 Laura Florea
Laura Florea Lavinia Caba
Lavinia Caba Eusebiu Vlad Gorduza
Eusebiu Vlad Gorduza- 1Department of Nephrology-Internal Medicine, Faculty of Medicine, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania
- 2Department of Medical Genetics, Faculty of Medicine, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania
Bartter syndrome (BS) is a rare tubulopathy that causes polyuria, hypokalemia, hypochloremic metabolic alkalosis, and normotensive hyperreninemic hyperaldosteronism. It is characterized by locus, clinical, and allelic heterogeneity. Types 1–4 of BS are inherited according to an autosomal recessive pattern, while type 5, which is transient, is X linked. There are specific correlations between the clinical expression and the molecular defect, but since it is a rare disease, such studies are rare. Therapeutic interventions are different, being correlated with types of BS.
Introduction
Bartter syndrome (BS) is a rare tubulopathy characterized by polyuria, hypokalemia, hypochloremic metabolic alkalosis, and normotensive hyperreninemic hyperaldosteronism (1). Incidence is 0.1/100,000 (2).
The main mechanism is the defective reabsorption of salt, mainly at the thick ascending limb (TAL) of the loop of Henle (3). TAL intervenes in the homeostasis of extracellular fluid by sodium reabsorption; homeostasis of calcium, magnesium, bicarbonate, and ammonium; and synthesis of uromodulin (Tamm-Horsfall protein) with a role in maintaining the composition of urinary proteins (4).
Hereditary diseases involving proteins of the TAL are classified into three categories according to the TAL function affected: sodium reabsorption, calcium and magnesium reabsorption, and uromodulin synthesis. The first category includes BS types 1–5 and HELIX syndrome (hypohidrosis, electrolyte imbalance, lacrimal gland dysfunction, ichthyosis, and xerostomia) (produced by mutations in CLDN10 gene). The second category includes familial hypomagnesemia with hypercalciuria and nephrocalcinosis (produced by mutations in CLDN16 or CLDN19 genes), familial hypercalcemia hypocalciuric types 1–3 (produced by mutations in CASR, GNA11, AP2S1 genes), and autosomal dominant hypocalcemia types 1–2 (produced by mutations in CASR, GNA11 genes). Hyperuricemic nephropathy, familial juvenile 1 (produced by UMOD gene) belongs to the third category (4).
Genes and Proteins in Bartter Syndrome
