
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 21 June 2022
Sec. Neonatology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.886627
This article is part of the Research TopicInsights in Neonatology: 2021View all 15 articles
 Zakia Boudar1
Zakia Boudar1 Sofia Sehli1
Sofia Sehli1 Sara El Janahi1
Sara El Janahi1 Najib Al Idrissi2
Najib Al Idrissi2 Salsabil Hamdi3
Salsabil Hamdi3 Nouzha Dini4
Nouzha Dini4 Hassan Brim5
Hassan Brim5 Saaïd Amzazi6
Saaïd Amzazi6 Chakib Nejjari7,8
Chakib Nejjari7,8 Michele Lloyd-Puryear9
Michele Lloyd-Puryear9 Hassan Ghazal1,10*
Hassan Ghazal1,10*Early infancy is critical for the development of an infant's gut flora. Many factors can influence microbiota development during the pre- and postnatal periods, including maternal factors, antibiotic exposure, mode of delivery, dietary patterns, and feeding type. Therefore, investigating the connection between these variables and host and microbiome interactions in neonatal development would be of great interest. As the “unculturable” era of microbiome research gives way to an intrinsically multidisciplinary field, microbiome research has reaped the advantages of technological advancements in next-generation sequencing, particularly 16S rRNA gene amplicon and shotgun sequencing, which have considerably expanded our knowledge about gut microbiota development during early life. Using omics approaches to explore the neonatal microbiome may help to better understand the link between the microbiome and newborn diseases. Herein, we summarized the metagenomics methods and tools used to advance knowledge on the neonatal microbiome origin and evolution and how the microbiome shapes early and late individuals' lives for health and disease. The way to overcome limitations in neonatal microbiome studies will be discussed.
The human microbiome is a set of microorganisms (bacteria, viruses, protozoa, and fungi) residing in our body in different types of commensal relationships. The gut microbiota is the most important and consequential of them, with approximately 1,000 species (1) and 100 trillion microorganisms, which is ten times more than the number of eukaryotic cells that make up our body (2). It plays a role in regulating nutrient intake, intestinal motility, and metabolic and immunological development (3). However, how the newborn gut microbiota forms remains an open question. The combination of neonatal (gestational age, genetic history), maternal (delivery mode, diet), and environmental (e.g., antibiotic exposures) variables are considered to impact microbial colonization (4, 5), although the precise processes remain unknown. Several studies have shown that the infant gut microbiome can be influenced by different factors, leading to many diseases in the infant or child, including necrotizing enterocolitis, obesity, and inflammatory diseases (6, 7). Thus it is critical to understand the mechanism of microbiome colonization in newborns and how various factors may influence this process.
With the development, sophistication, and sensitivity of metagenomics and culturomics, identifying fecal microbes is improving our current knowledge about gut microbiota development, particularly during the early days' post-delivery (8, 9). Two methods have been widely used to examine microbial diversity: 16S rRNA gene amplicon sequencing and whole-genome shotgun metagenomics sequencing (WGS). WGS metagenomics sequencing allows the characterization of microbial whole genomes, while 16S rRNA gene amplicon gives a depth description of the diversity of certain taxonomic groups (10, 11).
The purpose of this review is to summarize and discuss the available amplicon-based and WGS metagenomics methodologies and tools used to profile the newborn microbiome and study the in utero transmission of microorganisms from mother to newborn.
Conflicting results from various studies have unsettled the notion of the in utero gut microbiome. The “sterile uterus” theory maintains that the embryo grows in a sterile environment in utero, and except for intrauterine infections during pregnancy, microbial colonization begins after birth (9, 12). However, this theory of the sterile uterus has been called into question by recent studies using both metagenomics and culture techniques that revealed the presence of microbial community in the meconium, placenta, blood umbilical cord, and amniotic fluid (13, 14). In addition, other researchers have reported the presence of a unique microbiota in the placenta and amniotic fluid, as well as in healthy women at the time of elective cesarean section, associated with low diversity and a prevalence of Proteobacteria (15). Likewise, other studies have discovered microbes in the umbilical cord blood and amniotic fluid in healthy women and those with pregnancy complications (16, 17). Several studies have found microorganisms in the meconium, lending credence to the in utero colonization of the baby's gut theory. Staphylococcus was reported as the most frequent bacterium in meconium samples, followed by Enterobacteriaceae family, Enterococcus, Lactobacillus, and Bifidobacterium genera (18).
In contrast, supporters of the sterile uterus theory attribute the identified microbial components to external contamination (19) because there is no indication of bacterial colony survival (20). Likewise, attempts to grow viable bacteria from placental samples in healthy pregnancies have thus far been unsuccessful (21). Perez-Muñoz et al. (12), on the other hand, support both the “sterile uterus” theory and “in utero colonization” as a methodology-associated artifact (22). They concluded that the data stating a sterile uterine environment was more robust. They claim that methodological techniques in which contamination is fairly easy are responsible for the observed in utero colonization (12). In well-controlled studies, oral, vaginal, and placental samples were compared to contaminated controls. They determined that while there are unique microbial profiles in vaginal and oral samples, they did not discover a distinct placental microbiome, supporting the sterile environment theory (23). Therefore, firm conclusions remain elusive, and further research in this field is still required.
Several studies have shown that the gut microbiota has a role in the programming and development of the fetal immune system, metabolic programming, and preventing pathogen colonization of the gut, all of which have long-term consequences in infancy, early childhood, and adulthood (24–26). Colonization of the gut microbiome is a complicated process controlled by many variables (Figure 1) (27). Despite recent research on in utero colonization, the birth canal is still considered the baby's first bacterial encounter (28). The neonatal microbiota is dominated by Enterobacteria, Escherichia, and Shigella throughout the first few days (9, 29, 30). According to a new study (9), while the Firmicutes phylum dominates the meconium, Proteobacteria species dominate fecal samples from newborns throughout their 1st months of life. The newborn gut microbiota shows more significant fluctuations and minor variations in the early days of life. Nevertheless, as the infant ages, the bacterial communities increase in diversity and stability. It has been proven that by the age of two, the neonatal microbiota has stabilized to a level equivalent to that of adults (31). However, other scientists suggest that this process might take up to 5 years (32).
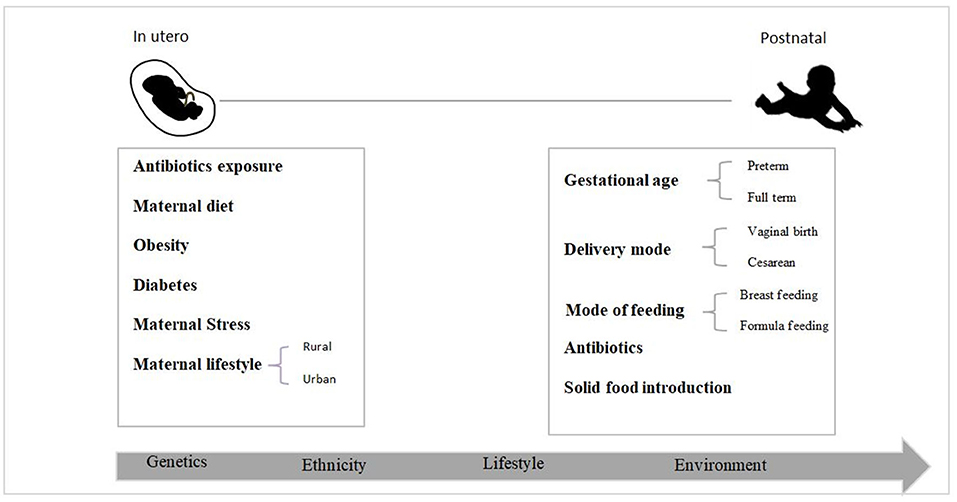
Figure 1. Early life factors that impact the newborn gut bacteria colonization process.
