine ratio, serum creatinine, or cystatin C to estimate GFR, while urinary sediment with phase-contrast microscopy under polarized light may be useful in those cases where leucocyte α-Gal A activity and GLA genotyping are not available. Children with Fabry disease and kidney involvement should receive enzyme replacement therapy and nephroprotective drugs (angiotensin-converting enzyme inhibitors or angiotensin receptor blockers) to prevent or slow the progressive loss of kidney functions. Early diagnosis of Fabry disease is important as enzyme replacement therapy reduces symptoms, improves clinical features and biochemical markers, and the quality of life. More importantly, early treatment could slow or stop progressive organ damage in later life.
The wide spectrum of signs and symptoms in Fabry disease represents a challenge in its management nowadays, and these include timely diagnosis and early therapy in children, the necessity for biomarkers that are correlated with the earliest changes in histology. Early enzyme-replacement therapy (ERT) and other therapeutic strategies may impact morbidity and mortality in Fabry disease patients. A delay in diagnosis of Fabry disease influences the quality of life and reduces the life expectancy in nontreated patients.
Fabry disease (FD), also known as Anderson-Fabry disease (OMIM #301500), is a multisystem and heterogenous lysosomal storage disease, with an X-linked inheritance pattern characterized by complete or partial deficiency of the lysosomal alpha-galactosidase A (α-Gal A) enzyme activity. The enzymatic defects result in subsequent accumulation of globotriaosylceramide (Gb3 or GL3) and glycosphingolipids within cellular lysosomes, plasma, and urine causing multiorgan damage with life-threatening manifestations (1).
Fabry disease is a multifaceted condition that begins during intrauterine life. Elleder et al. stated that storage material is already present in the fetal kidney (2), while Vedder et al. only investigated placental storage and speculated on storage in fetal organs (3). Usually, even if the affected infants look normal at birth, the clinical signs of the disease will develop gradually once undigested sphingolipids (SLs) such as globotriaosylceramide and globotriaosylsphingosine (lysoGb3 or lysoGL3) accumulate in the body, as a result of the degree of the enzyme deficiency and the severity of the toxic metabolites storage within organs. The clinical impact relies on the severity of the enzyme insufficiency and the tissues in which toxic nonmetabolized intermediates (such as Gb3) accumulate (4). Fabry disease recognizes two major phenotypes: “classic or early-onset” and a mild or “late-onset” phenotype. The classic forms usually occur in childhood or adolescence. Affected males with the late-onset type have residual α-Gal A activity, correlated with a later-onset cardiac and/or renal disease, and lack the major early-onset classical manifestations (5).
Even if a severe phenotype is more frequent in males vs. females, heterozygous women may also exhibit symptoms of varying severity depending on random inactivation of one of the two X chromosomes. The result of a random X chromosome inactivation is represented by a mosaic of cell populations, leading to variable phenotypes from asymptomatic to severely symptomatic heterozygous females (6).
Fabry disease patients require life-long follow-up to detect changes in signs and symptoms. It is characterized by progressive neurological, renal, cardiac, ocular, and dermatological manifestations (7). In Fabry classical disease, kidney involvement starts early, during intrauterine life by the Gb3 deposition. Even if chronic kidney disease (CKD) is discovered later in adult life in Fabry disease patients, a decline in glomerular filtration rate (GFR) can occur during adolescence. So, early and close monitoring of kidney and other organ functions is required. Early diagnosis of Fabry disease is important as enzyme replacement therapy reduces symptoms, and improves clinical features, biochemical markers, and the quality of life. More importantly, early treatment could slow or stop progressive organ damage in later life. Pulmonary involvement is usually mild and expressed by fatigue, persistent cough, obstructive lung disease, and impaired pulmonary function tests (8).
Fabry disease is a rare disorder with an estimated overall incidence varying from 1:17000 to 1:117000. The classic form of Fabry disease is estimated to have a prevalence of 1:22000 to 1:40000 in males, while the prevalence for atypical presentation is evaluated to be 1:1000 to 1:3000 in males and 1:6000 to 1:40000 in females (9). A recent meta-analysis of dialysis patients with FD revealed that the classic forms are more frequent than true late-onset forms if pathogenic GLA mutation is considered (10). Also, Choi et al. found a four times higher incidence for classical vs. late-onset Fabry disease (11).
Fabry disease prevalence in different geographical areas is presented in Table 1.
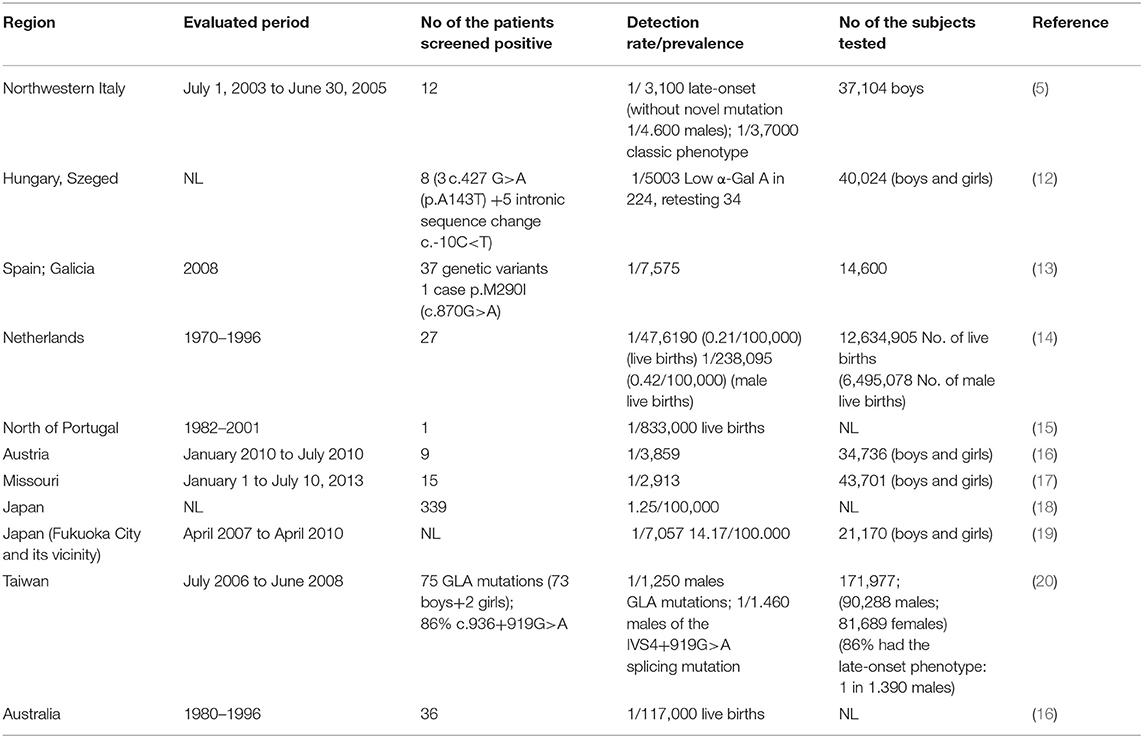
Table 1. Prevalence of Fabry disease according to region.
GLA gene (OMIM 300644) is located on the X chromosome (Xq22.1), consists of eight exons, and encodes for alpha-galactosidase A (enzyme commission number EC 3.2.1.22), a lysosomal enzyme. According to ClinVar, more than 900 variants in the GLA gene have been identified so far1.
GLA gene mutation will lead to the deficiency of the alpha-galactosidase A (α-Gal A) enzyme. The most frequent GLA mutations are missense followed by nonsense mutations, but also deletions, duplication, insertions, frameshift, and splice-site mutations are observed (11, 21).
It was suggested that classic forms of the Fabry disease are produced by mutations that lead to complete loss of function of the gene, whereas late-onset disease and milder phenotypes are produced by mutations that result in the amino acid change (11).
Based on different reports GLA p.R112C, p.L129P, p.C142W, p.P205L, p.H46R,p, M42V, pD266N, p.G271C, p.G274R, p.S297R, p.D322E, p.W349R, p.W226*, p.R220*, p.R227*, p.K240Efs*8, p.S345Rfs*28, p.T412Sfs*37. p.G266Vfs*8, p.L268fs*1, p.Q99fs*23, p.D61Efs*32 and p.L344fs *31 are considered classical variants; while p.M296I, p.R301Q, p.R112H, p.N215S, F113L, I91T, p.R363H, and L310V are considered late-onset variants (11, 22–28). GLA p.R112H and p.M296I are considered as variants of uncertain significance (29).
Some GLA variants may cause amino acid substitutions and/or low α-Gal A enzyme activity and it is still debated whether these mutations may cause Fabry symptoms and are considered variants of unknown significance at the moment for example p.E66Q (30, 31), p.D313Y (32–35), p.S126G (36), p.R118C (37, 38), and p.A143T (39, 40).
Detection of a known GLA pathogenic variant by sequencing analysis allows for a definitive Fabry disease diagnosis. If sequence analysis identifies a novel variant or variants of unknown significance a comprehensive examination is necessary and may include clinical symptoms, the course of disease, family history, α-Gal A enzyme activity level, lysoGb3 level in the blood, and renal biopsy in cases with additional symptoms compatible with Fabry disease (27) considering that not all result in Fabry disease, as many of them are benign ones or polymorphisms without clinical implications (41). Also, there is a wide phenotypic spectrum, even among family members confirmed with a similar genetic mutation (42) therefore a rigorous screening should be done.
Fabry disease has an X-linked pattern of inheritance, usually, the GLA mutation is transmitted to the boys through a heterozygous mother. A heterozygous female for GLA gene mutation may have affected boys (50%) and healthy boys (50%), and each daughter has a 50% chance of being a heterozygote. An affected father will not transmit the disorder to his son. The daughters of an affected father with Fabry disease will be heterozygotes. Negative family history of Fabry disease does not rule out the diagnosis.
The lysosomal hydrolase alpha-galactosidase A (α-Gal A) deficiency will lead to the systemic progressive lysosomal accumulation of complex glycosphingolipids with terminal α-galactosyl moieties, mainly globotriaosylceramide (Gb3) and its deacylated, amphiphilic metabolite, namely globotriaosylsphingosine (lysoGb3), and to a lesser extent, galactosylceramide and other derivatives (43).
Substrate accumulation within lysosomes in the cells of different tissues promotes various pathogenic mechanisms in which are implicated different mediators leading to multisystem lesions, resulting in clinical manifestations of the disease as well as the development of complications that reduce the quality of life (44).
Accumulation of Gb3 results in characteristic lysosomal deposits (Fabry inclusions) in different organs and cell types, known as myelin figures and zebra bodies (45) leading to cell death, with progression to fibrosis and irreversible organic damage and reducing the average life expectancy by 10 years at women and by 25 years at men (41).
It has been suggested that lysoGb3 may represent an important pathogenic factor in Fabry disease. LysoGb3 increases the proliferation of smooth muscle cells leading to increased intima-media thickness and arterial stiffness in patients with Fabry disease (44). In addition, the accumulation of lysoGb3 will deteriorate nociceptive neurons manifested as neuropathic pain (acroparasthesias), and intolerance to heat. Storage of Gb3 and glycosphingolipids in the peripheral nervous system causes dysregulation of vascular tone, tinnitus, and hearing loss (8). Small fiber neuropathy (SFN), a characteristic of FD, involves small myelinated and unmyelinated neurons. Thinly, myelinated Aδ fibers are particularly affected (45, 46). These Aδ fibers involve a cold sensation and mechanical pain sensitivity to pinprick stimuli. Later, thermal stimuli involve warmth perception and pain sensitivity to heat (C-fibers) (45). Aδ fibers are more susceptible to Gb3-induced damage. The SFN in Fabry disease is an age-dependent and disease severity-dependent neuropathy as well as gender-dependent, males being more severely affected (45). Fabry SFN is diagnosed with impaired cold sensation, neuropathic pain, and intraepidermal nerve fiber density (IENFD) < 5th percentile (45). Higher pain incidence was reported in male patients (60–80%) than in females (41–65%) (47, 48). Early therapy is important to reduce small nerve fiber damage, as they occur in the early stages of the disease. Enzyme replacement therapy (ERT) has been shown to reduce the overall pain scores and neuropathic pain in patients (49). Moreover, the accumulation of lysoGb3 contributes to the early damage of podocytes and fibrosis produced in epithelial cells (43). Deposition of lysoGb3 in podocytes (that primarily are loaded with glycolipids) will increase the expression of cytokine TGF-β1 which enhances the synthesis of an extracellular matrix, inhibiting matrix degradation, and altering cell-cell interaction. TGF β will lead to tissue fibrosis, TGF β being one of the main contributors to the development of renal fibrosis (50). TGF β1, one isoform of TGF β is a profibrotic mediator in various kidney diseases and causes both tubular and glomerular epithelial cell-to-mesenchymal transition. TGF-β signaling interacts with other signaling pathways (for example TGF-β/Smad) for mediating the fibrotic process (51). The function of podocytes is influenced by inflammatory cytokines level, which is influenced also by lysoGb3. The damage of podocytes will be followed by podocyte loss and glomerulus resulting in CKD with proteinuria and glomerulosclerosis. Chien et al. reported that accumulation of Gb3 in the kidney starts early during fetal development, as demonstrated by elevated urinary Gb3 levels at birth in male newborns with Fabry disease (52).
Also, renal tubular cells, glomerular endothelial, mesangial, and interstitial cells are affected. Furthermore, increased levels of lysoGb3 inhibit the endothelial nitric oxide synthase (eNOS) and thereby may be involved in the dysfunction of the endothelium (50, 53). Accumulation of Gb3 produces dysregulation of the enzyme nitric oxide endothelial synthase (eNOS) leading to the formation of oxidant species derived from nitric oxide, which may result in vasculopathy (50, 53). Storage of Gb3 and lysoGb3, within lysosomes, also involves cardiac cells (for example cardiomyocytes, valvular fibroblasts) (54). Increased levels of lysoGb3, will lead to cardiomyocyte hypertrophy and ischemia (54) causing a higher risk for cerebrovascular accidents and coronary microvascular disease; cardiac damage: dysrhythmias (tahy-/bradycardia), EKG abnormalities, hypertrophic cardiomyopathy, and valvular dysfunction (8).