Bartter syndrome is caused by mutations in genes encoding K+ channel (KCNJ1 gene), Cl– channel (CLCNKA and CLCNKB genes), their cotransporters (SLC12A1 gene), subunits of these channels (BSND gene), or regulators of the expression of certain transport channels (MAGE-D2 gene).
Table 1 summarizes the genes implied in the pathogeny of various types of BS. Figure 1 describes the gene expressions in different segments of the nephron and correspondent type of the BS.
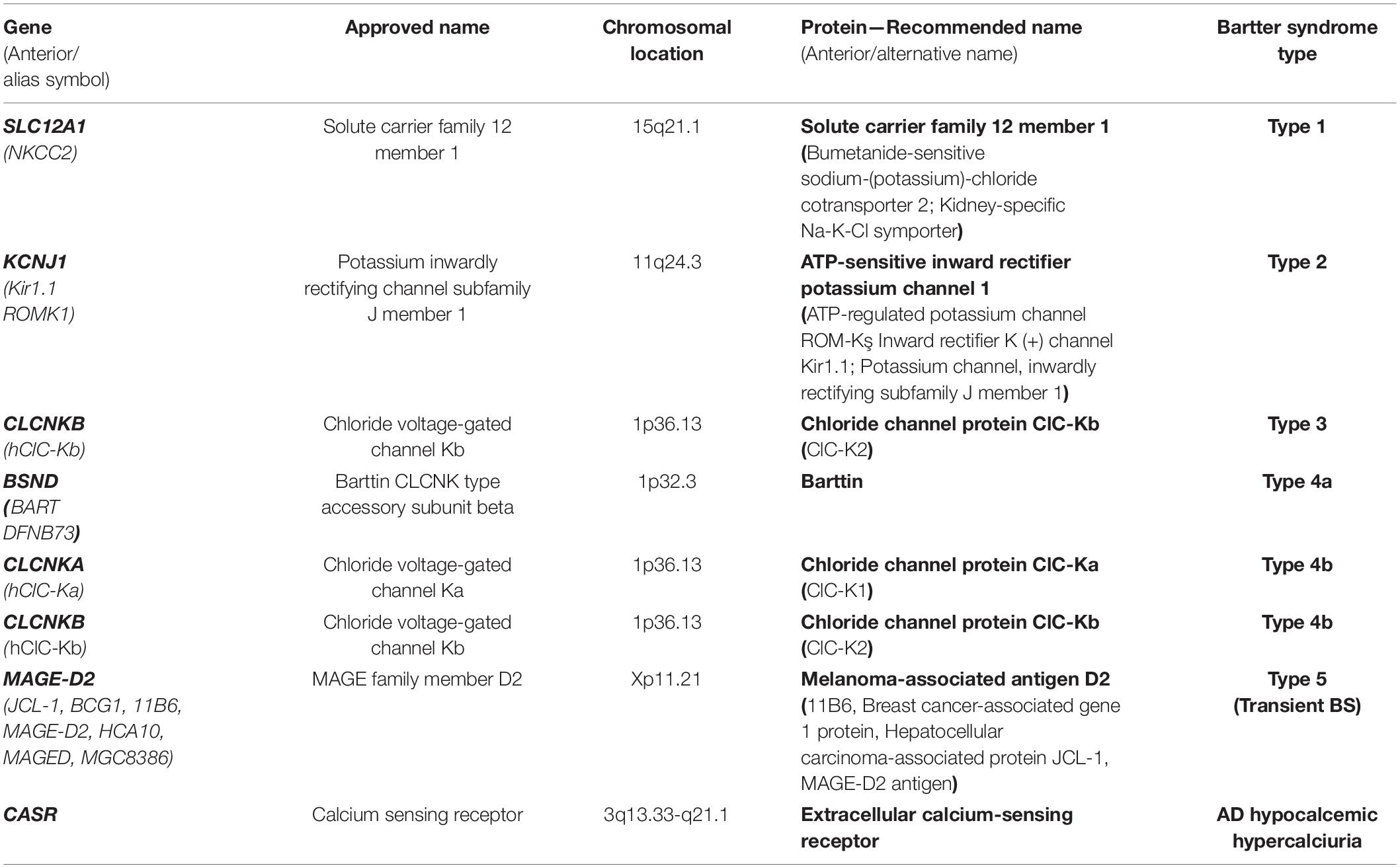
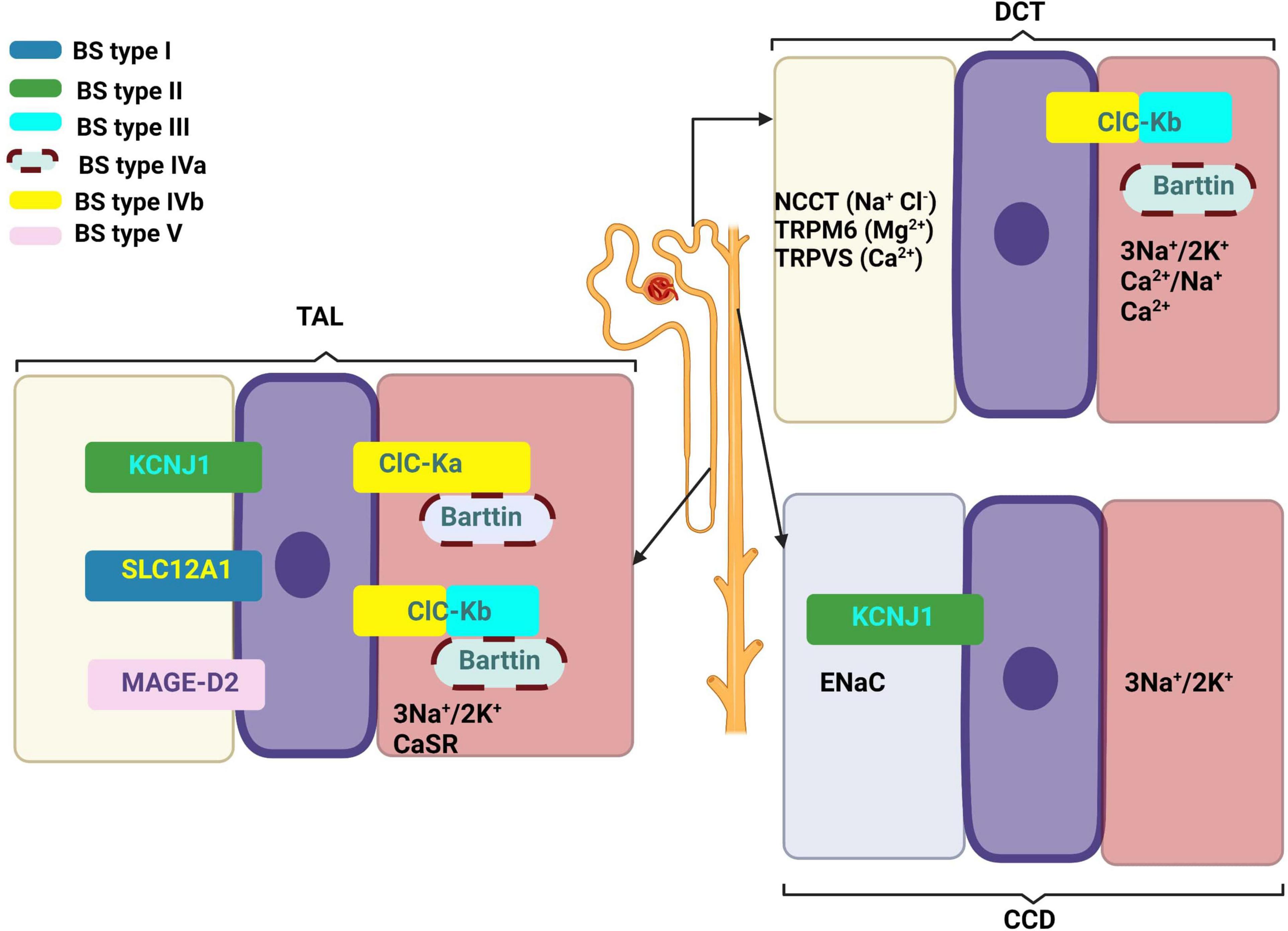
Figure 1. Gene expressions in different segments of the nephron and correspondent type of the Bartter syndrome. Created with BioRender.com.
Pathophysiology
At the descending limb of the loop of Henle, the urine becomes more and more hypertonic because this segment is permeable to water that passes extraluminal (passive transport) (8). At the level of the thin segment of the ascending limb, NaCl is reabsorbed according to the laws of osmosis. At the thick segment of the ascending limb and distal convoluted tubules (initial segment), Cl–, Na+, and K+ are actively reabsorbed (Figure 2). The result is a hypotonic urine. The collecting duct level is permeable to urea and water, and finally urine becomes hypertonic. These final changes at the nephron level are controlled by the antidiuretic hormone (ADH) that acts on the terminal segment of the distal tube and on the collecting duct (1). An important role in its regulation is played by the juxtaglomerular apparatus (JGA) which mediates the tubuloglomerular feedback (TGF). Under normal conditions, a decrease of intracellular concentration of Cl– in JGA cells indicates a reduced filtration which determines an activation of TGF, with the stimulation of renin synthesis and hyperfiltration. In patients with BS, TGF is uncoupled because Cl– is not reabsorbed into the macula densa. The cyclooxygenases are activated and large amounts of prostaglandin E2 are synthesized regardless of volume status, and this produces an excess of renin and aldosterone (1, 9).
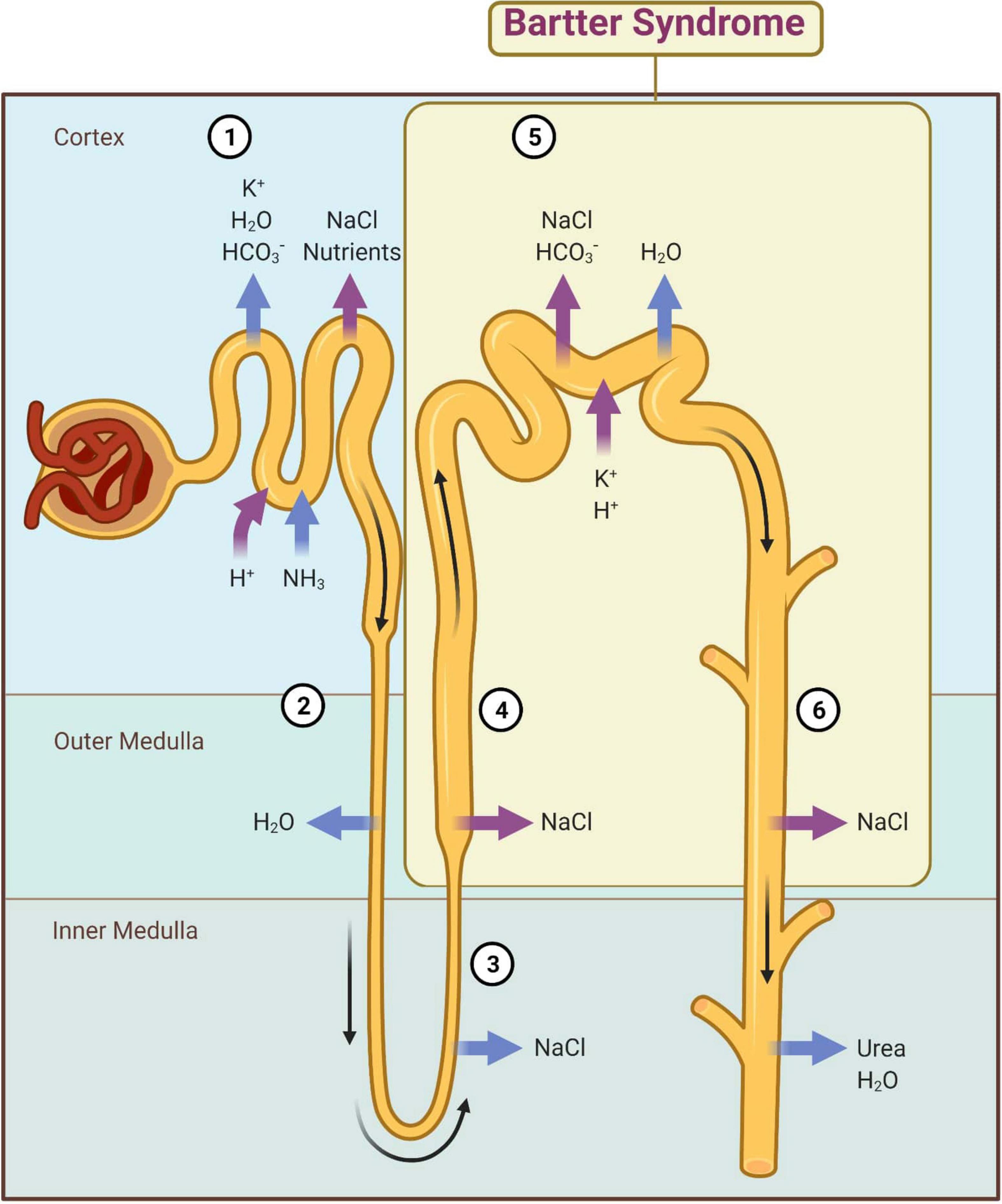
Figure 2. Main mechanisms of kidney reabsorption and secretion. 1—Proximal convoluted tubule; 2—Descending limb of loop of Henle; 3—Thin segment of ascending limb; 4—Thick segment of ascending limb; 5—Distal convoluted tubule; 6—Collecting duct; magenta arrow—active transport; blue arrow—passive transport. Adapted from “Kidney Reabsorption and Secretion,” by BioRender.com (2022). Retrieved from: https://app.biorender.com/biorender-templates (accessed on 22 March 2022).