It is well established that the delivery mode, either vaginal or cesarean section (CS), impacts gut microbiome colonization. Lactobacillaceae (Firmicutes) and Proteobacteria were initially characterized in vaginal-birth infants. Conversely, the gut of cesarean-delivered infants is dominated by Streptococcaceae and Staphylococcaceae (Firmicutes) (33, 34), with a low diversity during the first years of life (35). Interestingly, this difference in colonization disappears with age, and the microbiota of both vaginally and cesarean section-delivered infants become similar (36). Several studies have found that vaginal birth seeds have a more favorable and healthier microbiota than CS delivery seeds. Furthermore, it has been observed that the mode of delivery impacts the development of immunological responses that may lead to allergies and autoimmune diseases (37, 38). Extensive other studies demonstrated a link between cesarean delivery and a higher risk of asthma, obesity, and autoimmune disorders. However, more research needs to be done to provide a final answer to determine the relationship between mode of delivery and disease in later life (32).
Gestational age is an essential factor influencing the first colonization of an infant's gut. Several studies revealed differences in microbiota composition between term and preterm infants. The gut microbiota of full-term infants is generally dominated by Bifidobacterium and Lactobacillus, which are considered healthier bacteria (32, 39, 40), while preterm infants were found to have retarded colonization of Bifidobacterium and Lactobacillus in their early life. Instead, they are more likely to be colonized by potentially pathogenic bacteria, particularly E. coli, Staphylococcus, Enterobacteriaceae, and Bacteroides (41, 42). Premature infants are vulnerable to diseases including necrotizing enterocolitis (NEC) and sepsis, which are commonly caused by antimicrobial-resistant bacteria (38, 43, 44) due to their aberrant microbiota and immature gut immune systems. These diseases are rare in full-term babies, but they are severe and sometimes deadly in preterm babies. Moreover, gestational age has been revealed to influence milk composition, altering the metabolites that impact colonization, such as human milk oligosaccharides (HMOs) (15, 45).
Antibiotics are one of the most studied factors affecting microbiota, and they are the most prescribed treatment for infants. Antibiotic exposure and bacterial infections significantly affect microbiota composition in the postnatal and prenatal periods (46–48). Antibiotic treatment during pregnancy could affect the neonatal microbiota, leading to dysbiosis (48). Various studies have found a link between early gut microbiome dysbiosis and many diseases, such as asthma, immunological disorders (49), obesity, diabetes, and developmental disorders, such as autism, in later life. Early antibiotic exposure in infancy affects the composition and diversity of the infants' intestinal microbiota, with a reduction in Bifidobacteria and a marked increase in Proteobacteria. Furthermore, infants of mothers who received antibiotics before delivery showed the same microbiome changes as those seen in antibiotic-treated infants (48, 49). In addition, amoxicillin/clavulanic acid antibiotic medication was linked to a fourfold increased risk of newborn NEC (50).
Nutrition during infant development plays a significant role in early microbiota colonization. Healthy full-term infants born through vaginal delivery and exclusively breastfed are thought to have the most beneficial gut microbiota composition. Human milk (HM) represents the optimal natural food (51) and contains a mix of nutrients, commensal bacteria, and functional groups such as oligosaccharides. HM also contains antimicrobials (such as lactoferrin) that can prevent the colonization of enteropathogens and stimulate the growth of Bifidobacterium (52), which may promote neonatal health by decreasing the risk of obesity and NEC and promoting mental development in preterm infants (53). Based on recent data, HM contains an average of 106 bacterial cells/ml (54), dominated by Proteobacteria and Firmicutes phyla, and a minor component of Pseudomonas, Staphylococcus, and Streptococcus genera (51, 53).
The exact mechanism through which bacteria reach the mammary glands and are excreted into breast milk is still debated (55). One hypothesis is that HM mainly contains bacteria derived from the mother's skin and/or the infant's mouth (56). The other hypothesis, called the “Enteromammary pathway”, postulates that some bacteria migrate from the maternal gastrointestinal tract to the mammary glands during late gestation and lactation (57). Formula-fed infants are codominated by Bifidobacterium and Bacteroides with a low percentage of Escherichia coli and Clostridia (52, 58). A high level of Firmicutes with a low level of Bifidobacteria has been associated with a predisposition to obesity (59).
The second shift in the microbial colonization process after breastfeeding, is the introduction of solid food. During this period, the microbiota is rapidly changing, and it is characterized by bacteria such as the Ruminococcus, Blautia, Lachnospira, and Faecalibacterium genera that can digest mucin and glycans and produce bioactive molecules such as short-chain fatty acids (SCFAs) (32).
Genetics also plays a role in microbial colonization, but the mechanisms are still poorly understood. Ethnicity has been recognized as one of the factors affecting neonatal microbial colonization, even in infants from the same geographic locations (60). Studies showed that the impact of ethnicity on newborn microbiota was visible 3 months after birth, and that was before the introduction of supplemental foods. A longitudinal study assessed the gut microbiota composition of 106 infants of three Asian ethnicities (Malay, Indian, and Chinese) who lived in the same geographical region (Singapore) and revealed that ethnic impacts were visible at 3 months post-birth and remained significant until 12 months in Chinese and Indian infants. The microbiota of Indian newborns was characterized by increased abundances of Lactobacillus and Bifidobacterium, while Bacteroides and Akkermansia were more abundant in Chinese infants (60). As a result of these ethnic variations in microbiota composition, human genetics may have a role in the establishment of gut microbiota.
Other studies on twins have discovered heritable bacteria in the gut microbiota, including the Christensenellaceae family and methanogenic archaealarchaea (60). MZ twins share more microorganisms than DZ twins or non-twin pairs, according to a study of ten healthy twins, five monozygotic (MZ) and five dizygotic (DZ), whose ages ranged from 0 to 6 years (61).
Prior to the microbiome era, bacterial characterization was primarily focused on identifying pathogenic species. However, the revelation of the importance of the microbiome-host interaction in human health has resulted in the creation of novel microbe investigation technologies. These so-called metagenomics techniques have helped researchers better understand the microbial diversity contained in a sample. This approach directly uses the genetic material existing in an environmental sample without the requirement for culture. Table 1 lists the most commonly used methodologies for studying the microbiome, as well as its benefits and limitations.