The clinical symptoms of Fabry disease may present at any age, in children and adults (55, 56).
According to the residual GLA enzyme activity of normal value, it may be graded as residual (1–5% of normal values) or no residual (<1% of normal values), or nearly complete deficiency of α-Gal A activity (57). There are two different types of Fabry disease, the early-onset type and the late-onset type. Usually, the early-onset type occurs mostly in males with absent or nearly complete deficiency of α-Gal A activity, while the late-onset type occurs mainly in cases with residual α-Gal A activity (58). The occurrence of early symptoms during childhood is linked to the severity of α-Gal A deficiency. The early-onset type associated with classical phenotype for Fabry disease in male patients involves no residual α-Gal A enzyme activity and begins during childhood.
Clinical symptoms which appear in childhood are represented by gastrointestinal symptoms, neuropathic pain (pain attacks, chronic pain), acroparesthesia, angiokeratoma, hypohidrosis, and corneal opacities (cornea verticillata). Gastrointestinal and eye involvement was reported within the first decade of life. Also, the early median age at onset was observed in males vs. females (at least 2–5 years later in girls vs. boys) (59, 60). Cardiac, renal, and skin manifestations of Fabry disease occurred in the second decade of life (adolescence) (47, 59). Laney et al. (60) and Hopkin et al. (17) reported the mentioned signs and symptoms and their onset early in life (during the toddlerhood and early childhood period).
The main signs and symptoms of Fabry disease observed during childhood are pain (neuropathic pain most frequently localized in palms, soles, and fingertips or acroparesthesia that begin in early childhood) (60, 61), fatigue, gastrointestinal problems (most commonly abdominal pain and diarrhea) (60), reduced or absent sweating (hypohidrosis or anhidrosis), heat or cold or exercise intolerance and angiokeratoma that appears in children and young adolescents (43, 47, 60, 62, 63).
Some studies reported acroparesthesia as the most common initial symptom of Fabry disease with an early age of onset, often not recognized and therefore with a delaying diagnosis (8).
Other symptoms observed during childhood or adolescence may be the corneal sign (presence of corneal whorls or cornea verticillata observed upon slit-lamp examination (64, 65), hearing problems such as hearing loss, tinnitus (66), renal sign (pathological albuminuria or proteinuria, hyperfiltration), cardiac sign (valvular dysfunction, arrhythmias, conduction abnormalities). Also, it was reported delayed growth and it was noticed that the onset of puberty in boys may be affected (60). The signs and symptoms of Fabry disease in children stratified by age are presented in Table 2. The overall quality of life (QoL) of children with Fabry is often considerably reduced and characterized by anxiety, depression, and school absences. The signs and symptoms presented above should alert the pediatricians to the possibility of Fabry disease (62). FD patients require a multidisciplinary approach and organ-specific treatment since the most severe clinical effect is observed in the kidneys, heart, and central nervous system.
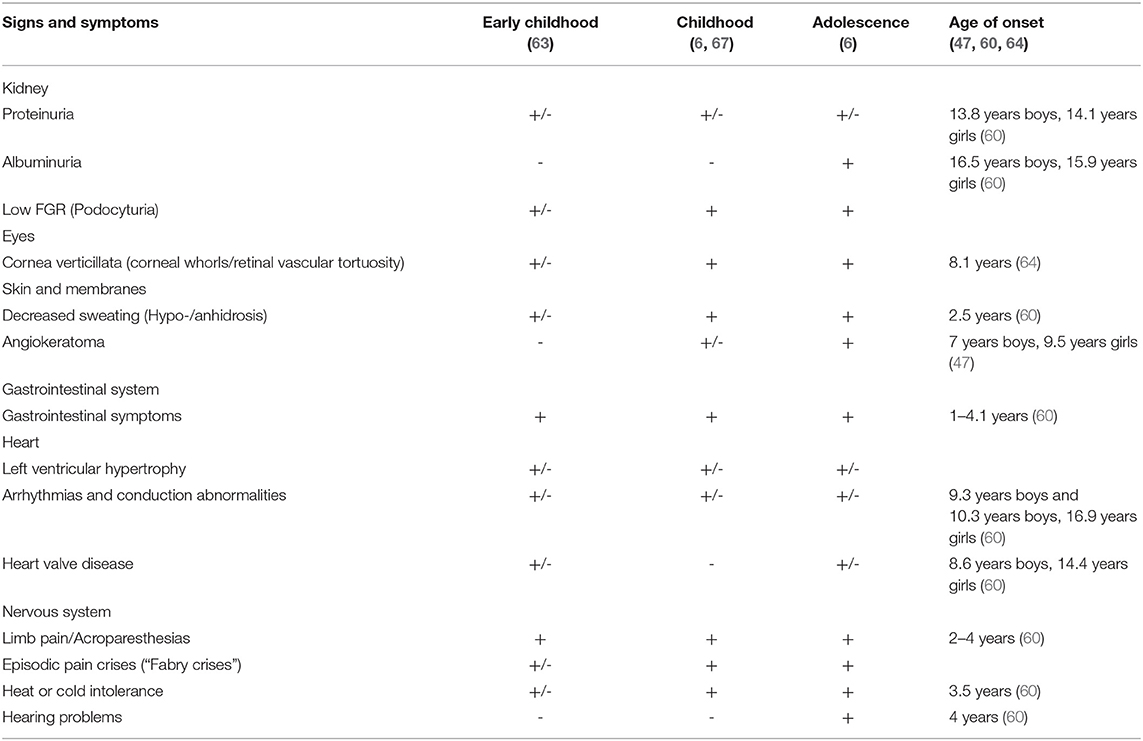
Table 2. Signs and symptoms of Fabry disease, stratified by age.
A recent paper reported 51 Romanian Fabry patients aged between 11 and 80 years (4). In this Romanian cohort, even if the onset was at about 13 years a significant delay in diagnosis was noticed after the appearance of irreversible damages. Only in two cases from affected families, the diagnosis was established in adolescence, before the onset of complications (4). In their study, Choi et al. reported an incidence of 94 FD cases (10 children-pediatric males) in 50 million people in South Korea, with a four times higher incidence of classical type Fabry disease compared with late-onset. They concluded that Fabry disease, especially the mild, late-onset type is underdiagnosed (11). The period between the mean age at onset and mean age at diagnosis was 1.5 years in the boys. Acroparesthesia was encountered in all children, starting with the age of five in the youngest child (11).
Kidney involvement in Fabry disease is common, its prevalence being 55%, according to Waldek et al. (68). The main cause of death in Fabry patients is cardiovascular disease, followed by cerebrovascular manifestations, with kidney failure or end-stage renal disease (ESRD) being the third most common cause of mortality. Furthermore, chronic kidney disease (CKD) is the main cause of kidney failure in untreated patients with the late-onset or classical type of Fabry (6). The earliest sign of Fabry nephropathy is proteinuria, which is the most significant existing biomarker for progressive decline in GFR (68). Patients with CKD without a clear cause of nephropathy plus concomitant symptoms or signs compatible with FD like neuropathic pain, gastrointestinal problems, angiokeratoma, etc, should trigger testing for Fabry disease.
Subclinical kidney damage appears during childhood and is associated with pathological albuminuria. CKD frequently affects adolescents and adults and is associated with a progressive decline of GFR (6). Increased levels of albuminuria and proteinuria, and a worsening in GFR indicate permanent nephron destruction, in glomerular as well as tubular regions, where interstitial fibrosis, glomerular and arteriolar sclerosis, besides tubular atrophy results in kidney injury (6).
A recent study raised the utility of abdominal ultrasound in patients with CKD of unknown etiology and a positive family history of kidney disease. The presence of parapelvic cysts on ultrasound in these patients can highlight suspicion of Fabry disease, especially since these cysts appear earlier than in the general population (69, 70). Also, Pisani et al. observed a high incidence of parapelvic cysts in Fabry disease patients (up to 43%). Therefore, in patients with a kidney disorder, CKD, or proteinuria of unknown etiology associated with other stigmata of the disease, the diagnosis of Fabry should be considered (57). Other ultrasound abnormalities, namely enlarged kidneys, and increased parenchymal echogenicity have been reported in these patients (71).
It was shown that Gb3 and lysoGb3 are progressively accumulated in different renal cells and to a large extent in podocytes and their quantity increases over time and leads to increased podocyte foot process width and urinary protein excretion, both consistent with morphological and functional podocyte alterations (67). Mauer et al. highlighted that mosaicism of podocytes (with or without Gb3 inclusions) in Fabry disease females is linked to podocyte injury. So, data about podocytes mosaicism are important as they may be useful to identify these patients with increased risk of progressive Fabry nephropathy development (72).
A recent study included 55 males (mean age 27) with classic Fabry disease from which 18 cases were under the age of 18 and revealed that Fabry disease has followed the podocyte injury (67). Previously, the same authors showed that in young patients (under the age of 19) with the classic type of Fabry disease the podocyte Gb3 volume proportion increases (73).
There is a physiological podocyte loss with aging, but this loss is more expressed in Fabry disease patients. In the beginning, the podocyte Gb3 accumulation rate is not compensated sufficiently by podocyte enlargement, causing an increasing podocyte Gb3 volume fraction in the first three decades of life. After this, Gb3 continues to accumulate concomitant with podocyte enlargement, along with podocyte loss (67). The same study emphasizes that the podocyte loss process starts quite early in Fabry disease, highlighting that early initiation of enzyme replacement therapy (ERT) is essential, especially before significant proteinuria occurrence (67). The progression of Fabry disease is concomitant with metabolite accumulation resulting in tissue involvement and progressive organs dysfunction as are depicted in Figure 1.
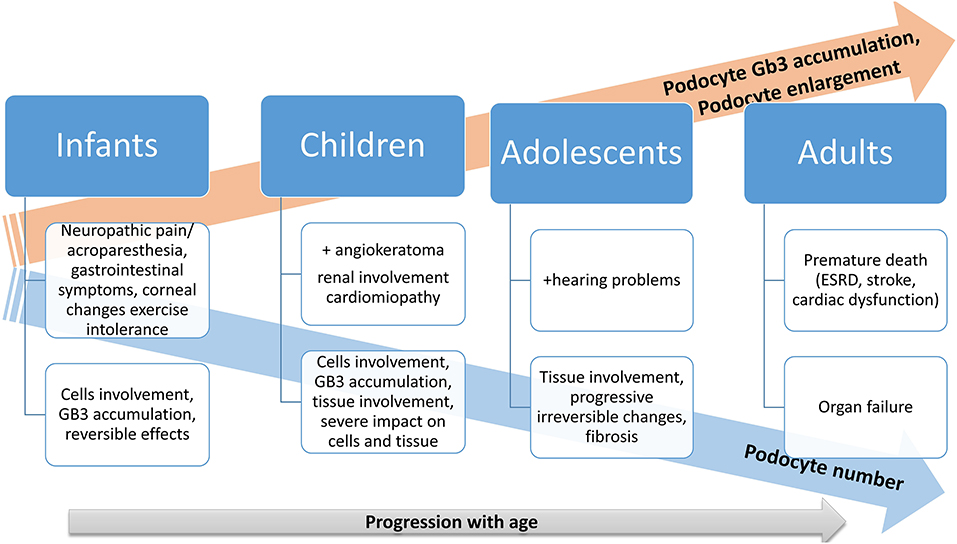
Figure 1. Progression of Fabry disease: a schematic illustration.
Also, podocyturia should be assessed in Fabry disease cases without renal involvement so far, as podocyturia precedes and is in direct relation to the magnitude of proteinuria (74). The same study suggested that podocyturia in children with Fabry disease may serve as an early biomarker of renal injury (74). In addition, it was reported that podocyturia correlates with the nephropathy's severity in Fabry disease (75–77).
“Podocyturia” evaluation involves an indirect immunofluorescence technique and it is performed only in specialized laboratories. Using special podocyte markers, positive synaptopodin is observed in podocytes in the urine sediment from FD patients (74).
A recent study found that glomerular filtration rate (GFR) and gender are not significant risk factors for pathological proteinuria occurrence, while age over 10 years and podocyturia are the two main risk factors associated with this (74).
Choi et al. investigated Fabry cases from South Korea and observed that all FD pediatric patients presented no proteinuria (94–106 mg/m2/day) and normal serum creatinine levels. Three pediatric male patients underwent kidney biopsy before ERT. In one case global sclerosis was observed, as seen on light microscopy (LM). The accumulation of Gb3 was observed in two cases in the mesangial cells on electron microscopy (11). All pediatric patients received ERT. The mean age at ERT initiation was 13.8 years old for boys, with a 3.4-year delay in ERT therapy after the onset of symptoms (11).
Fabry disease in Romanian children is rare and certainly underdiagnosed, compared to the worldwide level. Our Fabry pediatric patients with their main characteristics are presented in Table 3. For eGFR assessment updated Schwartz formula, eGFR=K x Height (cm)/Serum creatinine (mg/dl) k=0,413, was used (78). Within our cohort, one case was diagnosed in the ESRD stage in early life (11 years). A kidney biopsy was only performed in three (43%) cases, and a characteristic FD biopsy pattern was observed. In accordance with literature data, also in our cases, kidney involvement was proved by kidney biopsy before proteinuria occurrence or GFR decline (1 from three biopsies). In our female case, the first symptoms (acroparesthesia) appeared around the age of 11 years, followed by heat intolerance and abdominal pain at the age of 12 and 13, respectively. Non-opioid pain treatment was used only in one case and included most frequently acetaminophen but also neuromodulating anticonvulsant drugs such as gabapentin, as they proved to be efficient for neuropathic and central pain. Quality of life was evaluated in all patients with FD aged 8–17 with the Short Form 36 (SF-36) health-related quality of life survey and brief pain inventory form. ERT was proposed in four cases, but only three accepted it.