Hypovolemia activates the renin-aldosterone system, with elevation of renin and aldosterone levels in blood and urine (10). The effects of aldosterone on the distal tubule are as follows: the increase of Na+ reabsorption from the lumen in exchange with K+ and the stimulation of exchange between the intracellular H+ and the intraluminal K+. In the first case, Na+/K+-ATPase pumps increase the intracellular K+ which results in an increase in the gradient with respect to the lumen. In this way, K+ will leave the cell in the lumen and in the urine (10). In the second case, metabolic alkalosis increases due to the loss of H+ in urine (10).
Thick ascending limb cells are responsible for 25–30% of the reabsorbed NaCl in the kidneys (4, 11).
The segments of the Henle loop (proximal straight tubule, thin descending, thin ascending limb, and TAL) are differentiated by the thickness of the walls and the properties of the epithelium. The descending and ascending segments are placed in the renal medulla, while the TAL is located distally. In the kidney medulla, the Na+/K+-ATPase has low levels and low transport activity. There are also differences between different cells and segments in terms of Na+/K+-ATPase activity, SLC12A1 splicing, and phosphorylation (11).
SLC12A1
SLC12A1 is the major renal sodium, potassium, and chloride ion cotransporter expressed on the luminal membrane of renal epithelial cells of the TAL and macula densa. Its physiological functions are urine concentration, regulation of Ca2+ excretion, and reabsorption of NaCl in TAL (12). It is responsible for most of the NaCl absorption (6, 7).
All 12 solute carrier family proteins have some common properties: transport-coupled one cation per each transported anion (in the case of SLC12A1, for each one K+ and one Na+ that leave the cell enter two Cl–) which determines an electric balance; the anion is always the chlorine ion; their activity is modified by the changes in the cell volume; and the intracellular concentration of the chlorine ion; activity regulation is done by phosphorylation and dephosphorylation (12). It is positively regulated by WNK3 serine/threonine-protein kinase that activates SLC12A1 by phosphorylation and inhibits by dephosphorylation (12). This action is mediated by cation-chloride cotransporters (CCCs) dependent on cell tonicity (13). A decreased cell volume leads to phosphorylation of cotransporters by Serine/threonine-protein kinase STE20 (12, 14, 15).
Three full-length SLC12A1 isoforms exist in humans: isoform A, isoform B, and isoform F. Isoform A is the most intensely expressed (16, 17). However, there are differences in gene expression between different segments of TAL: isoforms A and F are mostly expressed in the outer stripe of the outer medulla, while isoform B is active in the cortex (16, 17). In addition, the affinity for Cl– of isoforms decreases in this order: isoform B, isoform F, and isoform A, a feature concordant with spatial localization of these isoforms. Thus, the isoforms with higher affinity are expressed in C-TAL where there is a higher concentration of Cl–, while in M-TAL, the lower-affinity isoforms are present and concordant with low levels of Cl– (16, 17).
CIC-K
ClC-Ka and ClC-Kb are expressed differently in the epithelial cells of the nephron, inner ear, and salivary glands (18). The ClC-K channels have the largest protopore conductance values. These values range from 15 to 22 ps (18).
The ClC-K channels inserted in the basolateral membrane of nephron epithelia require a barttin subunit for proper function. Lack of barttin causes BS type 4a with renal loss of NaCl (7). ClC-Kb inactivating mutations cause BS type 3 (7, 17). ClC-Ka is expressed mainly in the thin limb of the Henle loop, while ClC-Kb is expressed in the TAL, distal-convoluted tubules, and in collecting duct-intercalated cells (7, 19). ClC-Ka, ClC-Kb, and barttin are also expressed in the ear where they are involved in the endolymph secretion. Both modified isoforms or their common beta subunit (barttin) cause deafness in BS type 4 (18).
CLCNKA and CLCNKB
CLCNKA and CLCNKB are neighboring genes, present similar sequences, and probably result from the duplication of an ancestral gene. Deletions/non-sense mutations in CLCNKB produce loss of channel function which affects the response of the ClC-Kb complex to pH and Ca2+.
ROM-K
ROM-K is an ATP-sensitive channel with low conductance, located at the apical pole of the cells. There is no difference between the expression ROM-K in M-TAL and C-TAL; it is expressed from the inner stripe of the outer medulla to the macula densa (17).
MAGE-D2
MAGE-D2 is a nucleolar protein that increases SLC12A1 expression in the human TAL and in the distal convoluted tubules. MAGE-D2 interacts with HSP40 allowing the protection of proteins. Also, MAGE-D2 in association with Gs-alpha promotes the insertion of SLC12A1 and NCC (NaCl-co-transporter) into the plasma membrane. In Barter type 5, due to the existence of an abnormal MAGE-D2, protein degradation is amplified, associated with low levels of SLC12A1 and NCC (4, 20).
Clinical Signs
The typical symptoms in children with BS are polyhydramnios, premature delivery, polyuria, polydipsia, signs of hypovolemia, failure to thrive, and poor growth (21).
The age of presentation is different according to the type of mutation. For the BS types 4 and 5, polyhydramnios is earlier compared to BS types 1 and 2. The onset of BS type 3 is later in life and only rarely in the antenatal period (22).
There are rare cases of asymptomatic children diagnosed with BS. These cases presented hypokalemia metabolic alkalosis, normal blood pressure, and nephrocalcinosis, and usually such patients are discovered after a screening applied in a family with positive history of BS (23–26).
Antenatal Symptomatology
The most frequent feature is the polyhydramnios detected during pregnancy, starting at 22 weeks of gestation (3). The onset of polyhydramnios is earlier in fetuses with BS types 4 and 5 (BSND or MAGED2 variants) and represent the most severe forms. In BS type 1 (SLC12A1) and BS type 2 (KCNJ1), the onset of polyhydramnios is much tardive, concordant with a lower severity. In BS type 3 (CLCNKB), polyhydramnios is either absent or mild (3, 20).