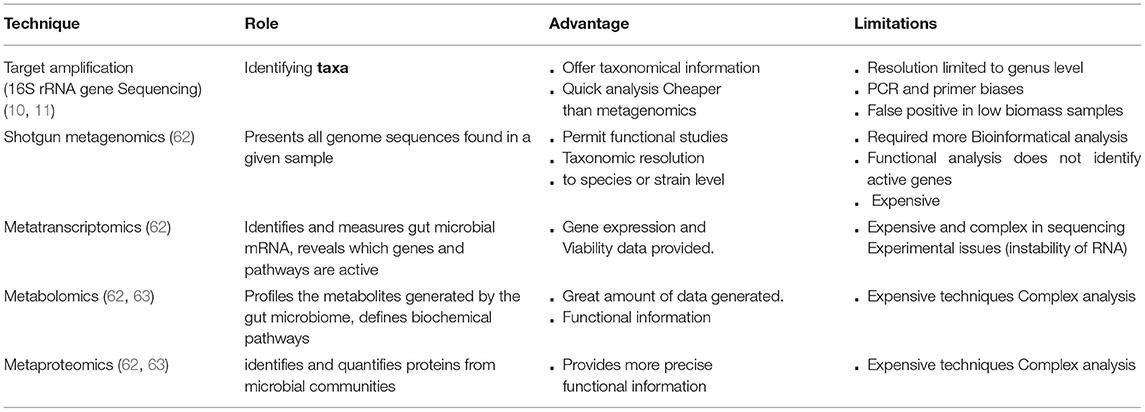
Table 1. Commonly used metagenomic techniques in microbiome analysis.
The most common methods to explore the human microbiome are 16S rRNA gene amplicon sequencing and shotgun metagenomics sequencing (WGS) (10). The 16S rRNA genes consist of both highly conserved and variable regions used for taxonomic classification. At the same time, WGS presents all genome sequences found in a given sample (10, 11). Meta-omic studies have been increasingly used to study the gut microbiome community to better understand taxonomic classification, metabolic pathways, and the essential proteins and metabolites implicated in a specific host phenotype (64). One of these methods is metabolomics, which is used to profile the metabolites generated by the gut microbiome and define metabolites and biochemical pathways (62). Metatranscriptomics is another method for identifying and measuring gut microbial mRNA that reveals which genes and pathways are active and play essential roles in health and diseases (62). Another powerful approach to identifying and quantifying proteins from microbial communities is metaproteomics (63). Using a single omics approach to explore the gut microbiota has its own set of limitations (62), which can be addressed by combining various omics techniques. Consequently, researchers may better understand the relationship between the microbiome and diseases (65, 66).
There are several bioinformatic tools known to analyze 16S rRNA gene amplicon sequencing data, including QIIME 2 (67), Mothur (68), amplicon sequence variants (ASV)-based DADA2 (69), MED (70), and UNOISE (71). Operational taxonomic unit (OTU)-based methods such as QIIME eliminate sequencing errors by clustering the sequences in OTUs using a similarity threshold (usually 97%) (72, 73). On the other hand, ASV-based tools predict and correct sequencing errors (denoising) before forming clusters, allowing resolving sequences differing by a single nucleotide (69).
Previous studies have contrasted these two techniques and concluded that OTUs give poorer taxonomic resolution than ASVs and that choosing between the two can have an influence on alpha diversity estimations (73, 74). Nevertheless, data quality and PCR errors have a significant impact on ASV approaches, resulting in the loss of a significant quantity of relevant information. As a result, when the data quality isn't good enough, an OTU-based strategy is required (75).
Prodan et al. compared the most common bioinformatics pipelines for 16S rRNA gene amplicon sequencing (including MOTHUR, QIIME-UCLUST, USEARCH-UPARSE, DADA2, USEARCH-UNOISE3, Qiime2, and Deblur) (76). They found that DADA2 had the best resolution and sensitivity, but USEARCH-UNOISE3 had the best overall performance, combining high sensitivity with excellent specificity. However, to produce more robust data, research in this field needs to move toward improved methods (77). Another study by the Almeida group found that when comparing QIIME, QIIME 2, MAPseq, and Mothur, QIIME 2 was the best tool for composition prediction, while MAPseq was more precise, with few genera being misallocated (78).
The reference database utilized, in addition to selecting the appropriate bioinformatics tool, is a crucial aspect in ensuring the most significant classification performance. The Ribosomal Database Project (RDP), SILVA, and Greengenes are the most important 16S databases (79). However, SILVA is updated more regularly than Greengenes, which was last updated in May 2013. In addition, SILVA contains rRNA sequences of different species, including eukaryotic organisms, archaea, and bacterial species (79).
The main metagenomic techniques and pipelines that have been used in neonatal microbiome studies are listed in Table 2.
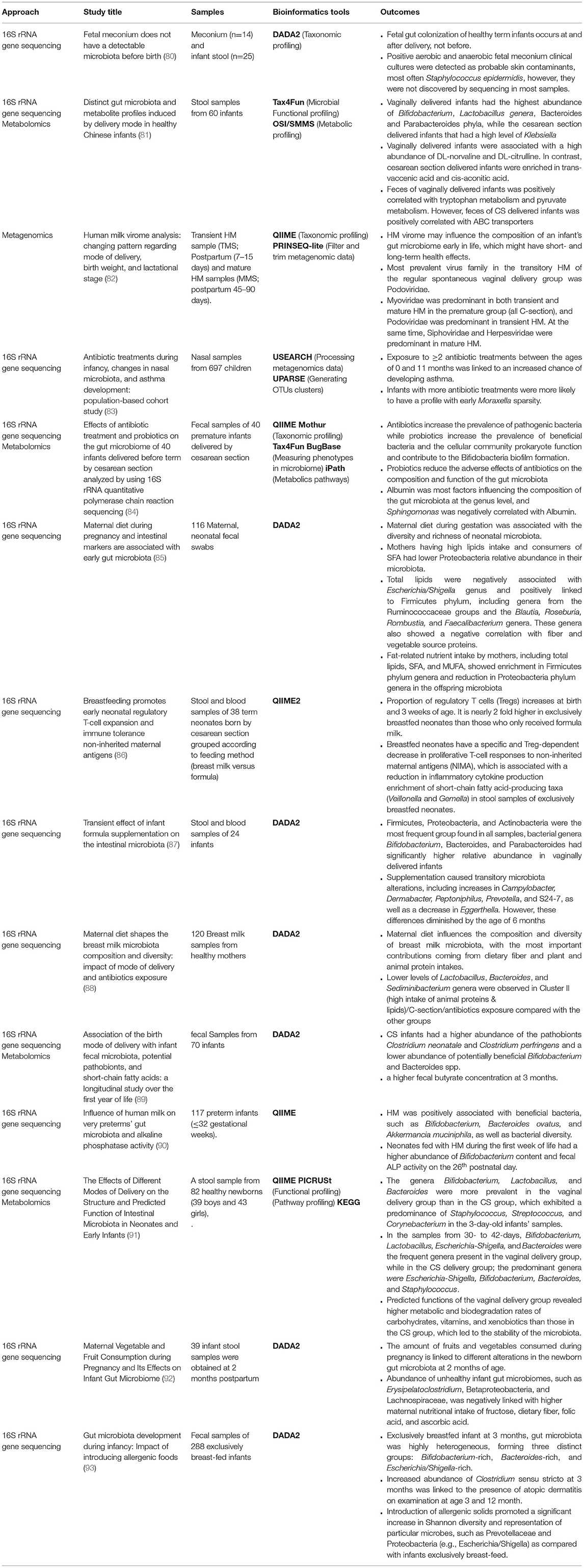
Table 2. Metaomics technologies in neonatal microbiome studies (02/2021–09/2021).