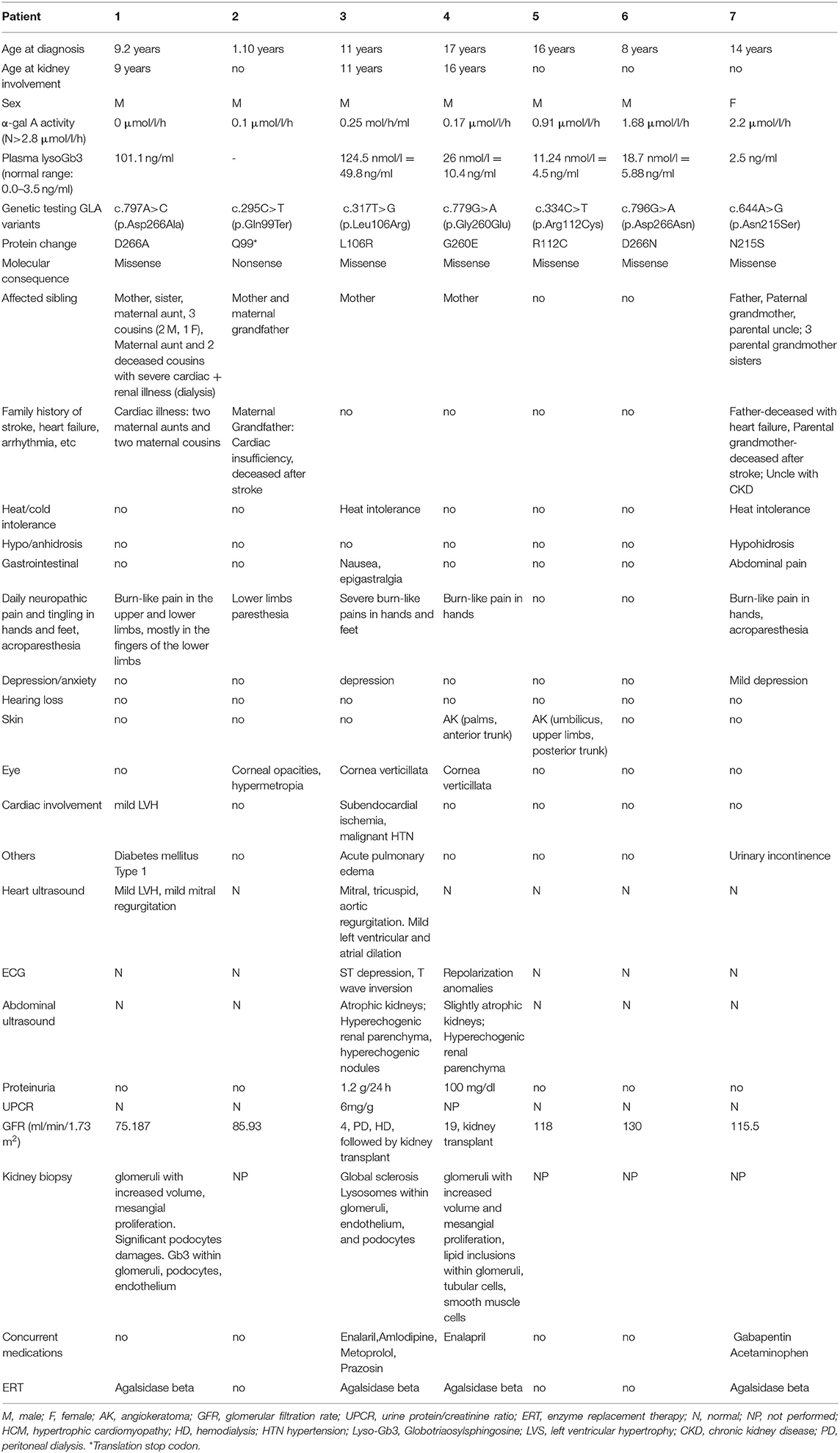
Table 3. Clinical characteristics and laboratory tests of the affected patients/individuals.
Different studies that included children and adolescents diagnosed with Fabry disease are listed in Table 4.
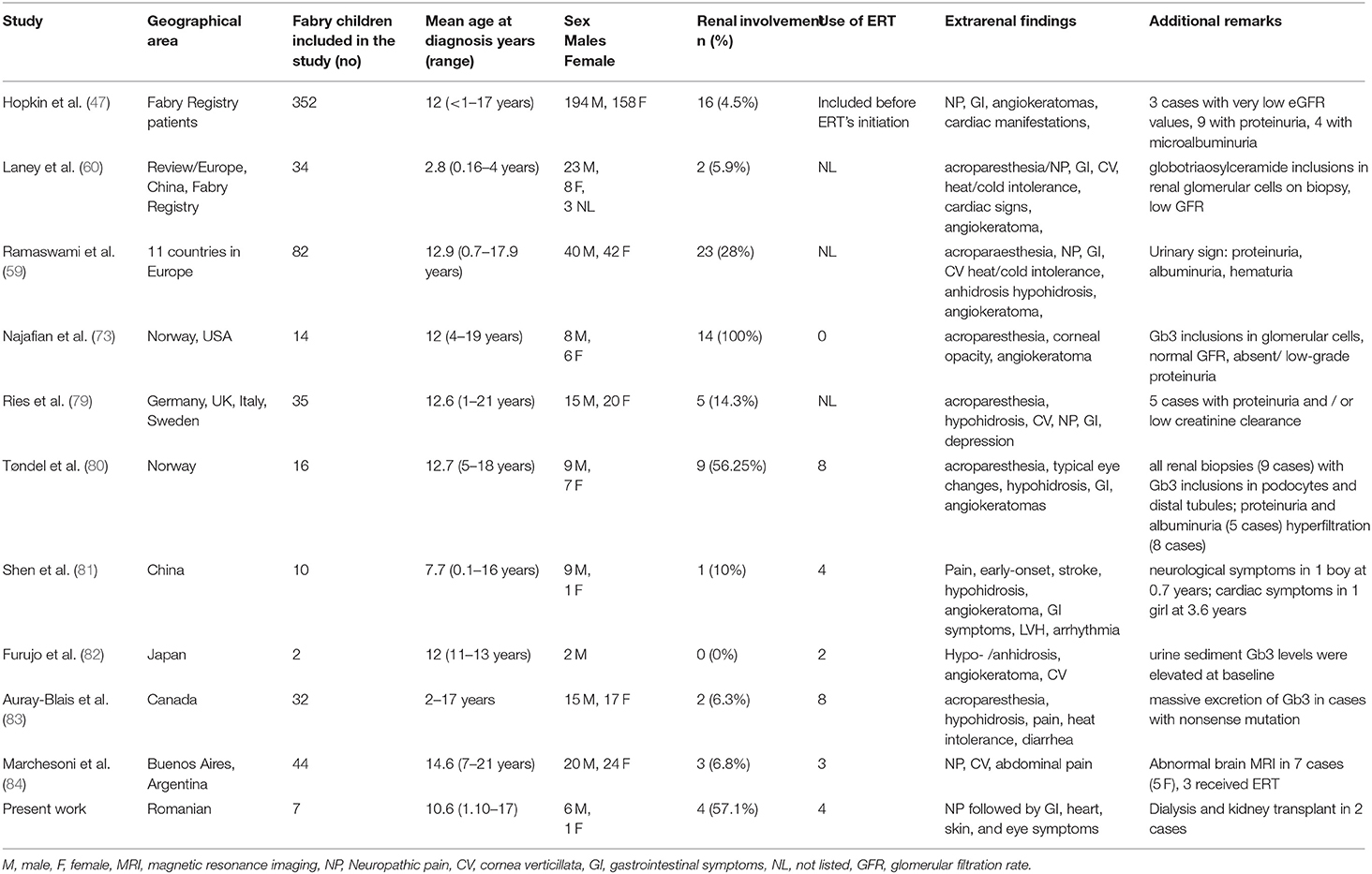
Table 4. Renal involvement in children and adolescents with Fabry disease in different studies.
Diagnosis of index cases of Fabry disease is usually delayed and rarely occurs during childhood, and this is due to the lack of specific symptoms (62, 85). The diagnosis of Fabry disease in a proband should include immediately a clinical examination, but also a biochemical and genetic investigation of the relatives both in males and females gender (8).
For boys with clinical signs and symptoms of Fabry disease, it is recommended to investigate α-Gal A activity, a value of < 1% being highly suggestive of Fabry disease and molecular testing for GLA gene mutation is necessary. Identification of a known mutation confirmed the diagnosis of Fabry. If a VUS is identified in the GLA gene, analysis of lysoGb3 is useful (1). If Fabry disease is suspected in girls GLA gene mutation analysis should be performed. In case of identification of a VUS in girls, lysoGb3 is recommended (1).
In their research, Wang et al. stated that heterozygous Fabry females should not be considered just carriers, as they may be symptomatic with severe organs involvement and risk of premature death similar to male Fabry disease patients (8). Similarly, Fernando et al. demonstrated a significant and severe affection for women with Fabry disease (86).
The activity of the α-Gal A enzyme may be measured by the liquid chromatography-tandem mass spectrometry (LC-MS) method or other methods from dried blood spots (DBSs), plasma, or serum (25, 87). The α-Gal A enzyme activity in blood is a sensitive analysis used for diagnosing affected males, but it has not been reliable for detecting manifesting heterozygous females which may have normal or only slightly decreased α-GAL A activity. The study of Stiles et al. showed that α-Gal A activity in DBS has high sensitivity (100%), but lower specificity (74%) for Fabry disease in males, as not all males with low α-Gal A activities were confirmed to have Fabry disease and recommended the enzyme analysis as the first-tier testing in males (87).
Detection of Gb3 in peripheral blood mononuclear cells (PBMCs) was proposed as a new tool for diagnosis and therapy monitoring of Fabry cases with classic form (88). Detection of Gb3 from PBMCs may represent a non-invasive, time-saving, and low-cost method (89). It may represent an alternative method in children, in whom invasive kidney biopsies are often refused (88). A reduction of Gb3 in PBMCs in Fabry patients was noticed after long-term enzyme replacement treatment (88). The response to ERT in FD was proposed as a promising method for monitoring (90), but Gb3 measurement in PBMCs did not represent the best alternative to investigate the Gb3 deposits in Fabry cases with missense mutations (89).
Analysis of lysoGb3, a degradation product of Gb3, represents an indicator of disease activity as it may reflect the overall total body substrate accumulation (91). Therefore, lysoGb3 is used for diagnostic and screening of Fabry disease, and also in monitoring treated patients, as its level decreases with ERT. It was reported that the measurement of lysoGb3 may be helpful for Fabry diagnosis, particularly in females with normal and/or borderline α-Gal A activity, and that associate non-specific symptoms (92, 93). The study of Stiles et al. showed that plasma lysoGb3 analysis and full gene sequencing are more efficient for the diagnosis of females with Fabry disease compared with α-Gal A enzyme activity (87).
It was suggested that lyso-Gb3 may be used also in the determination of the pathogenicity of a mutation (including heterozygous cases) (91). A recent study performed by Stiles et al. concluded that plasma lysoGb3 is a sensitive and specific biomarker for Fabry disease in males and females, and provides supportive diagnostic information when the results of GLA gene sequencing analysis are negative or inconclusive (87). Different studies highlighted the importance of plasma lysoGb3 in elucidating the impact of variants of uncertain clinical significance revealed by molecular investigations and assigning a disease classification (87, 94, 95). Normal lysoGb3 values cannot exclude Fabry disease in female cases but make the diagnosis of Fabry disease in men highly unlikely (96). It was reported that some variants, such as p.S126G, p. D313Y, p.A143T may not result in increased lysoGb3 levels (96). The lysoGb3 level may be measured from DBS by the LC-MS method (91).
Albuminuria and/or proteinuria, serum creatinine, glomerular filtration rate, and cystatin C, together with urinary microscopy and renal biopsy are often assessed for diagnostic, evaluation of kidney damage, monitoring of disease progression, and monitoring of treatment (97).
Tubular dysfunction is often underestimated in children with Fabry but it should be analyzed in routine clinical care. All children with Fabry disease should undergo a renal assessment: albuminuria, proteinuria (from 24-h urine collection), and GFR. Other parameters that should be considered are serum urea, creatinine, uric acid, and Cystatin C. For early detection of microalbuminuria, the measurement of the albumin/creatinine ratio in spot urine is recommended. In addition, the creatinine and cystatin C-based GFR-calculation is indicated for the estimation of renal function. Also, abdominal ultrasound should be carried out in these cases. These procedures should be evaluated at the initial clinical workup and follow-up monitoring (62). Considering that early kidney involvement is clinically silent and that early specific therapy is more likely to prevent the progressive damage of the kidney, alternative markers of renal dysfunction are required. Therefore, the research of biomarkers that are correlated with the earliest pathological findings is essential, as these biomarkers can become a non-invasive method of diagnosis in Fabry disease (98, 99).
Proteinuria (defined as the urine protein to urine creatinine ratio UPCR) even if it is not a sensitive biomarker for early kidney injury in Fabry nephropathy, is clinically one of the most often used biomarkers of Fabry nephropathy (68, 100). Based on the study of Mehta et al. that included 366 patients with Fabry from 11 European countries, the most frequently reported sign was proteinuria, which was found in 44% of hemizygous males and 33% of heterozygous female patients (101). The degree of proteinuria is an independent risk factor for the extent of declining renal function over time (68, 100). Increased levels of proteinuria before the starting of ERT may be used to predict the renal outcome. As it was reported that cases with more severe proteinuria progress more quickly to ESRD compared to those with mild proteinuria, therefore, treatment of Fabry nephropathy should aim to decrease proteinuria (100, 102). Progression of proteinuria, especially to levels > 1 g/day is a strong predictor of progression to ESRD in Fabry disease (73). It was reported that proteinuria may not be evident in all cases with advanced kidney disease, and not correlate with GFR decline (98). Nephrotic range proteinuria is rare in Fabry patients (100). Proteinuria may start during childhood, even in the first decade of life (60, 103). It was noticed that the responsiveness to ERT may be incomplete after proteinuria is manifest and the level of proteinuria usually does not reach normal values (73, 104). Loss of integrity of the glomerular basement membrane will lead to proteinuria that will have a significant contribution to the progression of renal injury toward tubulointerstitial fibrosis (50).