Antenatal genetic testing and biochemical analysis of amniotic fluid, the concentration of total protein and alpha-fetoprotein (decreased in BS), the Bartter index (which is defined corresponding to the multiplication of total protein and of AFP, both expressed in multiple of median—MoM) with 86% sensitivity and 84% specificity, can be used to confirm the diagnosis (27, 28).
Neonatal Symptomatology
All BS types, except BS type 3, have a neonatal onset with preterm birth (median gestational age between 29 and 33 weeks) and massive polyuria which lead to dehydration and rapid weight loss. Infants with BS type 4 have supplementary sensorineural hearing loss (22, 29).
The diagnosis can be confirmed using a next-generation sequencing (NGS) testing with gene panels that contain SLC12A1, KCNJ1, BSND, CLCNKA, CLCNKB, MAGED2, and SLC12A3 gene. These tests have 75% sensitivity and 90–100% specificity (30).
Childhood Symptomatology
Older infants and young children with BS type 3 fail to thrive, growth retardation, and present polyuria/polydipsia that lead to hypovolemia, persistent thirst, salt craving, constipation, unexplained fever, hypotonia, and recurrent vomiting (23–25).
Teenage Symptomatology
Older children and adolescents with BS type 3 can present thirst, salt craving, fatigue, muscle weakness, cramps, nocturia, constipation, poor growth, and pubertal delay.
In BS type 4, there is a risk for chronic kidney disease (CKD) and end stage renal disease (ESRD) (22).
Bartter syndrome type 5 (transient type) has some extrarenal clinical features: frontal bone cyst, dysmorphic facies, hydrocephalus and Chiari malformation, Marfanoid habitus with arachnodactyly and mitral insufficiency, pyloric stenosis, high blood pressure, interauricular communication, left ventricle hypertrophy, right aortic arch, retroesophageal left subclavian artery, moderate pulmonary stenosis, enteropathic acrodermatitis zinc deficiency type, angioma, thrombocytopenia, and deafness after the age of 2 (3, 4, 31).
The laboratory findings constant in all forms of BS are hypochloremic metabolic alkalosis, elevated renin and aldosterone levels, low to normal blood pressure due to chronic hypovolemia, low urine osmolality due to impaired concentrating ability, and hypokalemia (potassium levels that are less than 3 mmol/L). Transient neonatal hyperkalemia may be present only in patients with BS type 2. Hypercalciuria is present in patients with BS types 1 and 2 (associated with nephrocalcinosis) and also in BS type 5 (the nephrocalcinosis is rare and mild in this type). The normocalciuria is present in patients with BS types 3 and 4, and the hypocalciuria in patients with BS type 3. The mild hypomagnesemia may be present in some patients with BS type 3. Plasma Cl/Na ratio can be normal in BS types 1 and 2, decreased in BS types 3 and 4, and increased in BS type 5 (1, 21, 32–35).
Genetic Testing
Genetic testing is recommended in any clinical suspicion of BS. The European Rare Kidney Disease Reference Network Working Group for Tubular Disorders recommends a gene panel testing. This panel must contain not only all genes that cause BS and Gitelman syndrome but also genes that overlap phenotypically with BS and should be considered in differential diagnosis with BS: SLC12A1, KCNJ1, CLCNKB, CLCNKA, BSND, MAGED2, SLC12A3, CASR, KCNJ10, SLC26A3, CLDN10, SCNN1A, SCNN1B, SCNN1G, NR3C2, HSD11B2, CYP11B1, CLCN2, KCNJ5, and CACNA1H (1). Genetic testing is required for several reasons: confirmation of clinical diagnosis, adequate genetic counseling, adequate management of diseases with overlapping phenotypes, screening for deafness in BS type 4, and avoidance of aggressive treatment in transient BS type 5 (1).
Mutational heterogeneity is important in BS. According to the Human Gene Mutation Database Professional HGMD Professional 2021.4 (accessed in February 2022), 350 pathogenic variants were described. Figure 3 shows the implication in BS of different types of genetic modifications. The most common was missense/non-sense mutation, splicing substitution, small deletions, and gross deletion especially in SLC12A1, CLCNKB, and KCNJ1 genes.
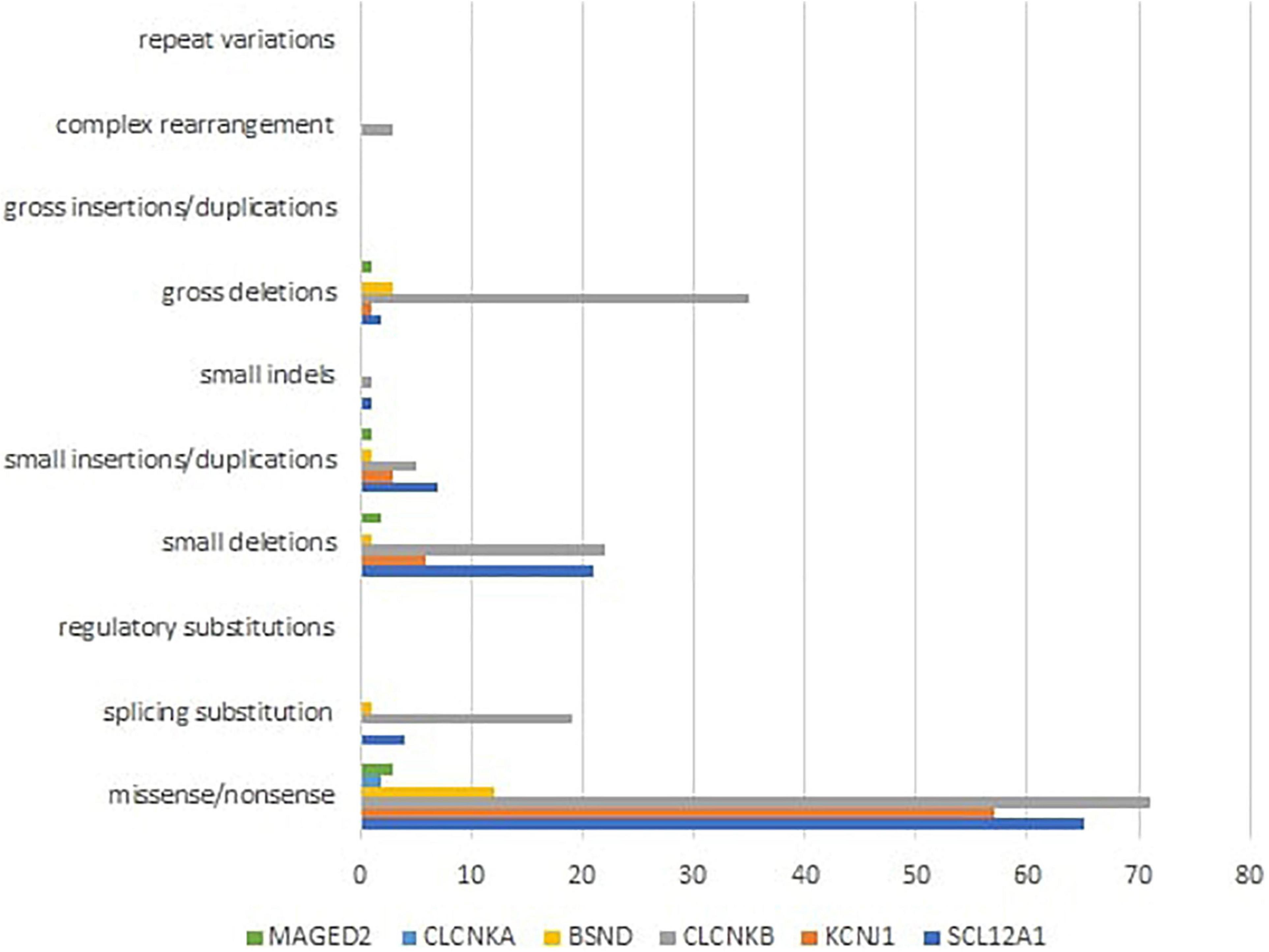
Figure 3. Different types of pathogenic variants in genes implied in Bartter syndrome (36).