Previous studies using 16S rRNA gene sequencing to explore the gut microbiome have allowed the identification and functional characterization of microbial communities and highlighted the associations between microbial composition wellbeing and illness (6, 7, 49). Many studies have found that different factors, such as delivery mode, antibiotic exposure, and milk diet, significantly impact the formation of gut microbiota. In a birth-cohort study in Finland, 697 children showed that early exposure to two or more antibiotics was linked to a higher risk of asthma (83). Another study by Pan et al. examined the impact of the delivery mode on the structure and predicted function of intestinal microbiota in neonates and early infants in the Chinese population and discovered that the genera Bifidobacterium, Lactobacillus, and Bacteroides were more common in the vaginal delivery group than in the cesarean section group, which had a predominance of Staphylococcus, Streptococcus, and Corynebacterium in the 3-day-old infants' samples. Additionally, the Bacteroides level was lower in the cesarean section group than that in the vaginal birth group, suggesting that the latter is associated with an increased risk of obesity (91).
16S rRNA gene amplicon sequencing represents the principal tool for the characterization of bacteria in tissues with low bacterial biomass (i.e., placenta and meconium samples). This approach is limited by challenges associated with polymerase chain reaction (PCR)-based short read length sequencing, including GC bias, sequencing errors, and difficulty identifying OTUs (66, 67). Although it is unable to distinguish viable taxa, it continues to have technical difficulties in achieving species-level precision in taxonomic profiling.
Shotgun metagenomics is the most informative technique for assessing taxonomic diversity present in a fecal sample (94). The findings of this analysis can be utilized to predict biological functions. The protein-coding sequences from the metagenomic readings are selected and compared to protein-coding sequences in a database to obtain functional profiling. This method may be used to provide a profile that describes the likely biological functions discovered in the sequenced metagenome (94, 95). For example, a recent study found that the HM virome may alter the makeup of an infant's gut microbiome early in infancy, thereby affecting both short- and long-term health (82).
Despite its numerous benefits, this technology has some limitations during DNA preparation, and post-analytical processing techniques suggest that this technology can yet be improved. The next point to consider is the high degree of expertise and high cost required to analyze such massive amounts of data (64, 69). Another technological limitation is that it is unable to make the difference between living and dead cells and thus unable to provide actual functional information (64, 69). Additionally, there are numerous incompletely annotated bacterial genomic sequences, as well as concerns regarding database correctness and coverage (96, 97).
Because metagenomics bioinformatics tools depend on the availability of annotated genomes, they are affected by reference sequence database restrictions. When evaluating metabolic potential, the absence of annotations for a large number of microbial species leads to a bias toward highly conserved pathways (housekeeping genes), even if there are major changes in taxonomic composition (64). Additionally, the lack of host DNA depletion kits makes metagenomics unreliable in tissues with low biomass such as placenta and meconium samples. Shotgun metagenomics (non-targeted sequencing) can quickly resolve species- and strain-level categorization, as well as reveal genome content, functional potential and partial genome assembly for organisms with low abundance. It is, however, still more costly than amplicon sequencing, is less tolerant of low biomass or contaminated materials, and requires more complicated and expensive analytic techniques.
Metabolomics studies on feces provide an essential analysis of the microbiome, including information on the host's metabolic profile, nutrition, and gut microbiota (62). This technique elucidates metabolites that mediate microbe-host interactions. In infancy, diet and mode of delivery have been found to have a significant connection with fecal metabolite composition (98). Human breast milk includes high levels of HMOs, which function as selective nutrients for particular bacterial groups (e.g., Bifidobacterium) in the production of SCFAs whose quantities change in relation to breastfeeding or formula diet. A recent study compared the gut microbiota samples of breastfed vs. formula-fed infants born by cesarean section (86). Breastfed neonates revealed a reduction in proliferative T-cell responses to non-inherited maternal antigens (NIMA) that was specific and Treg-dependent, as well as a decrease in inflammatory cytokine production (86). However, the fecal metabolome is thought to represent a functional output of the microbiome (99). Nevertheless, some of the metabolites will be shared by the gut microbiome and the host since feces include a combined metabolite output of both (62). However, because a large spectrum of metabolites is shared by the human host and intestinal microbes, metabolomics approaches are unable to differentiate them (100).
Although metabolomics is an excellent tool for investigating the role of bacteria in a variety of pathologies, including human intestinal disorders, and neurodegenerative diseases, it is limited by the inability to distinguish between host and bacterial metabolites (64), the complexity in associating the relative phylogenetic origin, and the lack of adequate reference databases (69, 70). Therefore, selecting suitable analytical methods and pipelines (equipment selection, sample processing, and statistical analysis) is a critical step in allowing relevant biological interpretation (64, 69, 70). Due to the large number and variety of metabolites discovered in a stool sample, as well as the heterogeneity of existing databases, data analysis may need manual integration of different databases by the user (100), which is a time-consuming and difficult phase that involves a significant chance of user-related mistakes.
Most microbiome research has focused on the gut, leading to the development of technologies that are better suited to samples with high microbial biomass (101). Samples from other body sites or, in comparison to the gut, have lower microbial biomass and are technically more difficult to explore. The main challenge with these samples is the amount of DNA, which comes from the environment. Microbial contamination has been presented as a real problem due to a lack of vigilance and understanding of technical problems while working with low microbial biomass samples (101–103). The publication of placental microbiota is an excellent illustration of this (101). This resulted in a false-positive finding, and unfortunately, other researchers have followed the same path.
Microbiome analysis has previously had problems due to a lack of controls. Initial research on the placental microbiota, for example, aimed to solve many issues about baby development and premature birth during pregnancy (13). Aagaard et al., on the other hand, did not take the essential procedures to monitor and reduce contamination. During the extractions, non-template controls were used, but only a fraction was sequenced. The sequences obtained in the negative controls were not compared to those found in the biological samples in any way. Furthermore, no environmental controls were collected or examined, preventing environmental contaminants from being detected.
Finally, the detection limit was not specified, prohibiting the researchers from determining whether a credible signal could be identified. Contaminant microbial species could not be reliably identified or analyzed in placenta samples, making it impossible to verify whether the placental microbial signature was genuinely endogenous. Lauder et al. (23) discovered that the microbial placenta profiles were similar to those of extraction blank control (EBC) and air samples, indicating that the placenta was likely sterile and lacked a varied microbial signature. Many other well-controlled investigations have now shown the absence of a placental microbiome (19–21). These studies emphasize the necessity of having solid processes and controls in place to avoid biased results.
As awareness of the aforementioned challenges of low microbial biomass sample examination, more researchers are now including controls in their data analysis. Nevertheless, this practice is still not standardized across the board, and researchers urgently need to learn from these past errors to improve microbiome research in the future, in particular for newborns. More researchers are increasingly integrating controls from sampling to data analysis as knowledge of the limitations of low microbial biomass sample inspection has increased. Nonetheless, this method is not uniform across the board, and researchers must learn from previous mistakes if they are to enhance microbiome research in the future.
Common microbiome procedures are not optimal for low microbial biomass samples, even in today's common microbiome research. Low biomass laboratory and analytical techniques have improved as a result of recent research, but there is always a potential for improvement (103, 104). To prevent contamination and biases, greater knowledge of when and how they arise is needed. DNA contamination and biases can occur at any step during the sample preparation and analysis process. Therefore, it is recommended to include controls from the samples of the professionals who collect and process samples, the collection room, the laboratory environment, equipment, and reagents. Moreover, all sample collectors and laboratory technicians should wear clothes that cover exposed skin, such as gloves and face masks (102, 103). Additionally, ultraviolet radiation can be used to minimize reagent and equipment contamination (101). It is also recommended that low biomass samples should be sequenced at a higher depth to capture a sufficient number of unique sequences (105).