Albuminuria is the most common clinical sign of renal involvement in patients with Fabry disease, often preceding a detectable loss of kidney function, and is related to glomerular and podocyte injuries (6, 97, 100, 105). Albuminuria is not a sensitive biomarker for early kidney damage as it did not discover subclinical phases, as it is detectable only if glomerular podocyte damage is present (6). The kidney biopsy revealed the presence of advanced kidney lesions in Fabry patients despite normal levels of albuminuria (80). Albuminuria is considered to be a more precise indicator of kidney damage in Fabry disease and represents an earlier pathological sign than proteinuria (6, 100). Even if microalbuminuria does not represent an accurate predictor in adults with Fabry, an earlier onset of microalbuminuria in children may have stronger predictive importance (106). Microalbuminuria represents a non-invasive evaluation tool that can be easily performed and that may be an early indicator of kidney disorder and is indicated to be monitored regularly and treated appropriately if present (100).
Serum creatinine or creatinine clearance may be used to assess renal function. Creatinine clearance is not a confident marker in the evaluation of glomerular filtration rate in children considering the muscle mass changes with age. For estimating the GFR to assess kidney function serum creatinine-based equations for routine clinical use are recommended the Bedside-Schwartz formula for children and The Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation for adults (78, 107). Fabry patients with rapid progression of nephropathy have a higher urinary protein to creatinine ratio (97). The study conducted by Madsen et al. indicated that high urine albumin–creatinine ratio (UACR > 300 mg/g) was associated with a faster reduction of kidney function, in both non-age-standardized and age-standardized analyses, independent of gender (108). Similarly, findings were previously reported by Wanner et al. (109), Germain et al. (110), and Nowak et al. (111).
Cystatin-C, a cysteine small protease inhibitor, comprised of 122 amino acids, is produced by all nucleated cells and is a biomarker of glomerular function related to estimated glomerular filtration rate (eGFR) (86). Cystatin-C has been proposed, as a sensitive and reliable marker of glomerular filtration rate of renal function in Fabry patients receiving ERT and it may be used for the estimation of the efficiency of the ERT (97, 112). It was shown that the level of Cystatin-C does not depend on age, sex, or muscle mass (100, 112). Cystatin-C is not widely used in clinical practice in the management of Fabry nephropathy due to higher costs than those for creatinine dosage and also due to the labor method used for its evaluation, and less available than creatinine (97, 113). In the study of Torralba-Cabeza et al. that included 178 subjects (89 Fabry patients and 89 healthy controls), Cystatin-C concentration was found to be a superior and more sensitive marker than serum creatinine for detection of early renal dysfunction and small decreases in glomerular filtration in both males and female Fabry patients (114).
Glomerular hyperfiltration may be a common and early sign in young Fabry disease and may represent a marker for Fabry nephropathy (97). The study of Riccio that included 87 Fabry patients revealed that hyperfiltration was correlated with young age, and low proteinuria levels (98). Glomerular hyperfiltration among Fabry patients might help in implementing therapeutic strategies. Different studies observed a decrease in glomerular hyperfiltration after the introduction of ERT, suggesting a positive effect in the long term (115, 116).
The level of serum lysoGb3 is increased not only in Fabry disease males but also in many Fabry disease heterozygotes. Serum lysoGb3 represents a biomarker that is easily measured for Fabry disease activity, for monitoring of ERT, and may be useful for treatment evaluation of heterozygotes females (96). Evidence shows that lysoGb3 promotes the proliferation of smooth muscle cells, damages nociceptive neurons, leads to podocyte loss and glomerulus fibrosis, and results in inhibition of endothelial nitric oxide synthase (53). In cases with non-specific Fabry symptoms (for example CKD or LVH) but without characteristic signs or biochemical changes of classical Fabry disease, increased levels of lysoGb3 are suggestive of a diagnosis of Fabry disease. Smid et al. suggested that a normal lysoGb3 cannot exclude Fabry disease in females but makes the diagnosis of Fabry in males highly unlikely (96). The study of Nowak et al. that included 66 genetically confirmed Fabry patients reported that serum lysoGb3 is a significant risk factor associated with important adverse clinical outcomes in a long-term study (68 months), and represents a useful biomarker for diagnosis in heterozygotes with normal α-Gal A enzyme activity (111).
Different studies recommend that kidney biopsy should be considered in selected pediatric cases, especially in children with significant proteinuria or a fast decline in renal function, a variant in the GLA gene, when the decision to start ERT is doubted and an uncertain diagnosis of Fabry disease (17, 97, 117). Kidney biopsy may be considered in selected pediatric cases, especially in children with significant proteinuria or a fast decline in renal function, when the decision to start ERT is doubted; or when it is necessary to rule out a second renal disorder (17). Kidney biopsy may be considered in patients where the diagnosis can be challenging and in those cases where there is uncertainty about whether to start ERT to identify the Gb3 accumulation (50). In the study of Choi et al. that investigated Fabry disease pediatric patients from South Korea, it was observed that all children presented no proteinuria and normal serum creatinine levels. Kidney biopsy performed in three pediatric male patients before ERT revealed global sclerosis (as seen on light microscopy), while in two cases the accumulation of Gb3 was observed in the mesangial cells on electron microscopy (11). Electron microscopy images may show characteristic/ pathognomonic zebra bodies lamellar deposits (lamellar lipid inclusion bodies) in podocyte cytoplasm and tubules (68).
It is very important to have an early diagnosis of Fabry nephropathy whereas the early initiation of treatment may stop or delay progressive renal dysfunction more effectively compared with the late therapy initiation. Different studies considered that the kidney biopsy with electron microscopy analysis represents the only diagnostic for confirmation or exclusion of Fabry disease nephropathy and recommend to be considered for all patients with CKD, a variant in the GLA gene, and an uncertain diagnosis of Fabry disease (97, 117). The study of Thurberg et al. observed after kidney biopsy in children higher storage of Gb3 in distal tubular epithelial cells and podocytes, with the widening of their foot processes (118).
Gb3 deposits may be identified in omiscated epoxy-embedded semithin sections stained with toluidine blue under light microscopy (119). Gb3 deposits emerge as dark blue, dens granules particularly in podocytes, epithelial and tubular cells as well as in smooth muscle and endothelial cells (41). This specific lipid staining examined by common light microscopy is a valuable method when electron microscopy is not available (58).
Light microscopy (periodic acid–Schiff) revealed a wide spectrum of morphological changes in the glomerular, tubulointerstitial, and vascular areas in mild albuminuric children (80). Furthermore, electron microscopy showed a heavy accumulation of Globotriaosylceramide (Gb3) inclusions in podocytes and distal tubular epithelial cells in all patients and lower amounts (slightly increased amounts) in endothelial and mesangial cells (80). Based on these findings, Tøndel et al. (80) endorsed a timely kidney biopsy in all albuminuric patients with FD trying to assess the magnitude of kidney damage (80). Another study documented early renal pathologic lesions by electron microscopy (EM) in young patients with Fabry disease. The same study showed an age-dependent progressive Gb3 inclusions density in podocytes and not in endothelial and mesangial cells (73). Moreover, this study documented the existence of podocytes damage in Fabry patients without albuminuria, therefore this parameter is not sensitive enough for a diagnosis of early Fabry nephropathy (73). In another study, the morphologic changes in the kidney biopsy samples in young patients with Fabry disease treated with agalsidase alfa or agalsidase beta for 5 years were evaluated (119). This study demonstrated a significant correlation between podocyte GL3 clearance and cumulative agalsidase dose in young patients with Fabry disease as well as concomitant reduction of podocyte effacement (119).
Considering that kidney biopsy is invasive and carries some risk for complications it is a controversial subject in children with Fabry disease (6). Recently it was recommended to take into account that kidney biopsy should only be selected in cases where it is under the question to initiate ERT, in those children with marked proteinuria, or those children with a rapid decrease in kidney function or for differential diagnosis (6). Electron microscopy images may show characteristic/ pathognomonic zebra bodies lamellar deposits (lamellar lipid inclusion bodies) in podocyte cytoplasm and tubules (68).
Targeted urinary microscopy, a non-invasive, accessible, affordable, and rapid diagnostic test that may be used, in those cases where leucocyte α-Gal A activity and GLA genotyping are not available. Different reports suggest that urine microscopy may be useful for the evaluation of the progression of the disorder (120) but its value is limited especially in heterozygous females and due to the fact that the majority of the findings are not specifically characteristic for Fabry disease. Examination of urinary specimens under phase-contrast microscopy using a polarized light filter may provide additional information by identifying birefringent Maltese cross patterns (MC, oval fat bodies) in the urine sediment of patients. Even though it is a valuable method is not a routine evaluation in most clinics evaluating Fabry patients. Selvarajah et al. observed characteristic MC particles inside the vacuolated urinary epithelial cells, with lamellated appearance with protrusions probably due to accumulation of Gb3, namely MC2 particles, and reported high sensitivity and specificity (100%) (120). In addition, Selvarajah et al. suggested that the number of characteristic urinary MC particles increases concomitantly with rising albuminuria, therefore, this investigation may be useful in assessing Fabry's nephropathy progression. The routine laboratory urine assessment by microscopy that does not include a polarized light examination may easily miss the MC particles (120). By using the proper microscopy method also podocyturia may be evaluated (121). Besides microscopy of stained cells, immunohistochemistry, or immunofluorescence methods different techniques may use to assess podocyturia (liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) method, or mRNA quantification (77, 120, 122, 123).
Elevated levels of podocyturia were observed by Trimarchi et al. in cases with Fabry disease compared to the normal population and suggested that podocyturia may precede proteinuria (77). In addition, Sanchez-Niño et al. showed that podocyturia may precede pathological albuminuria in Fabry disease (124). Fall et all, showed that Fabry disease is associated with increased podocyte loss and reported direct associations between podocyturia and severity of Fabry nephropathy. In addition, in the study of Fall et al. an inverse association was found between podocyturia and eGFR in male Fabry patients (76). It was shown that Fabry cases with ERT had a lower podocyturia, suggesting that ERT may reduce their irreversible loss by stabilizing podocytes' attachment, and suggested that monitoring of podocyturia may be useful for follow-up of Fabry patients on treatment in order to evaluate the treatment response (76, 77).
Increased levels of urinary Gb3 may be found in Fabry patients with no residual α-Gal A enzyme activity. Normal levels of urinary Gb3 were measured in heterozygous females and those cases with a major residual α-Gal A enzyme activity (3).
Vedder et al. reported that elevated levels of Gb3 in plasma or urine did not correlate with the severity of the disease nor with Fabry-related symptoms. According to the analysis of the Dutch Fabry cohort, it may be considered that urinary or plasma Gb3 levels have no value as surrogate disease markers (3).
The level of urinary Gb3 being reduced after the beginning of enzyme replacement therapy is an indicator of the metabolic effects of treatment. Increased level of urinary Gb3 is associated with the production of α-Gal A antibodies (3, 91, 125).
Simonetta et al. considered that there is no evidence to use urinary Gb3 for the prognostic role or for monitoring the response to enzyme replacement therapy on the nephrological outcome (91). The study of Auray-Blais et al. that included 110 children and adults with Fabry disease found no correlation between the estimated GFR and urinary Gb3 (126). Similarly, changes in urine Gb3 are not useful biomarkers for the prediction of Fabry disease-related changes in eGFR (125). A significant correlation between the levels of urinary excretion of Gb3/creatinine and types of mutations (p = 0.0007), sex (p < 0.0001), and treatment (p = 0.0011) in children and adults with Fabry disease was observed in the study performed by Auray-Blais et al. (126). The levels of urinary lysoGb3 correlate with proteinuria and albuminuria, but not with GFR, therefore it does not represent a good indicator of renal function (126).
Beta 2-microglobulin (β2M) represents a biomarker for the tubular reabsorption function (113). Even if it is not usually measured, β2M in serum samples was shown to be the most appropriate renal biomarker for the determination of renal impairment among Fabry disease patients. Considering that β2M is not influenced by muscle mass, it may represent a potential marker that may be used for the estimation of GFR more accurately in adults. Argyropoulos et al. considered that serum β2M as a measure of glomerular filtration function is not useful in children when compared with adults (127).
Uromodulin, also known as Tamm-Horsfall protein, is the most abundant protein in human urine, that is exclusively produced in the kidney and its daily secretion is about 50–150 mg (128). An important biomarker for the development of CKD may be represented by the urinary concentration of uromodulin (129). Uromodulin may be a potential urinary biomarker of the renal tubular reserve function in male and female Fabry patients with nephropathy (130).
Doykov et al. reported significantly lower uromodulin levels in Fabry disease cases with both kidney and cardiac involvement (p = 0.0045). Lower levels of uromodulin were observed in the late-stage patients presenting kidney, heart, and central nervous system involvement (p = 0.0049) (131). In a previous study, uromodulin excretion was reduced in the urine of FD patients (132), while Matafora found a higher urinary excretion of uromodulin in naive Fabry patients (130). Despite these conflicting results, both studies found that urinary excretion of uromodulin came to normal after ERT, with concentrations similar to those observed in healthy controls (130, 132). Based on these the authors (130, 132) concluded/speculated that this urinary protein may be used as a marker to monitor the response to ERT.