Differential Diagnosis
The differential diagnosis of BS should be made with conditions where the main signs are poyhydramnios, salt loss, salt loss with hypokalemic alkalosis, hypokalemic alkalosis without salt loss, and nephrocalcinosis (1, 22).
The polyhydramnios may be a sign present in aneuploidia (the karyotype is abnormal), different gastrointestinal malformations, and congenital chloride diarrhea (dilated intestinal loops present).
The salt loss can be a manifestation in pseudohypoaldosteronism type I, but in this case is associated with metabolic acidosis and hyperkalemia.
The salt loss with hypokalemic alkalosis can be a sign in congenital chloride diarrhea, pseudo-BS (in cystic fibrosis, for example), surreptitious vomiting, surreptitious laxative use (in these entities, the additional finding is low urinary chloride), Gitelman syndrome (associated hypocalciuria and hypomagnesemia), HNF1B (hepatocyte nuclear factor 1 beta), nephropathy (associates renal malformation, MODY5—maturity onset diabetes of the young type 5, hypomagnesemia), HELIX syndrome (hypercalcemia is present), autosomal dominant hypocalcemia, EAST/SeSAME syndrome (seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance), and surreptitious diuretic use (1, 22).
Hypokalemic alkalosis without salt loss associated hypertension with low renin/aldosterone and can be found in primary hyperaldosteronism, apparent mineralocorticoid excess, and Liddle syndrome (1, 22).
The nephrocalcinosis can also be found in distal renal tubular acidosis (metabolic acidosis), medical conditions without metabolic alkalosis (proximal tubular defects, familial hypomagnesemia/hypercalciuria), and apparent mineralocorticoid excess (1, 22).
Therapy of Bartter Syndrome
The antenatal therapy consists of repeated amniocentesis and/or maternal administration of NSAID (non-steroidal anti-inflammatory drug) in order to reduce the amniotic fluid volume. There are potential risks for the fetus (such as necrotizing enterocolitis or premature closure of the ductus arteriosus). The evidence is not sufficient to show that the benefit outweighs the potential adverse effects (1). The main therapeutic interventions are summarized in Table 2.
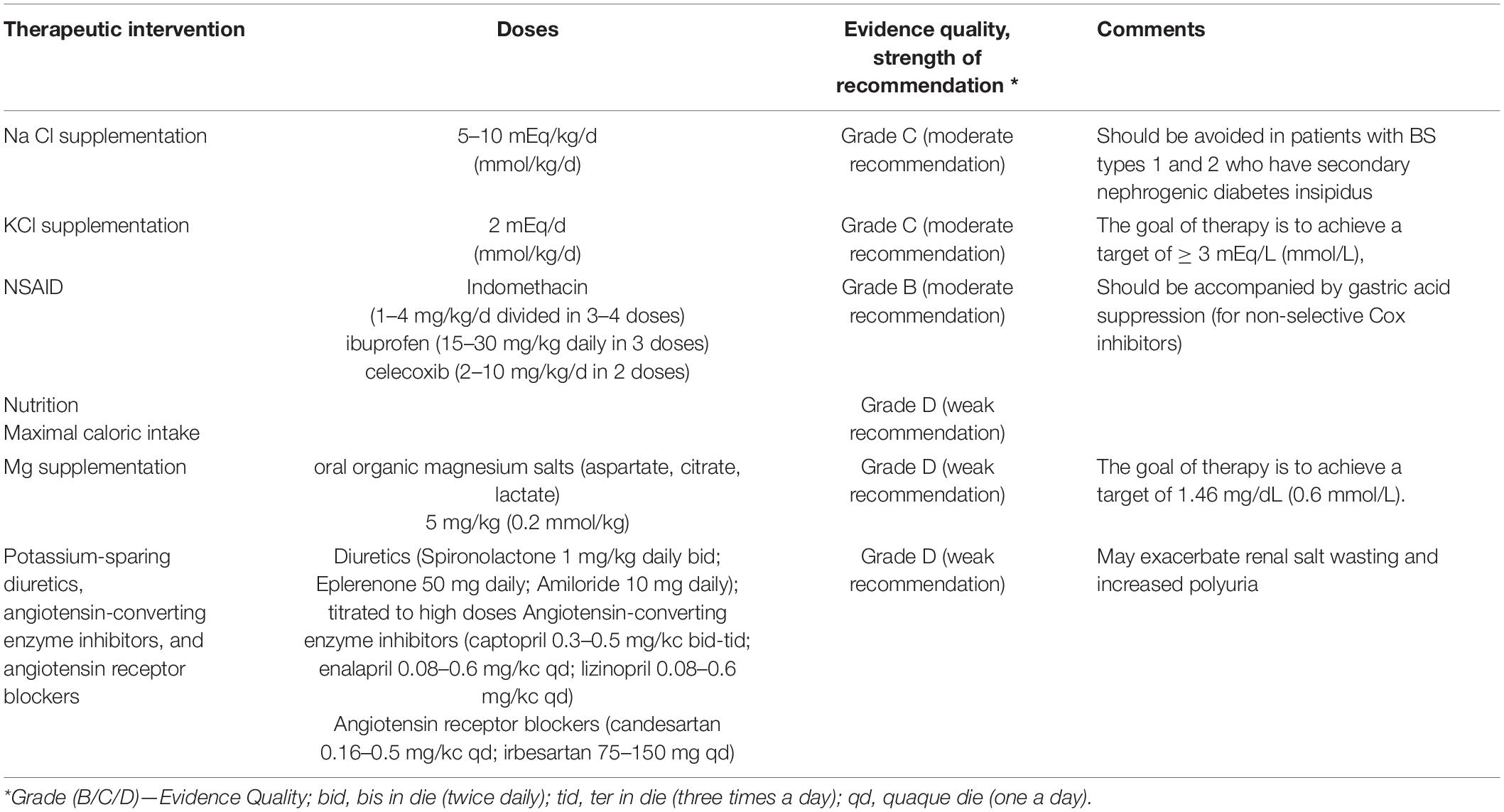
The postnatal therapy consists of maximal caloric intake in order to facilitate optimal growth (1), fluid repletion, sodium chloride (NaCl) supplementation of at least 5–10 mEq/kg/d, and potassium chloride (KCl) 1–2 mEq/d, spread out in frequent doses throughout the day, in order to compensate the urinary losses (1, 37).
Non-steroidal anti-inflammatory drugs that inhibit prostaglandin E2, which contributes to high urinary NaCl losses, are recommended in case of inadequate response in symptomatic patients with BS, especially in early childhood (38). Oral magnesium supplementation is recommended in case of hypomagnesemia [<1.70 mg/dl (0.7 mmol/L)] (37, 39). In case of symptomatic hypokalemia despite the supplements, potassium-sparing diuretics (such as spironolactone, eplerenone, or amiloride), angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers can be used (1, 37).