Several bioinformatic approaches have been developed to monitor and remove contaminated DNA that affects samples with low microbial biomass. First, as previously mentioned, the limit of detection can be achieved using positive and negative controls (101–103). Individual contaminated species may also be traced using their specific sequence from their source (i.e., equipment, reagents, environmental controls, etc.) and then eliminated or identified using publicly accessible software such as SourceTracker and Decontam (101, 106). While programs can be used to eliminate or track contaminants, they do not replace the need to monitor and investigate contaminants throughout the sampling and laboratory processes.
As the number of investigations into neonatal microbiome samples grows, it is critical that new protocols and methods are developed to reduce the impact of contamination and biases on these samples, as well as to fully understand the limitations when developing diagnostic tests based on the findings (101–103). To reduce some of these difficulties and better detect disease causes and consequences, new methods based on sensitive, high-throughput techniques that explore undiscovered microbes and microbial populations as a whole are necessary.
With the advent of metagenomics, we can now define the structure and function of the microbiome community at the pre- and post-delivery stages with high precision. Due to the metagenomics' potential, dogmas such as the “sterile womb” hypothesis are being challenged by the discovery of microbes in previously sterile tissues. However, if these bacteria are rucked as real occurrences and not just experimental artifacts/contaminants, it is unknown if they colonize the embryo or are only present for the purpose of priming the fetal immune system.
Our review of maternal and environmental factors influencing intestinal microbiota highlights the need to focus on these aspects during pregnancy. The gut microbiome plays a significant role in human growth and development, as well as in the establishment of immunological responses, indicating that balanced gut microbiota is essential for good health. Emerging data suggesting early-life gut microbiome establishment as a protective factor against gut dysbiosis–related diseases later in life supports the need for targeted therapies to restore the gut microbiome in early-life.
To keep up with the latest sequencing technology and Metagenomic methods, new bioinformatics tools are constantly being developed and updated. The most difficult issue for the user is not being confused on offered alternatives. Indeed, comparing the outcomes obtained through several Bioinformatic tools remains the best strategy. Furthermore, using frequent comparison studies and reviews to help the user is beneficial and should be read and well analyzed before making a final decision on which tool to use. Therefore, to understand the neonatal microbiome and the newborn-health link, it is necessary to choose efficient computational tools and methodologies to analyze the neonatal microbiome.
Even though microbiological techniques have advanced in the last years, certain aspects still need to be enhanced such as reducing contamination, which is a critical issue in microbiome studies, particularly for samples with low microbial mass, as well as the differences in sample collection, storage, DNA extraction protocols, sequencing methods, and bioinformatics tools, which could all be responsible for introducing biases into the results and variability between microbiome studies. Additionally, other sections of the infant gut microbiota composed of fungi and viruses require additional investigation. Furthermore, using fecal samples to study neonatal gut microbiota has limitations. It is not always representative of the gut microbiome and leaves out gut-adherent bacteria that affect colon epithelial physiology and functions.
Although dysbiosis has been associated with several diseases, such as inflammatory diseases, atopic diseases, and NEC, other conditions have also been linked to disruptions in the microbiome. Nevertheless, in most cases causal correlations have yet to be demonstrated. Moreover, is there a critical period of microbiome development that, if disturbed, leads to a disease state in cases where illness states have been connected to microbiome alterations? Are there some microorganisms that can protect you from diseases? What human host variables and/or environmental interactions are crucial for the development of a healthy newborn microbiome? Scientists working to determine the likely microbial etiologies of infant diseases must address these and other problems. By obtaining these responses, potential protective and therapeutic strategies can be created to modulate initial microbial colonization and decrease the risk of adverse health outcomes associated with unbalanced microbiota evolution.
HG, ZB, and ND contributed to conceptualization and writing. SS, SE, and NA contributed to writing. SH, HB, NA, ML-P, SA, and CN reviewed the manuscript. All authors read and approved the submitted version.
The Article Processing Charge (APC) paid by Mohammed VI University of Health Sciences (UM6SS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. (2010) 464:59–65. doi: 10.1038/nature08821
2. Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J. (2017) 474:1823–36. doi: 10.1042/BCJ20160510
3. Pickard JM, Zeng MY, Caruso R, Núñez G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. (2017) 279:70–89. doi: 10.1111/imr.12567
4. Kim H, Sitarik AR, Woodcroft K, Johnson CC, Zoratti E. Birth mode, breastfeeding, pet exposure, and antibiotic use: associations with the gut microbiome and sensitization in children. Curr Aller Asthma Rep. (2019) 19:1–9. doi: 10.1007/s11882-019-0851-9
5. Francino MP. Birth mode-related differences in gut microbiota colonization and immune system development. Ann Nutr Metabol. (2018) 73:12–6. doi: 10.1159/000490842
6. Young VB. The intestinal microbiota in health and disease. Curr Opin Gastroenterol. (2012) 28:63. doi: 10.1097/MOG.0b013e32834d61e9
7. Turroni F, Milani C, Duranti S, Lugli GA, Bernasconi S, Margolles A, et al. The infant gut microbiome as a microbial organ influencing host well-being. Ital J Pediatr. (2020) 46:1–3. doi: 10.1186/s13052-020-0781-0
8. D'Argenio V. Human microbiome acquisition and bioinformatic challenges in metagenomic studies. Int J Mol Sci. (2018) 19:383. doi: 10.3390/ijms19020383
9. Milani C, Duranti S, Bottacini F, Casey E, Turroni F, Mahony J, et al. The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol Molec Biol Rev. (2017) 81:e00036–17. doi: 10.1128/MMBR.00036-17