Recently, Steubl et al. observed that uromodulin was independently associated with ESRD or rapid loss of eGFR, and suggested that uromodulin might serve as a robust predictor of rapid kidney function decline (129). It was suggested that uromodulin up-regulation could be considered a very early marker of kidney damage at the tubular level, in cases with a normal level of creatinine and normal GFR, especially in heterozygous female Fabry patients, that may associate the normal value of α-Gal A activity (130, 133).
The excretion of prostaglandin H2 D-isomerase is increased in Fabry disease, probably due to tubular dysfunction present in affected patients (130). The study of Matafora et al. revealed that the prostaGLAndin H2 d-isomerase concentration decreased after ERT speculating that these proteins may be used as markers for monitoring the response to treatment (130).
Bikunin or urinary trypsin inhibitor (UTI) is a serine protease inhibitor, whose excretion increases in inflammations and accumulates in urine. In pathological conditions such as kidney disease, the concentrations of bikunin in plasma and urine are increased (97). The study of Lepedda et al. that included 24 Fabry cases and 43 healthy controls, showed that urine bikunin levels may be an early biomarker of renal deterioration in Fabry patients (134). The same study performed by Lepedda noticed no association between serum creatinine and urine bikunin levels, therefore, the direct involvement of the kidney in urine bikunin excretion in the case of Fabry's patients is not enough (134).
By its multisystemic character and progressive course, Fabry disease is characterized by a significant reduction in life quality and expectancy. Fabry disease requires a multidisciplinary approach and organ-specific treatment. Specific therapy for Fabry disease should not be initiated in individuals bearing non-pathogenic GLA variants.
Currently, available treatment options are represented by enzyme replacement therapy (ERT), and chaperon therapy. Increased efforts were made in the last years for a better therapy in Fabry patients and new treatments that include substrate reduction therapy (SRT) gene therapy, and mRNA based therapy are administrated in clinical trials.
Currently, available enzyme replacement therapies are agalsidase-alpha and agalsidase-beta approved in Europe since 2001. ERT aims to restitute defective α-GAL A (58). Agalsidase alfa (Replagal; Shire Human Genetic Therapies Inc., Lexington, MA, USA/ (Replagal; Takeda Pharmaceutical, Tokyo, Japan), with a certified dose of 0.2 mg/kg, in male pediatric patients over the age of 7, or agalsidase beta (Fabrazyme; Genzyme, a Sanofi company, St Germain en Laye, France) at the licensed dose at 1.0 mg/kg in pediatric patients ≥ 8 years of age. ERT is recommended to be administered every other week in a short intravenous infusion.
According to US consensus, the ERT should be considered in all symptomatic cases regardless of age or gender (17). In asymptomatic boys with Fabry disease with GLA mutation, based on US consensus, the ERT should be recommended around the age of 8–10 years (17). According to the European Fabry Working Group consensus in asymptomatic boys with classic mutation for Fabry, the ERT treatment should be started at the age of 16 (135).
The initiation of ERT therapy as early as possible gives the best clinical outcome and ERT's effect depends on the stage of the disease (85). Prevention and early specific therapy, ERT, are important, as they may slow progressive symptomatic organ complications, improve quality of life and in later life, improve both morbidity and mortality, and preserve life expectancy. ERT should be considered in the case of symptomatic boys and girls that present neuropathic pain, pathological albuminuria (≥3 mg/mmol creatinine), severe gastrointestinal symptoms, and abdominal pain or cardiac involvement. Initiation of ERT based only on the presence of angiokeratoma is not recommended in children (62).
Different studies with agalsidase-alfa showed decreased Gb3 accumulation in the liver and in tubular epithelial cells and a reduction of Gb3 excretion in urine, a notable reduction in podocytes Gb3 inclusions, and complete clearance of glomerular endothelial and mesangial inclusions (49, 119, 136).
It was shown that ERT reduced Gb3 in the kidneys, heart, and skin, being particularly effective in clearing the endothelial cells. On the other hand, podocytes, distal tubular cells, and smooth muscle cells showed a smaller reduction of Gb3 than that observed in other cell types therefore it seems to be more resistant to ERT (118, 136).
The clinical benefit of ERT is mainly observed in patients who start ERT before the presence of irreversible organ damage (137). Arends et al. observed that despite treatment with ERT disease progression is predicted by the presence of reduced renal function, and proteinuria at the time of therapy initiation (138).
Unfortunately, in response to ERT, immunoglobulin G antibodies may be generated (in about 40% of Fabry males with no α-Gal A activity) and lead to inhibition of enzyme activity that may negatively influence the clinical outcome of Fabry patients (139).
In the non-pathogenic GLA variant carriers (for example p.E66Q, p.R118C, p.S126G, p.A143T, and p.D313Y) ERT should not be started (62). Two new forms of ERT, with increased stability and lower immunogenicity, for the treatment of Fabry disease have been developed; Pegunigalsidase-alfa (PRX-102, Protalix Biotherapeutics, Israel and Chiesi Global Rare Diseases, USA) and moss-agalactosidase A (moss-aGal, Greenovation biopharmaceuticals, Germany). It was suggested that PRX-102 may stabilize renal function and that moss-aGal may target kidney cells (140, 141). Despite ERT treatments (agalsidase alfa and beta) that are associated with anti-drug antibody (ADA) development that is associated with reduced pharmacodynamic and clinical responses, the new ERT pegunigalsidase-alfa is associated with low immunogenicity, improvements in symptoms, and also with an important reduction in Gb3 deposition in the kidney and a reduction in plasma lysoGb3 level (140).
Chaperone therapy with Migalastat (Galafold, Amicus Therapeutics), was approved in 2016 in Europe and 2018 in the USA, respectively. In European Union, Migalastat was approved first for Fabry patients with an amenable mutation, older than 16 years. In 2021 Migalastat's (Galafold) approval was expanded to FD children with an amenable mutation, starting at age 12 and weighing at least 45 kilograms (99 pounds). Chaperone therapy may be used in patients with “amenable” variants (for example p. N215S) that are missense mutations with normal α-Gal A catalytic activity, but these mutations lead to a reduction in overall α-Gal A enzymatic activity due to strongly decreased stability of the mutated protein (142) Based on the review of Weidemann, it is important to classify the GAL gene mutation for amenability to treatment with Migalastat in each new Fabry case (142). Non-amenable GAL gene mutations (for example (large mutations, frameshift mutations, splicing mutations, insertions, truncations) do not pass the good laboratory practice (GLP) HEK assay for amenability developed by Benjamin et al. (143).
A study by Riccio et al. showed a significant decrease in proteinuria after 1-year of therapy with Migalastat (144). Migalastat is significantly eliminated by the kidneys and is not recommended in Fabry cases with GFR <30 ml/min/1.73 m2 or ESRD that requires dialysis. In the case of GFR >30 ml/min/1.73 m2 is not necessary the dose adjustment (41). Migalastat is the only oral treatment for Fabry, that may be used as first-line treatment in ERT-naive patients, or as an alternative to ERT (144). Migalastat represents an alternative to ERT in cases where ERT response is lost or in cases of antibody formation to ERT (42). Migalastat stabilizes α-GalA mutated protein, escaping from degradation and movement to the lysosomes (136). Migalastat as monotherapy has been shown to reduce the Gb3 accumulation but for achieving its effects, the drug requires at least some endogenous enzyme production. Thus, only a limited group of Fabry patients would respond to monotherapy with Migalastat. Migalastat is recommended for selected patients, without mutations that cause complex enzyme alterations (145). In the future, a possible approach for increasing the stability of the α-Gal A enzyme activity may be represented by the co-administration of Migalastat and ERT. The study of Warnock et al. revealed an increase of α-Gal A plasma level of 1.2–5.1 fold in cases that received combined Migalastat and ERT compared with those that received only ERT (146). The study of Benjamin et al. indicated that the co-administration of ERT and migalastat in Fabry mice resulted in an increased AGAL tissue uptake and improved Gb3 reduction (147).
Chaperones favor the proper folding of the mutated protein and increase its stability, which results in a reduction of Gb3 and its substrates. Fabry patients which are eligible for treatment with Migalastat are established by using an in vitro enzyme activity assay (122). Chaperone Migalastat is an inhibitor of α-GAL A, but in small doses, it may increase α-GAL A enzymatic activity for some GLA gene mutations (142).
Additional therapy is represented by substrate reduction therapy (SRT) which targets the glycosphingolipid synthesis to reduce the formation of metabolites that cannot be degraded. SRT is only available in clinical studies (139, 140) and it is not licensed to treat patients at the moment. In patients with residual enzyme activity, SRT may be enough as a single therapy for reducing the level of the substrate. SRT drugs that are used in clinical trials are Lucerastat and Venglustat.
Lucerastat or N-butyldeoxygalactonojirimycin (Idorsia Pharmaceutical Ltd, Allschwil, Switzerland) functions as an inhibitor of glucosylceramide synthase and thus will prevent the accumulation of Gb3 and lysoGb3 by restricting the quantity of ceramide that is metabolized in glycosphingolipid and is currently under evaluation in phase three clinical study for Fabry disease (139). Recently it was reported the reduction of sphingolipids and their metabolites after initiation of SRT at doses of 1,000 mg twice a day as single therapy (136). It was suggested that in addition to ERT may provide a new form of combination therapy that could be beneficial to this population.
Venglustat (Ibiglustat, Sanofi Genzyme, Cambridge, MA), an oral inhibitor of glucosylceramide synthase prevents the synthesis of glucosylceramide (GL1) and therefore reduces Gb3 (139) and is under investigation in phase two clinical study.
In the case of patients with Fabry disease with specific (“amenable”) mutations, treatment with chaperones may represent the appropriate approach while for the remaining Fabry patients, the combined therapy such as ERT with substrate reduction therapy, might have a benefic effect.
Gene therapy, a promising option for treatment, aims to correct the underlying genetic defect of Fabry disease. Theoretically, gene therapy could be more effective than current options in Fabry disease due to the fact that transduced cell populations will produce α-Gal A continuously. A pilot study (NCT02800070) that included five patients with type I Fabry disease used lentiviral vectors for carrying the coding sequence for α-Gal A. The patients received autologous transduced CD34+-selected cells that were genetically engineered to express α-Gal A and the transduced cells may provide the functional α-Gal A enzyme to sites that are not accessible to ERT therapy. The trial showed that the Fabry patients' recipients had plasma and leukocyte normal α-Gal A activity and associated reductions of plasma and urine Gb3 and lysoGb3 levels (148). Due to the previous studies that revealed the persistence of adeno-associated viruses for multiple years and considering that they lead to therapeutic levels of transgene expression in 2019 two clinical trials were approved for the treatment of Fabry disease. Those clinical trials utilized adeno-associated viruses-based gene therapy or adenovirus encoding GLA cDNA are administrated for cellular expression of αGal A in different organs such as kidneys, lives, heart, etc (149). Therefore, gene therapy may represent in the future an effective treatment option for patients with Fabry disease. Gene therapy is associated with some risks, the most significant are toxicity and immune reactions. Virus vectors used may induce inflammatory responses, stimulating the production of antibodies that may lead to a decreasing efficacy. The integration of the functional gene into the host genome may disrupt the host gene at the site of insertion or may activate o proto-oncogene and promote malignancy development (6, 150).
mRNA therapy, with drug-like properties, induces a prolonged human α-galactosidase protein production and subsequent secretion. Multi-component lipid nanoparticles were developed for the distribution of mRNA that encodes a human α-galactosidase protein (149). It was observed that administration of lipid nanoparticles mRNA for human α-galactosidase increased α-Gal A levels expressed in different tissues (kidney liver, heart) and led to an improved Gb3 clearance (151). Unfortunately, mRNA-based therapy has a transient effect therefore repeated administration is required. In the future, mRNA therapy may represent an alternative approach to the treatment of Fabry disease.
Simultaneously with specific therapy, supportive care should be offered to manage pain, gastrointestinal symptoms, arterial blood pressure, and, when kidney failure occurs, renal replacement therapy and transplantation (Table 5).
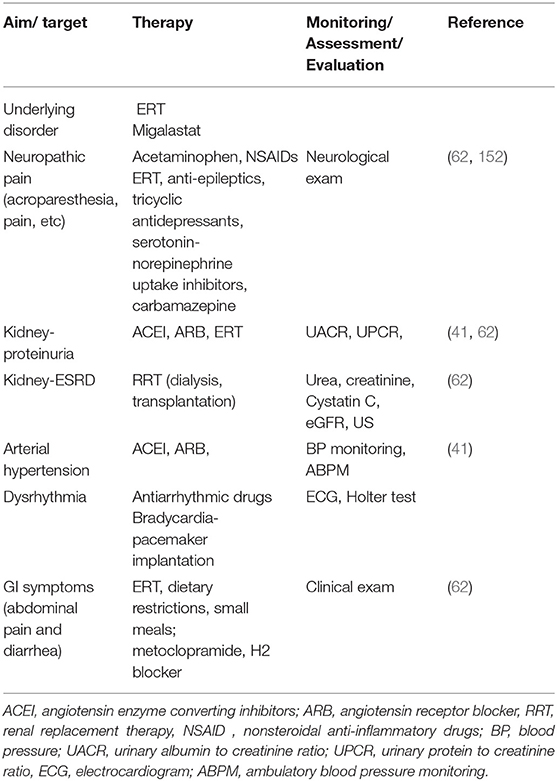
Table 5. Symptomatic therapy in pediatric patients with FD.
A low sodium diet is recommended in Fabry patients considering that sodium may reduce the effects of ACEI, and ARB and may predispose/increase the risk for ERDS in cases with proteinuria (139). Blood pressure should be monitored when starting ACEI or ARB treatment, and it is recommended to avoid the combination of both drugs as they may lead to additive drug-related adverse effects The ACEI or ARB administration should be given at bedtime in Fabry patients with low blood pressure (41). It is recommended for Fabry patients to avoid the use of nephrotoxic substances, non-steroidal anti-inflammatory drugs (NSAID) such as ibuprofen that may lead to acute kidney injury. Because of avoidance of sunlight exposure and malabsorptive gastrointestinal disease in Fabry patients, vitamin D deficiency should be checked and vitamin D supplementation should be provided if it is necessary. Vitamin D receptor activation reduces the inflammation associated with lysoGb3 and, therefore, Fabry patients should be screened for and started on the appropriate supplementation regimen if needed (153).