Follow-Up in Bartter Syndrome
The patients with BS should be followed in pediatric or adult centers experienced in tubular disorders. The recommended frequency of hospital visits is 3–6 months for infants and young children and 6–12 months for adults. All the recommendations are summarized in Table 3. A clinical work on hydration status, degree of polyuria, muscular weakness, growth, and psychomotor development is recommended at each follow-up visit (1, 22).
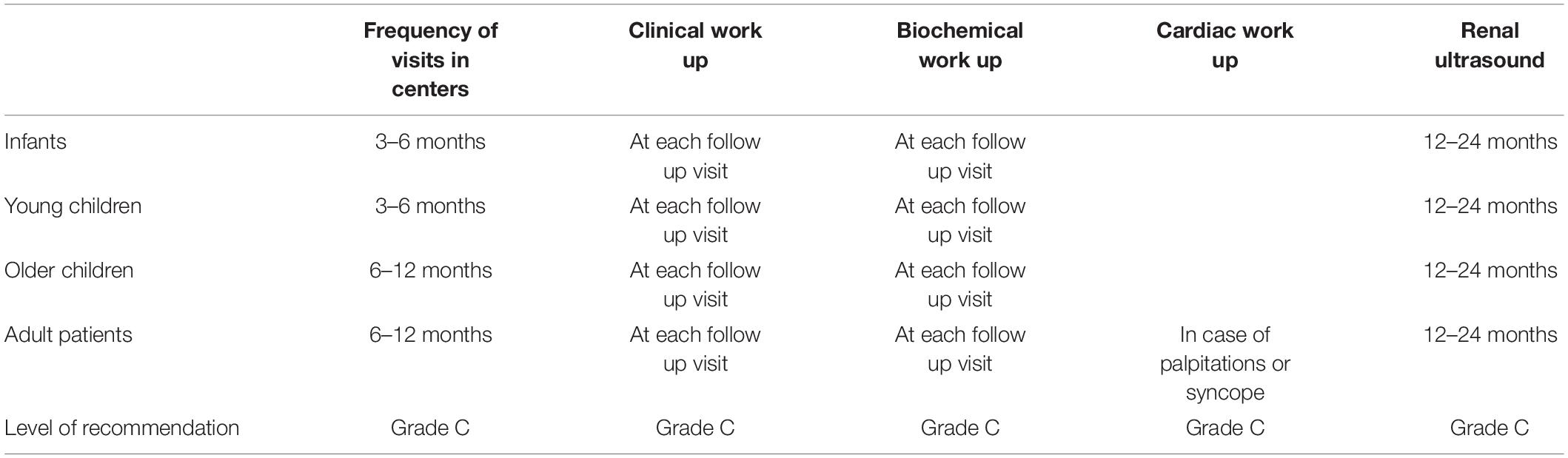
A biochemical work focused on metabolic acid-base status (either blood gas or by measurement of venous total CO2), serum electrolytes (natremia, potassium, chloride, magnesium, and bicarbonate levels), renal function, microalbuminuria, urinary calcium excretion, PTH, and urine osmolality for the detection of nephrogenic diabetes insipidus is recommended at each follow-up visit (1, 22).
Growth hormone deficiency may be considered for children with growth retardation despite treatment.
Renal ultrasound is recommended to be performed at least every 12–24 months to monitor the occurrence of kidney stones, nephrocalcinosis, and obstructive uropathy.
Cardiac work out is recommended for patients (particularly adults) with palpitations or syncope. The physician can use the drugs that slow the sinus rhythm (beta-blockers, calcium channel blockers as verapamil, diltiazem, digoxin) or drugs that influence the QT interval (proton pump inhibitors, fluoroquinolones, macrolides, gentamicin, or antiviral drugs). Electrocardiography, Holter, and stress electrocardiography are recommended to detect cardiac arrhythmias which are determined by a prolonged QT interval in the context of hypokalemia and hypomagnesemia (1, 22).
The quality of life is important to be assessed by using age-specific standardized questionnaires. The quality-of-life scores are related by biochemical parameters (potassium and aldosterone) (1, 41).
Sports can be performed with a good hydration and additional salt and electrolytes.
Anesthesia for patients with BS must be performed after improvement of hypokalemia and hypomagnesemia, which in addition with anesthetic agents can lead to neuromuscular blockage. Potassium levels > 3.0 mmol/L and magnesium > 0.5 mmol/are suggested by guidelines (42).
Kidney Disease
Renal disease is characterized by urolithiasis, obstructive uropathy, and nephrocalcinosis. Nephrotic-range proteinuria has also been reported in patients with BS (21, 24, 43). Renal biopsy can show diffuse glomerular and tubule-interstitial lesions with enlarged glomeruli and focal segmental glomerulosclerosis (23).
Only some patients have an evolution to end-stage kidney disease. CKD can occur as a late manifestation in patients with BS, particularly types 1, 4a, and 4b (21, 23, 44). The CKD in BS seems to have several mechanisms: chronic stimulation of the renin–angiotensin system, periodic dehydration, prematurity, nephrocalcinosis, long-term NSAID drug treatment, and chronic hypokalemia (4).
There are few reported cases of kidney transplants (23, 45–50). The electrolytes and urinary concentrating abnormalities were corrected. No evidence of recurrent disease was observed.
Few studies reported growth failure with growth hormone (GH) deficiency in patients with BS, particularly type 3 who have severe metabolic abnormalities. The poor growth explanation is unclear: acid-base or electrolyte disturbances in BS or whether it is an intrinsic part of the disorder. After metabolic control, recombinant human GH supplementation can be performed (21, 24, 51–53).
Conclusion
Knowledge of clinical heterogeneity in BS is important for initiating genetic investigations and proper management. Early molecular diagnosis is desirable for personalized therapy.
Data Availability Statement
Some data was obtained from Human Gene Mutation Database (HGMD® Professional 2021.4) available online: http://www.hgmd.cf.ac.uk/ac/all.php (accessed February 15, 2022).
Author Contributions
LF, LC, and EG: writing – original draft, review and editing. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CLDN10, claudin 10; CLDN16, claudin 16; CLDN19, claudin 19; CASR, calcium sensing receptor; AP2S1, adaptor related protein complex 2 subunit sigma 1; GNA11, G protein subunit alpha 11; UMOD, uromodulin; SLC12A1, solute carrier family 12 member 1; KCNJ1, potassium inwardly rectifying channel subfamily J member 1; CLCNKB, chloride voltage-gated channel Kb; CLCNKA, chloride voltage-gated channel Ka; BSND, barttin CLCNK type accessory subunit beta; MAGED2, MAGE family member D2; SLC12A3, solute carrier family 12 member 3; CASR, calcium sensing receptor; KCNJ10, potassium inwardly rectifying channel subfamily J member 10; SLC26A3, solute carrier family 26 member 3; CLDN10, claudin 10; SCNN1A, sodium channel epithelial 1 subunit alpha; SCNN1B, sodium channel epithelial 1 subunit beta; SCNN1G, sodium channel epithelial 1 subunit gamma; NR3C2, nuclear receptor subfamily 3 group C member 2; HSD11B2, hydroxysteroid 11-beta dehydrogenase 2; CYP11B1, cytochrome P450 family 11 subfamily B member 1; CLCN2, chloride voltage-gated channel 2; KCNJ5, potassium inwardly rectifying channel subfamily J member 5; CACNA1H, calcium voltage-gated channel subunit alpha1 H.