10. Wang Q, Wang K, Wu W, Giannoulatou E, Ho JWK Li L. Host and microbiome multiomics integration: applications and methodologies. Biophys Rev. (2019) 11:55–65. doi: 10.1007/s12551-018-0491-7
11. Abellan-Schneyder I, Matchado MS, Reitmeier S, Sommer A, Sewald Z, Baumbach J, et al. Primer, pipelines, parameters: issues in 16S rRNA gene sequencing. MSphere. (2021) 6:e01202–20. doi: 10.1128/mSphere.01202-20
12. Perez-Muñoz ME, Arrieta MC, Ramer-Tait AE, Walter J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome. (2017) 5:1–9. doi: 10.1186/s40168-017-0268-4
13. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Translat Med. (2014) 6:237ra65. doi: 10.1126/scitranslmed.3008599
14. He Q, Kwok LY Xi X, Zhong Z, Ma T, Xu H, et al. The meconium microbiota shares more features with the amniotic fluid microbiota than the maternal fecal and vaginal microbiota. Gut Microbes. (2020) 12:1794266. doi: 10.1080/19490976.2020.1794266
15. Cabrera-Rubio R, Mira-Pascual L, Mira A, Collado MC. Impact of mode of delivery on the milk microbiota composition of healthy women. J Dev Orig Health Dis. (2016) 7:54–60. doi: 10.1017/S2040174415001397
16. Romero R, Miranda J, Chaiworapongsa T, Chaemsaithong P, Gotsch F, Dong Z, et al. Sterile and microbial-associated intra-amniotic inflammation in preterm prelabor rupture of membranes. J Maternal-Fetal Neonatal Med. (2015) 28:1394–409. doi: 10.3109/14767058.2014.958463
17. Wang X, Buhimschi CS, Temoin S, Bhandari V, Han YW, Buhimschi IA. Comparative microbial analysis of paired amniotic fluid and cord blood from pregnancies complicated by preterm birth and early-onset neonatal sepsis. PLoS ONE. (2013) 8:e56131. doi: 10.1371/journal.pone.0056131
18. Mesa MD, Loureiro B, Iglesia I, Fernandez Gonzalez S, Llurba Olivé E, et al. The evolving microbiome from pregnancy to early infancy: a comprehensive review. Nutrients. (2020) 12:133. doi: 10.3390/nu12010133
19. Olomu IN, Pena-Cortes LC, Long RA, Vyas A, Krichevskiy O, Luellwitz R, et al. Elimination of “kitome” and “splashome” contamination results in lack of detection of a unique placental microbiome. BMC Microbiol. (2020) 20:1–9. doi: 10.1186/s12866-020-01839-y
20. Rackaityte E, Halkias J, Fukui EM, Mendoza VF, Hayzelden C, Crawford ED, et al. Viable bacterial colonization is highly limited in the human intestine in utero. Nat Med. (2020) 26:599–607. doi: 10.1038/s41591-020-0761-3
21. Kuperman AA, Zimmerman A, Hamadia S, Ziv O, Gurevich V, Fichtman B, et al. Deep microbial analysis of multiple placentas shows no evidence for a placental microbiome. Int J Obstetr Gynaecol. (2020) 127:159–69. doi: 10.1111/1471-0528.15896
22. Stinson LF, Keelan JA, Payne MS. Identification and removal of contaminating microbial DNA from PCR reagents: impact on low-biomass microbiome analyses. Lett Appl Microbiol. (2019) 68:2–8. doi: 10.1111/lam.13091
23. Lauder AP, Roche AM, Sherrill-Mix S, Bailey A, Laughlin AL, Bittinger K, et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome. (2016) 4:1–1. doi: 10.1186/s40168-016-0172-3
24. Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ. (2018) 361:36–34. doi: 10.1136/bmj.k2179
25. Fujimura KE, Slusher NA, Cabana MD, Lynch SV. Role of the gut microbiota in defining human health. Expert Rev Anti Infect Ther. (2010) 8:435–54. doi: 10.1586/eri.10.14
26. Flint HJ, cott KP, Louis P, Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. (2012) 9:577–89. doi: 10.1038/nrgastro.2012.156
27. Moossavi S, Sepehri S, Robertson B, Bode L, Goruk S, Field CJ, et al. Composition and variation of the human milk microbiota are influenced by maternal and early-life factors. Cell Host Microbe. (2019) 25:324–235. doi: 10.1016/j.chom.2019.01.011
28. Walker WA. The importance of appropriate initial bacterial colonization of the intestine in newborn, child, and adult health. Pediatr Res. (2017) 82:387–95. doi: 10.1038/pr.2017.111
29. Wang S, Ryan CA, Boyaval P, Dempsey EM, Ross RP, Stanton C. Maternal vertical transmission affecting early-life microbiota development. Trends Microbiol. (2020) 28:28–45. doi: 10.1016/j.tim.2019.07.010
30. Zimmermann P, Curtis N. Factors influencing the intestinal microbiome during the first year of life. Pediatr Infect Dis J. (2018) 37:e315–e335. doi: 10.1097/INF.0000000000002103
31. Derrien M, Alvarez AS, de Vos WM. The gut microbiota in the first decade of life. Trends Microbiol. (2019) 27:997–1010. doi: 10.1016/j.tim.2019.08.001
32. Tanaka M, Nakayama J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol Int. (2017) 66:515–22. doi: 10.1016/j.alit.2017.07.010
33. Mitchell C, Hogstrom L, Bryant A, Bergerat A, Cher A, Pochan S, et al. Delivery mode impacts newborn gut colonization efficiency. bioRxiv. (2020). doi: 10.1101/2020.01.29.919993
34. Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, Aagaard KM. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med. (2017) 23:314–26. doi: 10.1038/nm.4272
35. Princisval L, Rebelo F, Williams BL, Coimbra AC, Crovesy L, Ferreira AL, et al. Association between the mode of delivery and infant gut microbiota composition up to 6 months of age: a systematic literature review considering the role of breastfeeding. Nutr Rev. (2021) 80:113–27. doi: 10.1093/nutrit/nuab008
36. Lee E, Kim BJ, Kang MJ, Choi KY, Cho HJ, Kim Y, et al. Dynamics of gut microbiota according to the delivery mode in healthy Korean infants. Allergy Asthma Immunol Res. (2016) 8:471–7. doi: 10.4168/aair.2016.8.5.471
37. Cunnington AJ, Sim K, Deierl A, Kroll JS, Brannigan E, Darby J. “Vaginal seeding” of infants born by caesarean section. BMJ. (2016) 3352:i227. doi: 10.1136/bmj.i227
38. Sanidad KZ, Zeng MY. Neonatal gut microbiome and immunity. Curr Opin Microbiol. (2020) 56:30–7. doi: 10.1016/j.mib.2020.05.011
39. Tirone C, Pezza L, Paladini A, Tana M, Aurilia C, Lio A, et al. Gut and lung microbiota in preterm infants: immunological modulation and implication in neonatal outcomes. Front Immunol. (2019) 10:2910. doi: 10.3389/fimmu.2019.02910
40. Tauchi H, Yahagi K, Yamauchi T, Hara T, Yamaoka R, Tsukuda N, et al. Gut microbiota development of preterm infants hospitalised in intensive care units. Benef Microbes. (2019) 10:641–51. doi: 10.3920/BM2019.0003
41. Arboleya S, Binetti A, Salazar N, Fernández N, Solís G, Hernandez-Barranco A, et al. Establishment and development of intestinal microbiota in preterm neonates. FEMS Microbiol Ecol. (2012) 79:763–72. doi: 10.1111/j.1574-6941.2011.01261.x
42. Henderickx JGE, Zwittink RD, van Lingen RA, Knol J, Belzer C. The preterm gut microbiota: an inconspicuous challenge in nutritional neonatal care. Front Cell Infect Microbiol. (2019) 9:85. doi: 10.3389/fcimb.2019.00085
43. Dalby MJ, Hall LJ. Recent advances in understanding the neonatal microbiome. F1000Research. (2020) 9:422. doi: 10.12688/f1000research.22355.1