It is recommended to monitor boys at least once a year and girls at least every 2–3 years. In the case of asymptomatic children from families with members diagnosed with Fabry disease, Germain et al. recommend that the baseline assessment of organ involvement should begin at 5 years of age for boys and at 12–15 years of age for girls (62).
FD is a multisystemic and multifaceted disease that starts early in life, with symptoms occurring during childhood with progressive evolution that worsens throughout adulthood. Nowadays with early diagnosis of kidney involvement in FD and new proposed therapies a better outcome is expected.
CM and CB: ideas, coordination of the study, writing and editing the manuscript, analysis of the data, and writing up the results. CM, IS, and CS: coordination of the study between three centers and writing the manuscript. CM, IS, CS, and CB: write and revised the manuscript. All authors have read and agreed with the final form of the manuscript.
This was partly supported by a project financed by the Romanian Ministry of Education and Research, CNCS—UEFISCDI, project no PN-III-P4-ID-PCE-2020-1928, within the PNCDI III, contract no. PCE 72/2021.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors want to thank Dr. Mihaela Munteanu for her involvement in diagnosis and monitoring children with Fabry disease and kidney involvement.
1. ^Available online at: https://www.ncbi.nlm.nih.gov/clinvar?term=300644[MIM].
1. Vardarli I, Rischpler C, Herrmann K, Weidemann F. Diagnosis and screening of patients with fabry disease. Ther Clin Risk Manag. (2020) 16:551–8. doi: 10.2147/TCRM.S247814
2. Elleder M, Poupetová H, Kozich V. Fetální patologie Fabryho nemoci a mukopolysacharidózy I [Fetal pathology in Fabry's disease and mucopolysaccharidosis type I]. Cesk Patol. (1998) 34:7–12.
3. Vedder AC, Strijland A, vd Bergh Weerman MA, Florquin S, Aerts JM, Hollak CE. Manifestations of Fabry disease in placental tissue. J Inherit Metab Dis. (2006) 29:106–11. doi: 10.1007/s10545-006-0196-0
4. Alkhzouz C, Miclea D, Bucerzan S, Lazea C, Nascu I, Sido PG. Early clinical signs in lysosomal diseases. Med Pharm Rep. (2021) 94:43–6. doi: 10.15386/mpr-2228
5. Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. (2006) 79:31–40. doi: 10.1086/504601
6. Chimenz R, Chirico V, Cuppari C, Ceravolo G, Concolino D, Monardo P, et al. Fabry disease and kidney involvement: starting from childhood to understand the future. Pediatr Nephrol. (2022) 37:95–103. doi: 10.1007/s00467-021-05076-x
7. Militaru S, Adam R, Ismail G, Rusu E, Dulămea A, Jurcut R. Demographic and clinical characteristics of the full 2015-2018 cohort of Romanian Fabry Disease patients. Curr Health Sci J. (2019) 45:272–7. doi: 10.12865/CHSJ.45.03.04
8. Wang RY, Lelis A, Mirocha J, Wilcox WR. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med. (2007) 9:34–45. doi: 10.1097/GIM.0b013e31802d8321
9. Bokhari SRA, Zulfiqar H, Hariz A. Fabry Disease. In: StatPearls. Treasure Island (FL): StatPearls Publishing (2022). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK435996/ (accessed January 7, 2022)
10. Capuano I, Garofalo C, Buonanno P, Pinelli M, Di Risi T, Feriozzi S, et al. Identifying Fabry patients in dialysis population: prevalence of GLA mutations by renal clinic screening, 1995-2019. J Nephrol. (2020) 33:569–81. doi: 10.1007/s40620-019-00663-6
11. Choi JH, Lee BH, Heo SH, Kim GH, Kim YM, Kim DS, et al. Clinical characteristics and mutation spectrum of GLA in Korean patients with Fabry disease by a nationwide survey: Underdiagnosis of late-onset phenotype. Medicine. (2017) 96:e7387. doi: 10.1097/MD.0000000000007387
12. Wittmann J, Karg E, Turi S, Legnini E, Wittmann G, Giese AK, et al. Newborn screening for lysosomal storage disorders in hungary. JIMD Rep. (2012) 6:117–25. doi: 10.1007/8904_2012_130
13. Colon C, Ortolano S, Melcon-Crespo C, Alvarez JV, Lopez-Suarez OE, Couce ML, et al. Newborn screening for Fabry disease in the north-west of Spain. Eur J Pediatr. (2017) 176:1075–81. doi: 10.1007/s00431-017-2950-8
14. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. (1999) 105:151–6. doi: 10.1007/s004399900075
15. Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H, et al. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet. (2004) 12:87–92. doi: 10.1038/sj.ejhg.5201044
16. Mechtler TP, Stary S, Metz TF, De Jesús VR, Greber-Platzer S, Pollak A. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet. (2012) 379:335–41. doi: 10.1016/S0140-6736(11)61266–X
17. Hopkin RJ, Jefferies JL, Laney DA, Lawson VH, Mauer M, Taylor MR, et al. Fabry Pediatric Expert Panel. The management and treatment of children with Fabry disease: a United States-based perspective. Mol Genet Metab. (2016) 117:104–13. doi: 10.1016/j.ymgme.2015.10.007
18. Koto Y, Sakai N, Lee Y, Kakee N, Matsuda J, Tsuboi K, et al. Prevalence of patients with lysosomal storage disorders and peroxisomal disorders: A nationwide survey in Japan. Mol Genet Metab. (2021) 133:277–88. doi: 10.1016/j.ymgme.2021.05.004
19. Inoue T, Hattori K, Ihara K, Ishii A, Nakamura K, Hirose S. Newborn screening for Fabry disease in Japan: prevalence and genotypes of Fabry disease in a pilot study. J Hum Genet. (2013) 58:548–52. doi: 10.1038/jhg.2013.48
20. Hwu WL, Chien YH, Lee NC, Chiang SC, Dobrovolny R, Huang AC, et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c936+919G>A (IVS4+919G>A). Hum Mutat. (2009) 30:1397–405. doi: 10.1002/humu.21074
21. Zhang J, Zhang C, Gao E, Zhou Q. Next-generation sequencing-based genetic diagnostic strategies of inherited kidney diseases. Kidney Dis. (2021) 7425–37. doi: 10.1159/000519095
22. Mitobe S, Togawa T, Tsukimura T, Kodama T, Tanaka T, Doi K, et al. Mutant α-galactosidase A with M296I does not cause elevation of the plasma globotriaosylsphingosine level. Mol Genet Metab. (2012) 107:623–6. doi: 10.1016/j.ymgme.2012.07.003
23. Germain DP, Shabbeer J, Cotigny S, Desnick RJ. Fabry disease: twenty novel alpha-galactosidase A mutations and genotype-phenotype correlations in classical and variant phenotypes. Mol Med. (2002) 8:306–12. doi: 10.1007/BF03402156
24. Sakuraba H, Tsukimura T, Togawa T, Tanaka T, Ohtsuka T, Sato A, et al. Fabry disease in a Japanese population-molecular and biochemical characteristics. Mol Genet Metab Rep. (2018) 17:73–9. doi: 10.1016/j.ymgmr.2018.10.004
25. Sawada T, Kido J, Sugawara K, Matsumoto S, Takada F, Tsuboi K, et al. Detection of novel Fabry disease-associated pathogenic variants in Japanese patients by newborn and high-risk screening. Mol Genet Genomic Med. (2020) 8:e1502. doi: 10.1002/mgg3.1502
26. Yamamoto S, Nagasawa T, Sugimura K, Kanno A, Tatebe S, Aoki T, et al. Clinical diversity in patients with Anderson-Fabry disease with the R301Q mutation. Intern Med. (2019) 58:603–7. doi: 10.2169/internalmedicine.0959-18
27. Sawada T, Kido J, Sugawara K, Nakamura K. High-Risk screening for Fabry disease: a nationwide study in Japan and Literature Review. Diagnostics. (2021) 11:1779. doi: 10.3390/diagnostics11101779
28. Oliveira JP, Nowak A, Barbey F, Torres M, Nunes JP, Teixeira-E-Costa F, et al. Fabry disease caused by the GLA pPhe113Leu (pF113L) variant: Natural history in males. Eur J Med Genet. (2020) 63:103703. doi: 10.1016/j.ejmg.2019.103703
29. Hung CL, Wu YW, Lin CC, Lai CH, Jyh-Ming Juang J, Chao TH, et al. 2021 TSOC expert consensus on the clinical features, diagnosis, and clinical management of cardiac manifestations of Fabry Disease. Acta Cardiol Sin. (2021) 37:337–54. doi: 10.6515/ACS.202107_37(4).20210601A
30. Togawa T, Tsukimura T, Kodama T, Tanaka T, Kawashima I, Saito S, et al. Fabry disease: biochemical, pathological and structural studies of the α-galactosidase A with E66Q amino acid substitution. Mol Genet Metab. (2012) 105:615–20. doi: 10.1016/j.ymgme.2012.01.010
31. Peng H, Xu X, Zhang L, Zhang X, Peng H, Zheng Y, et al. GLA variation pE66Q identified as the genetic etiology of Fabry disease using exome sequencing. Gene. (2016) 575:363–7. doi: 10.1016/j.gene.2015.09.088
32. du Moulin M. Muschol N. pD313Y is more than just a polymorphism in Fabry disease. Clin Genet. (2018) 93:1258. doi: 10.1111/cge.13233
33. Froissart R, Guffon N, Vanier MT, Desnick RJ, Maire I. Fabry disease: D313Y is an alpha-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol Genet Metab. (2003) 80:307–14. doi: 10.1016/S1096-7192(03)00136-7
34. Niemann M, Rolfs A, Giese A, Mascher H, Breunig F, Ertl G, et al. Lyso-Gb3 indicates that the alpha-galactosidase A mutation D313Y is not clinically relevant for Fabry disease. JIMD Rep. (2013) 7:99–102. doi: 10.1007/8904_2012_154
35. Oder D, Wanner C, Nordbeck P. The D313Y genotype-pathogenic mutation or polymorphism? Clin Genet. (2018) 93:1257. doi: 10.1111/cge.13237
36. Lukas J, Giese AK, Markoff A, Grittner U, Kolodny E, Mascher H, et al. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in Fabry disease. PLoS Genet. (2013) 9:e1003632. doi: 10.1371/journal.pgen.1003632
37. Ferreira S, Ortiz A, Germain DP, Viana-Baptista M, Caldeira-Gomes A, Camprecios M, et al. The alpha-galactosidase A pArg118Cys variant does not cause a Fabry disease phenotype: data from individual patients and family studies. Mol Genet Metab. (2015) 114:248–58. doi: 10.1016/j.ymgme.2014.11.004
38. Talbot A, Nicholls K. Elevated Lyso-Gb3 Suggests the R118C GLA Mutation Is a Pathological Fabry Variant. JIMD Rep. (2019) 45:95–8. doi: 10.1007/8904_2018_146
39. Terryn W, Vanholder R, Hemelsoet D, Leroy BP, Van Biesen W, De Schoenmakere G, et al. Questioning the Pathogenic Role of the GLA pAla143Thr “Mutation” in Fabry Disease: Implications for Screening Studies and ERT. JIMD Rep. (2013) 8:101–8. doi: 10.1007/8904_2012_167
40. Lenders M, Weidemann F, Kurschat C, Canaan-Kühl S, Duning T, Stypmann J, et al. Alpha-Galactosidase A pA143T, a non-Fabry disease-causing variant Orphanet. J Rare Dis. (2016) 11:54. doi: 10.1186/s13023-016-0441-z
41. Silva CAB, Moura-Neto JA, Dos Reis MA, Vieira Neto OM, Barreto FC. Renal manifestations of Fabry disease: a narrative review. Can J Kidney Health Dis. (2021) 8:2054358120985627. doi: 10.1177/2054358120985627
42. McCloskey S, Brennan P, Sayer JA. Variable phenotypic presentations of renal involvement in Fabry disease: a case series. F1000Res. (2018) 7:356. doi: 10.12688/f1000research.13708.1
43. Olivera-González S, Josa-Laorden C, Torralba-Cabeza MA. The pathophysiology of Fabry disease. Rev Clin Esp. (2018) 218:22–8. doi: 10.1016/j.rceng.2017.09.001
44. Nowicki M, Bazan-Socha S, Błazejewska-Hyzorek B, Gellert R, Imiela J, Kazmierczak J, et al. Enzyme replacement therapy in Fabry disease in Poland: a position statement. Pol Arch Intern Med. (2020) 130:91–7. doi: 10.20452/pamw.15117
45. Biegstraaten M, Hollak CE, Bakkers M, Faber CG, Aerts JM, van Schaik IN. Small fiber neuropathy in Fabry disease. Mol Genet Metab. (2012) 106:135–41. doi: 10.1016/j.ymgme.2012.03.010
46. Politei JM, Durand C, Schenone AB. Small fiber neuropathy in Fabry disease: a review of pathophysiology and treatment. J Inborn Errors Metab. (2016) 4:e160002. Available from: doi: 10.1177/2326409816661351
47. Hopkin RJ, Bissler J, Banikazemi M, Clarke L, Eng CM, Germain DP, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry registry. Pediatr Res. (2008) 64:550–5. doi: 10.1203/PDR.0b013e318183f132