References
1. Konrad M, Nijenhuis T, Ariceta G, Bertholet-Thomas A, Calo LA, Capasso G, et al. Diagnosis and management of bartter syndrome: executive summary of the consensus and recommendations from the European rare kidney disease reference network working group for tubular disorders. Kidney Int. (2021) 99:324–35. doi: 10.1016/j.kint.2020.10.035
2. Orphanet Report Series. «Prevalence of Rare Diseases: Bibliographic Data», Orphanet Report Series, Rare Diseases Collection, Janvier 2022. (2022). Available online at: https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf (accessed March 15, 2022).
3. Legrand A, Treard C, Roncelin I, Dreux S, Bertholet-Thomas A, Broux F, et al. Prevalence of novel MAGED2 mutations in antenatal Bartter syndrome. Clin J Am Soc Nephrol. (2018) 13:242–50. doi: 10.2215/CJN.05670517
4. Vargas-Poussou R. Pathophysiological aspects of the thick ascending limb and novel genetic defects: helix syndrome and transient antenatal Bartter syndrome. Pediatr Nephrol. (2021) 37:239–52. doi: 10.1007/s00467-021-05019-6
5. HGNC, HUGO Gene Nomenclature Committee. The Resource for Approved Human Gene Nomenclature. (2022). Available online at: http://www.genenames.org/ (accessed March 28, 2022).
6. UNIPROT. The Mission of UniProt is to Provide the Scientific Community with a Comprehensive, High-Quality and Freely Accessible Resource of Protein Sequence and Functional Information. (2022). Available online at: https://www.uniprot.org/ (accessed March 25, 2022).
7. da Silva Cunha T, Heilberg IP. Bartter syndrome: causes, diagnosis, and treatment. Int J Nephrol Renovasc Dis. (2018) 11:291. doi: 10.2147/IJNRD.S155397
8. Covic M, Covic A, Ardeleanu Ş. Anatomy and physiology of the kidneys. In: Covic A editor. Nephrology. Theoretical and Practical Principles. (Iasi: Demiurg Press) (2011). p. 21–35.
9. Reinalter SC, Jeck N, Brochhausen C, Watzer B, Nüsing RM, Seyberth HW, et al. Role of cyclooxygenase-2 in hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int. (2002) 62:253–60. doi: 10.1046/j.1523-1755.2002.00435.x
10. Amirlak I, Dawson KP. Bartter syndrome: an overview. QJM. (2000) 93:207–15. doi: 10.1093/qjmed/93.4.207
11. Dimke H, Schnermann J. Axial and cellular heterogeneity in electrolyte transport pathways along the thick ascending limb. Acta Physiol (Oxf). (2018) 223:e13057. doi: 10.1111/apha.13057
12. de Los Heros P, Pacheco-Alvarez D, Gamba G. Role of WNK kinases in the modulation of cell volume. Curr Top Membr. (2018) 81:207–35. doi: 10.1016/bs.ctm.2018.08.002
13. Gamba G. The thiazide-sensitive Na-Cl cotransporter: molecular biology, functional properties, and regulation by WNKs. Am J Physiol Renal Physiol. (2009) 297:F838–48. doi: 10.1152/ajprenal.00159.2009
14. Kahle KT, Rinehart J, Giebisch G, Gamba G, Hebert SC, Lifton RP. A novel protein kinase signaling pathway essential for blood pressure regulation in humans. Trends Endocrinol Metab. (2008) 19:91–5. doi: 10.1016/j.tem.2008.01.001
15. Delpire E, Gagnon KB. SPAK and OSR1: STE20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. Biochem J. (2008) 409:321–31. doi: 10.1042/BJ20071324
16. Carota I, Theilig F, Oppermann M, Kongsuphol P, Rosenauer A, Schreiber R, et al. Localization and functional characterization of the human NKCC2 isoforms. Acta Physiol (Oxf). (2010) 199:327–38. doi: 10.1111/j.1748-1716
17. Bankir L, Figueres L, Prot-Bertoye C, Bouby N, Crambert G, Pratt JH, et al. Medullary and cortical thick ascending limb: similarities and differences. Am J Physiol Renal Physiol. (2020) 318:F422–42. doi: 10.1152/ajprenal.00261.2019
18. Jentsch TJ, Pusch M. CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol Rev. (2018) 98:1493–590. doi: 10.1152/physrev.00047.2017
19. Hennings JC, Andrini O, Picard N, Paulais M, Huebner AK, Cayuqueo IKL, et al. The ClC-K2 chloride channel is critical for salt handling in the distal nephron. J Am Soc Nephrol. (2017) 28:209–17. doi: 10.1681/ASN.2016010085
20. Laghmani K, Beck BB, Yang SS, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnios, transient antenatal Bartter’s syndrome, and MAGED2 mutations. N Engl J Med. (2016) 374:1853–63. doi: 10.1056/NEJMoa1507629
21. Walsh PR, Tse Y, Ashton E, Iancu D, Jenkins L, Bienias M, et al. Clinical and diagnostic features of Bartter and Gitelman syndromes. Clin Kidney J. (2018) 11:302–9. doi: 10.1093/ckj/sfx118
22. Konrad M, Nijenhuis T, Ariceta G, Bertholet-Thomas A, Calo LA, Capasso G, et al. Diagnosis and Management of Bartter Syndrome: Consensus and Recommendations from the ERKNet Working Group for Tubular Disorders. (2020). Available online at: https://www.erknet.org/guidelines-pathways/tubulopathies/bartter-syndrome (accessed April 11, 2022).