44. Leggett RM, Alcon-Giner C, Heavens D, Caim S, Brook TC, Kujawska M, et al. Rapid MinION profiling of preterm microbiota and antimicrobial-resistant pathogens. Nat Microbiol. (2020) 5:430–42. doi: 10.1038/s41564-019-0626-z
45. Khodayar-Pardo P, Mira-Pascual L, Collado MC, Martínez-Costa C. Impact of lactation stage, gestational age and mode of delivery on breast milk microbiota. J Perinatol. (2014) 34:599–605. doi: 10.1038/jp.2014.47
46. Schwartz DJ, Langdon AE, Dantas G. Understanding the impact of antibiotic perturbation on the human microbiome. Genome Med. (2020) 12:1–2. doi: 10.1186/s13073-020-00782-x
47. Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H, et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Translat Med. (2016) 8:343ra82. doi: 10.1126/scitranslmed.aad7121
48. Tamburini S, Shen N, Wu HC, Clemente JC. The microbiome in early life: implications for health outcomes. Nat Med. (2016) 22:713–22. doi: 10.1038/nm.4142
49. Stiemsma LT, Michels KB. The role of the microbiome in the developmental origins of health and disease. Pediatrics. (2018) 141:e20172437. doi: 10.1542/peds.2017-2437
50. Seelbach-Goebel B. Antibiotic therapy for premature rupture of membranes and preterm labor and effect on fetal outcome. Geburtshilfe Frauenheilkunde. (2013) 73:1218–27. doi: 10.1055/s-0033-1360195
51. Fernández L, Langa S, Martín V, Maldonado A, Jiménez E, Martín R, et al. The human milk microbiota: origin and potential roles in health and disease. Pharmacol Res. (2013) 69:1–10. doi: 10.1016/j.phrs.2012.09.001
52. Vega-Bautista A, de la Garza M, Carrero JC, Campos-Rodríguez R, Godínez-Victoria M, Drago-Serrano ME. The impact of lactoferrin on the growth of intestinal inhabitant Bacteria. Int J Molec Sci. (2019) 20:4707. doi: 10.3390/ijms20194707
53. Quigley M, Embleton ND, McGuire W. Formula versus donor breast milk for feeding preterm or low birth weight infants. Cochrane Datab System Rev. (2018) 6:CD002971. doi: 10.1002/14651858.CD002971.pub4
54. Boix-Amorós A, Collado MC, Mira A. Relationship between milk microbiota, bacterial load, macronutrients, and human cells during lactation. Front Microbiol. (2016) 7:492. doi: 10.3389/fmicb.2016.00492
55. Gopalakrishna KP, Hand TW. Influence of maternal milk on the neonatal intestinal microbiome. Nutrients. (2020) 12:823. doi: 10.3390/nu12030823
56. Mira A, Rodríguez JM. The origin of human milk bacteria. In: McGuire M, McGuire MA, Bode L, editors. Prebiotics and Probiotics in Human Milk: Origins and Functions of Milk-Borne Oligosaccharides and Bacteria. Cambridge, MA: Academic Press (2017) p. 349–64. doi: 10.1016/B978-0-12-802725-7.00013-0
57. Rodríguez JM. The origin of human milk bacteria: is there a bacterial entero-mammary pathway during late pregnancy and lactation? Adv Nutr. (2014) 5:779–84. doi: 10.3945/an.114.007229
58. Wang Z. Comparing Gut Microbiome Virome in the Breast Milk-and Formula-fed Late Preterm Infants. Electronic Theses Dissertations (2019). p. 3375. Available online at: https://openprairie.sdstate.edu/etd/3375 (accessed October 18, 2021).
59. Gurnani M, Birken C, Hamilton J. Childhood obesity: causes, consequences, and management. Pediatr Clin. (2015) 62:821–40. doi: 10.1016/j.pcl.2015.04.001
60. Xu J, Lawley B, Wong G, Otal A, Chen L, Ying TJ, et al. Ethnic diversity in infant gut microbiota is apparent before the introduction of complementary diets. Gut Microbes. (2020) 11:1362–73. doi: 10.1080/19490976.2020.1756150
61. Zhou S, Xu R, He F, Zhou J, Wang Y, Zhou J, et al. Diversity of Gut Microbiota Metabolic Pathways in 10 Pairs of Chinese Infant Twins. PLoS ONE. (2016) 11:e0161627. doi: 10.1371/journal.pone.0161627
62. Daliri EB, Ofosu FK, Chelliah R, Lee BH, Oh DH. Challenges and Perspective in Integrated Multi-Omics in Gut Microbiota Studies. Biomolecules. (2021) 11:300. doi: 10.3390/biom11020300
63. Vernocchi P, Del Chierico F, Putignani L. Gut microbiota profiling: metabolomics based approach to unravel compounds affecting human health. Front Microbiol. (2016) 7:1144. doi: 10.3389/fmicb.2016.01144
64. Beck LC, Granger CL, Masi AC, Stewart CJ. Use of omic technologies in early life gastrointestinal health and disease: from bench to bedside. Expert Rev Proteomics. (2021) 18:247–59. doi: 10.1080/14789450.2021.1922278
65. Zhang X, Li L, Butcher J, Stintzi A, Figeys D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome. (2019) 7:1–2. doi: 10.1186/s40168-019-0767-6
66. Peters DL, Wang W, Zhang X, Ning Z, Mayne J, Figeys D. Metaproteomic and Metabolomic Approaches for Characterizing the Gut Microbiome. Proteomics. (2019) 19:e1800363. doi: 10.1002/pmic.201800363
67. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME2. Nat Biotechnol. (2019) 37:852–7. doi: 10.1038/s41587-019-0209-9
68. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. (2009) 75:7537–41. doi: 10.1128/AEM.01541-09
69. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. (2016) 13:581–3. doi: 10.1038/nmeth.3869
70. Eren AM, Morrison HG, Lescault PJ, Reveillaud J, Vineis JH, Sogin ML. Minimum entropy decomposition: unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. ISME J. (2015) 9:968–79. doi: 10.1038/ismej.2014.195
71. Edgar RC. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv. (2016) 081257. doi: 10.1101/081257
72. Moossavi S, Atakora F, Fehr K, Khafipour E. Biological observations in microbiota analysis are robust to the choice of 16S rRNA gene sequencing processing algorithm: case study on human milk microbiota. BMC Microbiol. (2020) 20:1–9. doi: 10.1186/s12866-020-01949-7
73. Bharti R, Grimm DG. Current challenges and best-practice protocols for microbiome analysis. Brief Bioinform. (2021) 22:178–93. doi: 10.1093/bib/bbz155
74. Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. (2017) 11:2639–43. doi: 10.1038/ismej.2017.119
75. Pérez-Cobas AE, Gomez-Valero L, Buchrieser C. Metagenomic approaches in microbial ecology: an update on whole-genome and marker gene sequencing analyses. Microbial Genom. (2020) 6:8. doi: 10.1099/mgen.0.000409
76. Prodan A, Tremaroli V, Brolin H, Zwinderman AH, Nieuwdorp M, Levin E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE. (2020) 15:e0227434. doi: 10.1371/journal.pone.0227434
77. Marizzoni M, Gurry T, Provasi S, Greub G, Lopizzo N, Ribaldi F, et al. Comparison of bioinformatics pipelines and operating systems for the analyses of 16S rRNA gene amplicon sequences in human fecal samples. Front Microbiol. (2020) 11:1262. doi: 10.3389/fmicb.2020.01262
78. Matias Rodrigues JF, Schmidt TSB, Tackmann J, von Mering C. MAPseq: highly efficient k-mer search with confidence estimates, for rRNA sequence analysis. Bioinformatics. (2017) 33:3808–10. doi: 10.1093/bioinformatics/btx517