48. Hoffmann B, Beck M, Sunder-Plassmann G, Borsini W, Ricci R, Mehta A, FOS European Investigators. Nature and prevalence of pain in Fabry disease and its response to enzyme replacement therapy–a retrospective analysis from the Fabry Outcome Survey. Clin J Pain. (2007) 23:535–42. doi: 10.1097/AJP.0b013e318074c986
49. Schiffmann R, Kopp JB, Austin HA 3rd, Sabnis S, Moore DF, Weibel T, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. (2001) 285:2743–9. doi: 10.1001/jama.285.21.2743
50. Carnicer-Cáceres C, Arranz-Amo JA, Cea-Arestin C, Camprodon-Gomez M, Moreno-Martinez D, Lucas-Del-Pozo S, et al. Biomarkers in Fabry Disease. Implications for Clinical Diagnosis and Follow-up. J Clin Med. (2021) 10:1664. doi: 10.3390/jcm10081664
51. Gu YY, Liu XS, Huang XR, Yu XQ, Lan HY. Diverse Role of TGF-β in Kidney Disease. Front Cell Dev Biol. (2020) 8:123. doi: 10.3389/fcell.2020.00123
52. Chien YH, Olivova P, Zhang XK, Chiang SC, Lee NC, Keutzer J, et al. Elevation of urinary globotriaosylceramide (GL3) in infants with Fabry disease. Mol Genet Metab. (2011) 102:57–60. doi: 10.1016/j.ymgme.2010.08.023
53. Kok K, Zwiers KC, Boot RG, Overkleeft HS, Aerts JMFG, Artola M. Fabry disease: molecular basis, pathophysiology, diagnostics and potential therapeutic directions. Biomolecules. (2021) 11:271. doi: 10.3390/biom11020271
54. Hagège A, Réant P, Habib G, Damy T, Barone-Rochette G, Soulat G, et al. Fabry disease in cardiology practice: literature review and expert point of view. Arch Cardiovasc Dis. (2019) 112:278–87. doi: 10.1016/j.acvd.2019.01.002
55. Klingelhöfer D, Braun M, Seeger-Zybok RK, Quarcoo D, Brüggmann D, Groneberg DA. Global research on Fabry's disease: demands for a rare disease. Mol Genet Genomic Med. (2020) 8:e1163. doi: 10.1002/mgg3.1163
56. Ries M, Gupta S, Moore DF, Sachdev V, Quirk JM, Murray GJ, et al. Pediatric Fabry disease. Pediatrics. (2005) 115:e344–55. doi: 10.1542/peds.2004-1678
57. Pisani A, Petruzzelli Annicchiarico L, Pellegrino A, Bruzzese D, Feriozzi S, Imbriaco M, et al. Parapelvic cysts, a distinguishing feature of renal Fabry disease. Nephrol Dial Transplant. (2018) 33:318–23. doi: 10.1093/ndt/gfx009
59. Ramaswami U, Whybra C, Parini R, Pintos-Morell G, Mehta A, Sunder-Plassmann G, et al. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta Paediatr. (2006) 95:86–92. doi: 10.1080/08035250500275022
60. Laney DA, Peck DS, Atherton AM, Manwaring LP, Christensen KM, Shankar SP, et al. Fabry disease in infancy and early childhood: a systematic literature review. Genet Med. (2015) 17:323–30. doi: 10.1038/gim.2014.120
61. Burand AJ Jr, Stucky CL. Fabry disease pain: patient and preclinical parallels. Pain. (2021) 162:1305–21. doi: 10.1097/j.pain.0000000000002152
62. Germain DP, Fouilhoux A, Decramer S, Tardieu M, Pillet P, Fila M, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet. (2019) 96:107–17. doi: 10.1111/cge.13546
63. Luna PC, Boggio P, Larralde M. Dermatologic aspects of Fabry disease. J Inborn Errors Metab Screen. (2016) 4:1–7. doi: 10.1177/2326409816661353
64. Allen LE, Cosgrave EM, Kersey JP, Ramaswami U. Fabry disease in children: correlation between ocular manifestations, genotype and systemic clinical severity. Br J Ophthalmol. (2010) 94:1602–5. doi: 10.1136/bjo.2009.176651
65. Kalkum G, Pitz S, Karabul N, Beck M, Pintos-Morell G, Parini R, et al. Paediatric Fabry disease: prognostic significance of ocular changes for disease severity. BMC Ophthalmol. (2016) 16:202. doi: 10.1186/s12886-016-0374-2
66. Suntjens E, Dreschler WA, Hess-Erga J, Skrunes R, Wijburg FA, Linthorst GE, et al. Hearing loss in children with Fabry disease. J Inherit Metab Dis. (2017) 40:725–31. doi: 10.1007/s10545-017-0051-5
67. Najafian B, Tøndel C, Svarstad E, Gubler MC, Oliveira JP, Mauer M. Accumulation of globotriaosylceramide in podocytes in Fabry nephropathy is associated with progressive podocyte loss. J Am Soc Nephrol. (2020) 31:865–75. doi: 10.1681/ASN.2019050497
68. Waldek S, Feriozzi S. Fabry nephropathy: a review-how can we optimize the management of Fabry nephropathy? BMC Nephrol. (2014) 15:72. doi: 10.1186/1471-2369-15-72
69. Azambuja Neves RFC, Varela P, Fernandes DE, Riguetti MTP, Geraldini S, Mata GF, et al. Renal ultrasound contributes to Fabry Disease Diagnosis. J Rare Dis Res Treat. (2020) 5:19–23. doi: 10.29245/2572-9411/2020/2.1195
70. GLAss RB, Astrin KH, Norton KI, Parsons R, Eng CM, Banikazemi M, et al. Fabry disease: renal sonographic and magnetic resonance imaging findings in affected males and carrier females with the classic and cardiac variant phenotypes. J Comput Assist Tomogr. (2004) 28:158–68. doi: 10.1097/00004728-200403000-00002
71. Aksoy GK, Çomak E, Akkaya B, Koyun M, Akman S. Proteinuria in a male adolescent with hearing loss: questions. Pediatr Nephrol. (2018) 33:1159–60. doi: 10.1007/s00467-017-3822-1
72. Mauer M, Glynn E, Svarstad E, Tøndel C, Gubler MC, West M, et al. Mosaicism of podocyte involvement is related to podocyte injury in females with Fabry disease. PLoS ONE. (2014) 9:e112188. doi: 10.1371/journal.pone.0112188
73. Najafian B, Svarstad E, Bostad L, Gubler MC, Tøndel C, Whitley C, et al. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. (2011) 79:663–70. doi: 10.1038/ki.2010.484
74. Liern M, Collazo A, Valencia M, Fainboin A, Isse L, Costales-Collaguazo C, et al. Podocyturia in paediatric patients with Fabry disease. Nefrologia. (2019) 39:177–83. doi: 10.1016/j.nefroe.2019.03.001
75. Lenders M, Brand E. Precision medicine in Fabry disease. Nephrol Dial Transplant. (2021) 36:14–23. doi: 10.1093/ndt/gfab038
76. Fall B, Scott CR, Mauer M, Shankland S, Pippin J, Jefferson JA, et al. Urinary Podocyte Loss Is Increased in Patients with Fabry Disease and Correlates with Clinical Severity of Fabry Nephropathy. PLoS ONE. (2016) 11:e0168346. doi: 10.1371/journal.pone.0168346
77. Trimarchi H, Canzonieri R, Schiel A, Politei J, Stern A, Andrews J, et al. Podocyturia is significantly elevated in untreated vs treated Fabry adult patients. J Nephrol. (2016) 29:791–7. doi: 10.1007/s40620-016-0271-z
78. Schwartz GJ, Muñoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol. (2009) 20:629–37. doi: 10.1681/ASN.2008030287
79. Ries M, Ramaswami U, Parini R, Lindblad B, Whybra C, Willers I, et al. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr. (2003) 162:767–72. doi: 10.1007/s00431-003-1299-3
80. Tøndel C, Bostad L, Hirth A, Svarstad E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. (2008) 51:767–76. doi: 10.1053/j.ajkd.2007.12.032
81. Shen Q, Liu J, Chen J, Zhou S, Wang Y, Yu L, et al. Multidisciplinary approach to screening and management of children with Fabry disease: practice at a Tertiary Children's Hospital in China. Orphanet J Rare Dis. (2021) 16:509. doi: 10.1186/s13023-021-02136-1
82. Furujo M, Kubo T, Kobayashi M, Ohashi T. Enzyme replacement therapy in two Japanese siblings with Fabry disease, and its effectiveness on angiokeratoma and neuropathic pain. Mol Genet Metab. (2013) 110:405–10. doi: 10.1016/j.ymgme.2013.07.005
83. Auray-Blais C, Cyr D, Ntwari A, West ML, Cox-Brinkman J, Bichet DG, et al. Urinary globotriaosylceramide excretion correlates with the genotype in children and adults with Fabry disease. Mol Genet Metab. (2008) 93:331–40. doi: 10.1016/j.ymgme.2007.10.001
84. Marchesoni C, Cisneros E, Pfister P, Yáñez P, Rollan C, Romero C, et al. Brain MRI findings in children and adolescents with Fabry disease. J Neurol Sci. (2018) 395:131–4. doi: 10.1016/j.jns.2018.10.009
85. Duicu C. Genetic testing in pediatrics-a narrative essay of challenges and possibilities in Romania. Rev Romana Med Lab. (2019) 27:355–9. doi: 10.2478/rrlm-2019-0041
86. Perretta F, Antongiovanni N, Jaurretche S. Major organic involvement in women with Fabry disease in Argentina. ScientificWorldJournal. (2018) 2018:6515613. doi: 10.1155/2018/6515613
87. Stiles AR, Zhang H, Dai J, McCaw P, Beasley J, Rehder C, et al. A comprehensive testing algorithm for the diagnosis of Fabry disease in males and females. Mol Genet Metab. (2020) 130:209–14. doi: 10.1016/j.ymgme.2020.04.006
88. Üçeyler N, Böttger J, Henkel L, Langjahr M, Mayer C, Nordbeck P, et al. Detection of blood Gb3 deposits as a new tool for diagnosis and therapy monitoring in patients with classic Fabry disease. J Intern Med. (2018) 284:427–38. doi: 10.1111/joim.12801
89. Cairns T, Müntze J, Gernert J, Spingler L, Nordbeck P, Wanner C. Hot topics in Fabry disease. Postgrad Med J. (2018) 94:709–13. doi: 10.1136/postgradmedj-2018-136056
90. Pereira EM, Silva ASD, Silva RND, Monte Neto JT, Nascimento FFD, Sousa JLM, et al. CD77 levels over enzyme replacement treatment in Fabry Disease Family (V269M). J Bras Nefrol. (2018) 40:333–8. doi: 10.1590/2175-8239-jbn-3910
91. Simonetta I, Tuttolomondo A, Daidone M, Pinto A. Biomarkers in Anderson-Fabry Disease. Int J Mol Sci. (2020) 21:8080. doi: 10.3390/ijms21218080
92. Nowak A, Mechtler TP, Desnick RJ, Kasper DC. Plasma LysoGb3: a useful biomarker for the diagnosis and treatment of Fabry disease heterozygotes. Mol Genet Metab. (2017) 120:57–61. doi: 10.1016/j.ymgme.2016.10.006
93. Sakuraba H, Togawa T, Tsukimura T, Kato H. Plasma lyso-Gb3: a biomarker for monitoring Fabry patients during enzyme replacement therapy. Clin Exp Nephrol. (2018) 22:843–9. doi: 10.1007/s10157-017-1525-3
94. Maruyama H, Miyata K, Mikame M, Taguchi A, Guili C, Shimura M, et al. Effectiveness of plasma lyso-Gb3 as a biomarker for selecting high-risk patients with Fabry disease from multispecialty clinics for genetic analysis. Genet Med. (2019) 21:44–52. doi: 10.1038/gim.2018.31
95. Duro G, Zizzo C, Cammarata G, Burlina A, Burlina A, Polo G, et al. Mutations in the GLA Gene and LysoGb3: is it really Anderson-Fabry Disease? Int J Mol Sci. (2018) 19:3726. doi: 10.3390/ijms19123726
96. Smid BE, van der Tol L, Biegstraaten M, Linthorst GE, Hollak CE, Poorthuis BJ. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J Med Genet. (2015) 52:262–8. doi: 10.1136/jmedgenet-2014-102872
97. Levstek T, Vujkovac B, Trebusak Podkrajsek K. Biomarkers of Fabry nephropathy: review and future perspective. Genes. (2020) 11:1091. doi: 10.3390/genes11091091
98. Riccio E, Sabbatini M, Bruzzese D, Annicchiarico Petruzzelli L, Pellegrino A, Spinelli L, et al. Glomerular hyperfiltration: an early marker of nephropathy in Fabry disease. Nephron. (2019) 141:10–7. doi: 10.1159/000493469
99. Riccio E, Sabbatini M, Capuano I, Pisani A. Early biomarkers of Fabry nephropathy: a review of the literature. Nephron. (2019) 143:274–81. doi: 10.1159/000502907
100. Kurschat CE. Fabry disease-what cardiologists can learn from the nephrologist: a narrative review. Cardiovasc Diagn Ther. (2021) 11:672–82. doi: 10.21037/cdt-20-981
101. Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. (2004) 34:236–42. doi: 10.1111/j.1365-2362.2004.01309.x
102. Germain DP, Waldek S, Banikazemi M, Bushinsky DA, Charrow J, Desnick RJ, et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol. (2007) 18:1547–57. doi: 10.1681/ASN.2006080816
103. Sheth KJ, Roth DA, Adams MB. Early renal failure in Fabry's disease. Am J Kidney Dis. (1983) 2:651–4. doi: 10.1016/S0272-6386(83)80047-X
104. Schiffmann R, Askari H, Timmons M, Robinson C, Benko W, Brady RO, et al. Weekly enzyme replacement therapy may slow decline of renal function in patients with Fabry disease who are on long-term biweekly dosing. J Am Soc Nephrol. (2007) 18:1576–83. doi: 10.1681/ASN.2006111263
105. Sestito S, Falvo F, Sallemi A, Petrisano M, Scuderi MG, Tarsitano F, et al. Renal involvement in paediatric Fabry disease. J Biol Regul Homeost Agents. (2019) 33:59–63.