23. Seys E, Andrini O, Keck M, Mansour-Hendili L, Courand PY, Simian C, et al. Clinical and genetic spectrum of Bartter syndrome type 3. J Am Soc Nephrol. (2017) 28:2540–52. doi: 10.1681/ASN.2016101057
24. Bettinelli A, Borsa N, Bellantuono R, Syrèn ML, Calabrese R, Edefonti A, et al. Patients with biallelic mutations in the chloride channel gene CLCNKB: long-term management and outcome. Am J Kidney Dis. (2007) 49:91–8. doi: 10.1053/j.ajkd.2006.10.001
25. Konrad M, Vollmer M, Lemmink HH, Van Den Heuvel LP, Jeck N, Vargas-Poussou R, et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol. (2000) 11:1449–59. doi: 10.1681/ASN.V1181449
26. Brochard K, Boyer O, Blanchard A, Loirat C, Niaudet P, Macher MA, et al. Phenotype–genotype correlation in antenatal and neonatal variants of Bartter syndrome. Nephrol Dial Transplant. (2009) 24:1455–64. doi: 10.1093/ndt/gfn689
27. Allaf B, Dreux S, Schmitz T, Czerkiewicz I, Le Vaillant C, Benachi A, et al. Amniotic fluid biochemistry in isolated polyhydramnios: a series of 464 cases. Prenat Diagn. (2015) 35:1331–5. doi: 10.1002/pd.4700
28. Garnier A, Dreux S, Vargas-Poussou R, Oury JF, Benachi A, Deschênes G, et al. Bartter syndrome prenatal diagnosis based on amniotic fluid biochemical analysis. Pediatr Res. (2010) 67:300–3. doi: 10.1203/PDR.0b013e3181ca038d
29. Birkenhäger R, Otto E, Schürmann MJ, Vollmer M, Ruf EM, Maier-Lutz I, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. (2001) 29:310–4. doi: 10.1038/ng752
30. Ashton EJ, Legrand A, Benoit V, Roncelin I, Venisse A, Zennaro MC, et al. Simultaneous sequencing of 37 genes identified causative mutations in the majority of children with renal tubulopathies. Kidney Int. (2018) 93:961–7. doi: 10.1016/j.kint.2017.10.016
31. Arthuis CJ, Nizon M, Komhoff M, Beck BB, Riehmer V, Bihouee T, et al. A step towards precision medicine in management of severe transient polyhydramnios: MAGED2 variant. J Obstet Gynaecol. (2018) 39:395–7. doi: 10.1080/01443615.2018.1454415
32. Peters M, Jeck N, Reinalter S, Leonhardt A, Tönshoff B, ünter Klaus G, et al. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am J Med. (2002) 112:183–90. doi: 10.1016/S0002-9343(01)01086-5
33. Castrop H, Schießl IM. Physiology and pathophysiology of the renal Na-K-2Cl cotransporter (NKCC2). Am J Physiol Renal Physiol. (2014) 307:F991. doi: 10.1152/ajprenal.00432.2014
34. Oppermann M, Mizel D, Kim SM, Chen L, Faulhaber-Walter R, Huang Y, et al. Renal function in mice with targeted disruption of the A isoform of the Na-K-2Cl co-transporter. J Am Soc Nephrol. (2007) 18:440–8. doi: 10.1681/ASN.2006091070
35. Oppermann M, Mizel D, Huang G, Li C, Deng C, Theilig F, et al. Macula densa control of renin secretion and preglomerular resistance in mice with selective deletion of the B isoform of the Na. K, 2Cl co-transporter. J Am Soc Nephrol. (2006) 17:2143–52. doi: 10.1681/ASN.2006040384
36. Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Qual Life Res. (2017) 136:665–77. doi: 10.1007/s00439-017-1779-6
37. Kömhoff M, Reinalter SC, Gröne HJ, Seyberth HW. Induction of microsomal prostaglandin E2 synthase in the macula densa in children with hypokalemic salt-losing tubulopathies. Pediatr Res. (2004) 55:261. doi: 10.1203/01.PDR.0000101747.09626.6B
38. Ranade VV, Somberg JC. Bioavailability and pharmacokinetics of magnesium after administration of magnesium salts to humans. Am J Ther. (2001) 8:345. doi: 10.1097/00045391-200109000-00008
39. Blanchard A, Bockenhauer D, Bolignano D, Calo LA, Cosyns E, Devuyst O, et al. Gitelman syndrome: consensus and guidance from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. (2017) 91:24–33. doi: 10.1016/j.kint.2016.09.046
40. Bockenhauer D, van’T Hoff W, Dattani M, Lehnhardt A, Subtirelu M, Hildebrandt F, et al. Secondary nephrogenic diabetes insipidus as a complication of inherited renal diseases. Nephron Physiol. (2010) 116:23–9. doi: 10.1159/000320117
41. Eder M, Darmann E, Haller MC, Bojic M, Peck-Radosavljevic M, Huditz R, et al. Markers of potassium homeostasis in salt losing tubulopathies—associations with hyperaldosteronism and hypomagnesemia. BMC Nephrol. (2020) 21:256. doi: 10.1186/s12882-020-01905-7
42. Gallagher H, Soar J, Tomson C. New guideline for perioperative management of people with inherited salt-wasting alkaloses. Br J Anaesth. (2016) 116:746–9. doi: 10.1093/bja/aew102
43. Puricelli E, Bettinelli A, Borsa N, Sironi F, Mattiello C, Tammaro F, et al. Long-term follow-up of patients with Bartter syndrome type I and II. Nephrol Dial Transplant. (2010) 25:2976–81. doi: 10.1093/ndt/gfq119
44. Jeck N, Reinalter SC, Henne T, Marg W, Mallmann R, Pasel K, et al. Hypokalemic salt-losing tubulopathy with chronic renal failure and sensorineural deafness. Pediatrics. (2001) 108:E5. doi: 10.1542/peds.108.1.e5
45. Blethen SL, van Wyk JJ, Lorentz WB, Jennette JC. Reversal of Bartter’s syndrome by renal transplantation in a child with focal, segmental glomerular sclerosis. Am J Med Sci. (1985) 289:31–6. doi: 10.1097/00000441-198501000-00006
46. Takahashi M, Yanagida N, Okano M, Ishizaki A, Meguro J, Kukita K, et al. A first report: living related kidney transplantation on a patient with Bartter’s syndrome. Transplant Proc. (1996) 28:1588.
47. Kim JY, Kim GA, Song JH, Lee SW, Han JY, Lee JS, et al. A case of living-related kidney transplantation in Bartter’s syndrome. Yonsei Med J. (2000) 41:662–5. doi: 10.3349/ymj.2000.41.5.662
48. Ueki R, Okutani R, Fukushima A, Tsuji T, Ohata H, Tashiro C. Perioperative endocrinological findings in a patient with Bartter’s syndrome and living-related renal transplantation. J Anesth. (2000) 14:105–8. doi: 10.1007/s005400050076
49. Kitanaka S, Sato U, Maruyama K, Igarashi T. A compound heterozygous mutation in the BSND gene detected in Bartter syndrome type IV. Pediatr Nephrol. (2006) 21:190–3. doi: 10.1007/s00467-005-2091-6
50. Lee SE, Han KH, Jung YH, Lee HK, Kang HG, Moon KC, et al. Renal transplantation in a patient with Bartter syndrome and glomerulosclerosis. Korean J Pediatr. (2011) 54:36–9. doi: 10.3345/kjp.2011.54.1.36
51. Akil I, Ozen S, Kandiloglu AR, Ersoy B. A patient with Bartter syndrome accompanying severe growth hormone deficiency and focal segmental glomerulosclerosis. Clin Exp Nephrol. (2010) 14:278–82. doi: 10.1007/s10157-009-0262-7
52. Gil-Peña H, Mejia N, Alvarez-Garcia O, Loredo V, Santos F. Longitudinal growth in chronic hypokalemic disorders. Pediatr Nephrol. (2010) 25:733–7. doi: 10.1007/s00467-009-1330-7
Keywords: Bartter syndrome, tubulophathy, genetic heterogeneity, channel disease, rare disease
Citation: Florea L, Caba L and Gorduza EV (2022) Genetic Heterogeneity in Bartter Syndrome: Clinical and Practical Importance. Front. Pediatr. 10:908655. doi: 10.3389/fped.2022.908655
Received: 30 March 2022; Accepted: 21 April 2022;
Published: 03 June 2022.
Edited by:
Claudia Banescu, University of Medicine, Pharmacy, Sciences and Technology of Târgu Mureş, RomaniaReviewed by:
Carmen Muntean (Duicu), George Emil Palade University of Medicine, Pharmacy, Sciences and Technology of Târgu Mureş, RomaniaCristina Skrypnyk, Arabian Gulf University, Bahrain
Copyright © 2022 Florea, Caba and Gorduza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lavinia Caba, bGF2aW5pYV96YW5ldEB5YWhvby5jb20=