106. Ramaswami U, Najafian B, Schieppati A, Mauer M, Bichet DG. Assessment of renal pathology and dysfunction in children with Fabry disease. Clin J Am Soc Nephrol. (2010) 5:365–70. doi: 10.2215/CJN.08091109
107. Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. (2009) 150:604–12. doi: 10.7326/0003-4819-150-9-200905050-00006
108. Madsen CV, Granqvist H, Petersen JH, Rasmussen ÅK, Lund AM, Oturai P, et al. Age-related renal function decline in Fabry disease patients on enzyme replacement therapy: a longitudinal cohort study. Nephrol Dial Transplant. (2019) 34:1525–33. doi: 10.1093/ndt/gfy357
109. Wanner C, Oliveira JP, Ortiz A, Mauer M, Germain DP, Linthorst GE, et al. Prognostic indicators of renal disease progression in adults with Fabry disease: natural history data from the Fabry Registry. Clin J Am Soc Nephrol. (2010) 5:2220–8. doi: 10.2215/CJN.04340510
110. Germain DP, Charrow J, Desnick RJ, Guffon N, Kempf J, Lachmann RH, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. (2015) 52:353–8. doi: 10.1136/jmedgenet-2014-102797
111. Nowak A, Koch G, Huynh-Do U, Siegenthaler M, Marti HP, Pfister M. Disease progression modeling to evaluate the effects of enzyme replacement therapy on kidney function in adult patients with the classic phenotype of Fabry disease. Kidney Blood Press Res. (2017) 42:1–15. doi: 10.1159/000464312
112. Feriozzi S, Germain DP, Di Vito R, Legrand A, Ricci R, Barbey F. Cystatin C as a marker of early changes of renal function in Fabry nephropathy. J Nephrol. (2007) 20:437–43.
113. Braga MC, Fonseca FLA, Marins MM, Gomes CP, Bacci MR, Martins AM, et al. Evaluation of Beta 2-Microglobulin, Cystatin C, and Lipocalin-2 as Renal Biomarkers for Patients with Fabry Disease. Nephron. (2019) 143:217–27. doi: 10.1159/000500570
114. Torralba-Cabeza MÁ, Olivera S, Hughes DA, Pastores GM, Mateo RN, Pérez-Calvo JI. Cystatin C and NT-proBNP as prognostic biomarkers in Fabry disease. Mol Genet Metab. (2011) 104:301–7. doi: 10.1016/j.ymgme.2011.06.021
115. West M, Nicholls K, Mehta A, Clarke JT, Steiner R, Beck M, et al. Agalsidase alfa and kidney dysfunction in Fabry disease. J Am Soc Nephrol. (2009) 20:1132–9. doi: 10.1681/ASN.2008080870
116. Ries M, Clarke JT, Whybra C, Timmons M, Robinson C, Schlaggar BL, et al. Enzyme-replacement therapy with agalsidase alfa in children with Fabry disease. Pediatrics. (2006) 118:924–32. doi: 10.1542/peds.2005-2895
117. van der Tol L, Svarstad E, Ortiz A, Tøndel C, Oliveira JP, Vogt L, et al. Chronic kidney disease and an uncertain diagnosis of Fabry disease: approach to a correct diagnosis. Mol Genet Metab. (2015) 114:242–7. doi: 10.1016/j.ymgme.2014.08.007
118. Thurberg BL, Rennke H, Colvin RB, Dikman S, Gordon RE, Collins AB, et al. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. (2002) 62:1933–46. doi: 10.1046/j.1523-1755.2002.00675.x
119. Tøndel C, Bostad L, Larsen KK, Hirth A, Vikse BE, Houge G, et al. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol. (2013) 24:137–48. doi: 10.1681/ASN.2012030316
120. Selvarajah M, Nicholls K, Hewitson TD, Becker GJ. Targeted urine microscopy in Anderson-Fabry disease: a cheap, sensitive and specific diagnostic technique. Nephrol Dial Transplant. (2011) 26:3195–202. doi: 10.1093/ndt/gfr084
121. Trimarchi H. Podocyturia: Potential applications and current limitations. World J Nephrol. (2017) 6:221–8. doi: 10.5527/wjn.v6.i5.221
122. Garovic VD, Craici IM, Wagner SJ, White WM, Brost BC, Rose CH, et al. Mass spectrometry as a novel method for detection of podocyturia in pre-eclampsia. Nephrol Dial Transplant. (2013) 28:1555–61. doi: 10.1093/ndt/gfs074
123. Wickman L, Afshinnia F, Wang SQ, Yang Y, Wang F, Chowdhury M, et al. Urine podocyte mRNAs, proteinuria, and progression in human glomerular diseases. J Am Soc Nephrol. (2013) 24:2081–95. doi: 10.1681/ASN.2013020173
124. Sanchez-Niño MD, Perez-Gomez MV, Valiño-Rivas L, Torra R, Ortiz A. Podocyturia: why it may have added value in rare diseases. Clin Kidney J. (2018) 12:49–52. doi: 10.1093/ckj/sfy081
125. Schiffmann R, Ries M, Blankenship D, Nicholls K, Mehta A, Clarke JT, et al. Changes in plasma and urine globotriaosylceramide levels do not predict Fabry disease progression over 1 year of agalsidase alfa. Genet Med. (2013) 15:983–9. doi: 10.1038/gim.2013.56
126. Auray-Blais C, Ntwari A, Clarke JT, Warnock DG, Oliveira JP, Young SP, et al. How well does urinary lyso-Gb3 function as a biomarker in Fabry disease? Clin Chim Acta. (2010) 411:1906–14. doi: 10.1016/j.cca.2010.07.038
127. Argyropoulos CP, Chen SS, Ng YH, Roumelioti ME, Shaffi K, Singh PP, et al. Rediscovering Beta-2 microglobulin as a biomarker across the spectrum of kidney diseases. Front Med. (2017) 4:73. doi: 10.3389/fmed.2017.00073
128. Devuyst O, Olinger E, Rampoldi L. Uromodulin: from physiology to rare and complex kidney disorders. Nat Rev Nephrol. (2017) 13:525–44. doi: 10.1038/nrneph.2017.101
129. Steubl D, Block M, Herbst V, Nockher WA, Schlumberger W, Kemmner S, et al. Urinary uromodulin independently predicts end-stage renal disease and rapid kidney function decline in a cohort of chronic kidney disease patients. Medicine. (2019) 98:e15808. doi: 10.1097/MD.0000000000015808
130. Matafora V, Cuccurullo M, Beneduci A, Petrazzuolo O, Simeone A, Anastasio P, et al. Early markers of Fabry disease revealed by proteomics. Mol Biosyst. (2015) 11:1543–51. doi: 10.1039/C4MB00707G
131. Doykov ID, Heywood WE, Nikolaenko V, Spiewak J, Hällqvist J, Clayton PT, et al. Rapid, proteomic urine assay for monitoring progressive organ disease in Fabry disease. J Med Genet. (2020) 57:38–47. doi: 10.1136/jmedgenet-2019-106030
132. Vylet'al P, Hulková H, Zivná M, Berná L, Novák P, Elleder M, et al. Abnormal expression and processing of uromodulin in Fabry disease reflects tubular cell storage alteration and is reversible by enzyme replacement therapy. J Inherit Metab Dis. (2008) 31:508–17. doi: 10.1007/s10545-008-0900-3
133. Rampoldi L, Scolari F, Amoroso A, Ghiggeri G, Devuyst O. The rediscovery of uromodulin (Tamm-Horsfall protein): from tubulointerstitial nephropathy to chronic kidney disease. Kidney Int. (2011) 80:338–47. doi: 10.1038/ki.2011.134
134. Lepedda AJ, Fancellu L, Zinellu E, De Muro P, Nieddu G, Deiana GA, et al. Urine bikunin as a marker of renal impairment in Fabry's disease. Biomed Res Int. (2013) 2013:205948. doi: 10.1155/2013/205948
135. Biegstraaten M, Arngrímsson R, Barbey F, Boks L, Cecchi F, Deegan PB, et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphanet J Rare Dis. (2015) 10:36. doi: 10.1186/s13023-015-0253-6
136. Guérard N, Oder D, Nordbeck P, Zwingelstein C, Morand O, Welford RWD, et al. Lucerastat, an iminosugar for substrate reduction therapy: tolerability, pharmacodynamics, and pharmacokinetics in patients with fabry disease on enzyme replacement. Clin Pharmacol Ther. (2018) 103:703–11. doi: 10.1002/cpt.790
137. van der Veen SJ, Hollak CEM, van Kuilenburg ABP, Langeveld M. Developments in the treatment of Fabry disease. J Inherit Metab Dis. (2020) 43:908–21. doi: 10.1002/jimd.12228
138. Arends M, Biegstraaten M, Hughes DA, Mehta A, Elliott PM, Oder D, et al. Retrospective study of long-term outcomes of enzyme replacement therapy in Fabry disease: analysis of prognostic factors. PLoS ONE. (2017) 12:e0182379. doi: 10.1371/journal.pone.0182379
139. Felis A, Whitlow M, Kraus A, Warnock DG, Wallace E. Current and investigational therapeutics for Fabry disease. Kidney Int Rep. (2019) 5:407–13. doi: 10.1016/j.ekir.2019.11.013
140. Schiffmann R, Goker-Alpan O, Holida M, Giraldo P, Barisoni L, Colvin RB, et al. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: A 1-year Phase 1/2 clinical trial. J Inherit Metab Dis. (2019) 42:534–44. doi: 10.1002/jimd.12080
141. Hennermann JB, Arash-Kaps L, Fekete G, Schaaf A, Busch A, Frischmuth T. Pharmacokinetics, pharmacodynamics, and safety of moss-aGalactosidase A in patients with Fabry disease. J Inherit Metab Dis. (2019) 42:527–33. doi: 10.1002/jimd.12052
142. Weidemann F, Jovanovic A, Herrmann K, Vardarli I. Chaperone therapy in Fabry disease. Int J Mol Sci. (2022) 23:1887. doi: 10.3390/ijms23031887
143. Benjamin ER, Della Valle MC, Wu X, Katz E, Pruthi F, Bond S, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. (2017) 19:430–8. doi: 10.1038/gim.2016.122
144. Riccio E, Zanfardino M, Ferreri L, Santoro C, Cocozza S, Capuano I, et al. Switch from enzyme replacement therapy to oral chaperone migalastat for treating fabry disease: real-life data. Eur J Hum Genet. (2020) 28:1662–8. doi: 10.1038/s41431-020-0677-x
145. Müntze J, Gensler D, Maniuc O, Liu D, Cairns T, Oder D, et al. Oral chaperone therapy migalastat for treating fabry disease: enzymatic response and serum biomarker changes after 1 year. Clin Pharmacol Ther. (2019) 105:1224–33. doi: 10.1002/cpt.1321
146. Warnock DG, Bichet DG, Holida M, Goker-Alpan O, Nicholls K, Thomas M, et al. Oral migalastat HCl leads to greater systemic exposure and tissue levels of active α-galactosidase A in Fabry patients when co-administered with infused agalsidase. PLoS ONE. (2015) 10:e0134341. doi: 10.1371/journal.pone.0134341
147. Benjamin ER, Khanna R, Schilling A, Flanagan JJ, Pellegrino LJ, Brignol N, et al. Co-administration with the pharmacological chaperone AT1001 increases recombinant human α-galactosidase A tissue uptake and improves substrate reduction in Fabry mice. Mol Ther. (2012) 20:717–26. doi: 10.1038/mt.2011.271
148. Khan A, Barber DL, Huang J, Rupar CA, Rip JW, Auray-Blais C, et al. Lentivirus-mediated gene therapy for Fabry disease. Nat Commun. (2021) 12:1178. doi: 10.1038/s41467-021-21371-5
149. Domm JM, Wootton SK, Medin JA, West ML. Gene therapy for Fabry disease: progress, challenges, and outlooks on gene-editing. Mol Genet Metab. (2021) 134:117–31. doi: 10.1016/j.ymgme.2021.07.006
150. Spiniello G, Verrillo F, Ricciolino R, Prozzo D, Tuccillo A, Caiazza M, et al. Gene therapy in Anderson-Fabry disease. State of the Art and Future Perspectives Cardiogenetics. (2020) 10:9075. doi: 10.4081/cardiogenetics.2020.9075
151. DeRosa F, Smith L, Shen Y, Huang Y, Pan J, Xie H, et al. Improved efficacy in a Fabry disease model using a systemic mRNA liver depot system as compared to enzyme replacement therapy. Mol Ther. (2019) 27:878–89. doi: 10.1016/j.ymthe.2019.03.001
152. Schuller Y, Linthorst GE, Hollak CE, Van Schaik IN, Biegstraaten M. Pain management strategies for neuropathic pain in Fabry disease–a systematic review. BMC Neurol. (2016) 16:25. doi: 10.1186/s12883-016-0590-7
Keywords: Fabry, kidney, GLA gene, biomarker, therapy
Citation: Muntean C, Starcea IM, Stoica C and Banescu C (2022) Clinical Characteristics, Renal Involvement, and Therapeutic Options of Pediatric Patients With Fabry Disease. Front. Pediatr. 10:908657. doi: 10.3389/fped.2022.908657
Received: 30 March 2022; Accepted: 12 May 2022;
Published: 01 June 2022.
Edited by:
Lars Pape, Essen University Hospital, GermanyReviewed by:
Sandro Feriozzi, Azienda Sanitaria Locale di Viterbo, ItalyCopyright © 2022 Muntean, Starcea, Stoica and Banescu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmen Muntean, ZHVpY3VjYXJtZW5AeWFob28uY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.