79. Balvočiute M, Huson DH. SILVA, RDP, Greengenes, NCBI and OTT - how do these taxonomies compare? BMC Genomics. (2017) 18:1–8. doi: 10.1186/s12864-017-3501-4
80. Kennedy KM, Gerlach MJ, Adam T, Heimesaat MM, Rossi L, Surette MG, et al. Fetal meconium does not have a detectable microbiota before birth. Nature Microbiology. (2021) 6:1–9. doi: 10.1101/2021.02.17.431710
81. Li N, Liang S, Chen Q, Zhao L, Li B, Huo G. Distinct gut microbiota and metabolite profiles induced by delivery mode in healthy Chinese infants. J Proteomics. (2021) 232:104071. doi: 10.1016/j.jprot.2020.104071
82. Dinleyici M, Pérez-Brocal V, Arslanoglu S, Aydemir O, Sevuk Ozumut S, Tekin N, et al. Human milk virome analysis: changing pattern regarding mode of delivery, birth weight, and lactational stage. Nutrients. (2021) 13:1779. doi: 10.3390/nu13061779
83. Toivonen L, Schuez-Havupalo L, Karppinen S, Waris M, Hoffman KL, Camargo CA, et al. Antibiotic treatments during infancy, changes in nasal microbiota, and asthma development: population-based cohort study. Clin Infect Dis. (2021) 72:1546–54. doi: 10.1093/cid/ciaa262
84. Gong C, Yang L, Liu K, Shen S, Zhang Q, Li H, et al. Effects of Antibiotic Treatment and Probiotics on the Gut Microbiome of 40 Infants Delivered Before Term by Cesarean Section Analysed by Using 16S rRNA Quantitative Polymerase Chain Reaction Sequencing. Int Med J Exper Clin Res. (2021) 27:e928467–1. doi: 10.12659/MSM.928467
85. Selma-Royo M, García-Mantrana I, Calatayud M, Parra-Llorca A, Martínez-Costa C, Collado MC. Maternal diet during pregnancy and intestinal markers are associated with early gut microbiota. J Nutr. (2021) 60:1429–42. doi: 10.1007/s00394-020-02337-7
86. Wood H, Acharjee A, Pearce H, Quraishi MN, Powell R, Rossiter A, et al. Breastfeeding promotes early neonatal regulatory T-cell expansion and immune tolerance of non-inherited maternal antigens. Allergy. (2021) 76:2447–60. doi: 10.1111/all.14736
87. Chin N, Méndez-Lagares G, Taft DH, Laleau V, Kieu H, Narayan NR, et al. Transient effect of infant formula supplementation on the intestinal microbiota. Nutrients. (2021) 13:807. doi: 10.3390/nu13030807
88. Cortes-Macías E, Selma-Royo M, García-Mantrana I, Calatayud M, González S, Martínez-Costa C, et al. Maternal diet shapes the breast milk microbiota composition and diversity: impact of mode of delivery and antibiotics exposure. J Nutr. (2021) 151:330–40. doi: 10.1093/jn/nxaa310
89. Mueller NT, Differding MK, Østbye T, Hoyo C, Benjamin-Neelon SE. Association of birth mode of delivery with infant faecal microbiota, potential pathobionts, and short chain fatty acids: a longitudinal study over the first year of life. Int J Obstetr Gynaecol. (2021) 128:1293–303. doi: 10.1111/1471-0528.16633
90. Morais J, Marques C, Faria A, Teixeira D, Barreiros-Mota I, Durão C, et al. Influence of human milk on very preterms' gut microbiota and alkaline phosphatase activity. Nutrients. (2021) 13:1564. doi: 10.3390/nu13051564
91. Pan K, Zhang C, Tian J. The effects of different modes of delivery on the structure and predicted function of intestinal microbiota in neonates and early infants. Polish J Microbiol. (2021) 70:45–55. doi: 10.33073/pjm-2021-002
92. Fan HY, Tung YT, Yang YC, Hsu JB, Lee CY, Chang TH, et al. Maternal vegetable and fruit consumption during pregnancy and its effects on infant gut microbiome. Nutrients. (2021) 13:1559. doi: 10.3390/nu13051559
93. Marrs T, Jo JH, Perkin MR, Rivett DW, Witney AA, Bruce KD, et al. Gut microbiota development during infancy: Impact of introducing allergenic foods. J Allergy Clin Immunol. (2021) 147:613–2. doi: 10.1016/j.jaci.2020.09.042
94. Didelot X, Walker AS, Peto TE, Crook DW, Wilson DJ. Within-host evolution of bacterial pathogens. Nat Rev Microbiol. (2016) 14:150–62. doi: 10.1038/nrmicro.2015.13
95. Sharpton TJ. An introduction to the analysis of shotgun metagenomic data. Front Plant Sci. (2014) 5:209. doi: 10.3389/fpls.2014.00209
96. Oulas A, Pavloudi C, Polymenakou P, Pavlopoulos GA, Papanikolaou N et al. Metagenomics: tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform Biol Insights. (2015) 9:BBI-S12462. doi: 10.4137/BBI.S12462
97. Segal JP, Mullish BH, Quraishi MN, Acharjee A, Williams HRT et al. The application of omics techniques to understand the role of the gut microbiota in inflammatory bowel disease. Therap Adv Gastroenterol. (2019) 12:1756284818822250. doi: 10.1177/1756284818822250
98. Tang ZZ, Chen G, Hong Q, Huang S, Smith HM, Shah RD, et al. Multi-omic analysis of the microbiome and metabolome in healthy subjects reveals microbiome-dependent relationships between diet and metabolites. Front Genet. (2019) 10:454. doi: 10.3389/fgene.2019.00454
99. Pettersen VK, Antunes LCM, Dufour A, Arrieta MC. Inferring early-life host and microbiome functions by mass spectrometry-based metaproteomics and metabolomics. Comput Struct Biotechnol J. (2021) 20:274–286. doi: 10.1016/j.csbj.2021.12.012
100. Cambiaghi A, Ferrario M, Masseroli M. Analysis of metabolomic data: tools, current strategies and future challenges for omics data integration. Briefings Bioinform. (2017) 18:498–510. doi: 10.1093/bib/bbw031
101. Selway CA, Eisenhofer R, Weyrich LS. Microbiome applications for pathology: challenges of low microbial biomass samples during diagnostic testing. J Pathol Clin Res. (2020) 6:97–106. doi: 10.1002/cjp2.151
102. Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. (2019) 27:105–17. doi: 10.1016/j.tim.2018.11.003
103. Minich JJ, Zhu Q, Janssen S, Hendrickson R, Amir A, Vetter R, et al. KatharoSeq Enables High-Throughput Microbiome Analysis from Low-Biomass Samples. mSystems. (2018) 3:e00218–17. doi: 10.1128/mSystems.00218-17
104. Zinter MS, Mayday MY, Ryckman KK, Jelliffe-Pawlowski LL, DeRisi JL. Towards precision quantification of contamination in metagenomic sequencing experiments. Microbiome. (2019) 7:1–5. doi: 10.1186/s40168-019-0678-6
105. Chafee M, Maignien L, Simmons SL. The effects of variable sample biomass on comparative metagenomics. Environ Microbiol. (2015) 17:2239–53. doi: 10.1111/1462-2920.12668
Keywords: neonatal microbiota, microbiome, metagenomics, dysbiosis, colonization, womb sterile, delivery, breastfeeding
Citation: Boudar Z, Sehli S, El Janahi S, Al Idrissi N, Hamdi S, Dini N, Brim H, Amzazi S, Nejjari C, Lloyd-Puryear M and Ghazal H (2022) Metagenomics Approaches to Investigate the Neonatal Gut Microbiome. Front. Pediatr. 10:886627. doi: 10.3389/fped.2022.886627
Received: 28 February 2022; Accepted: 23 May 2022;
Published: 21 June 2022.
Edited by:
Charles Christoph Roehr, University of Oxford, United KingdomReviewed by:
Alain Cuna, Children's Mercy Hospital, United StatesCopyright © 2022 Boudar, Sehli, El Janahi, Al Idrissi, Hamdi, Dini, Brim, Amzazi, Nejjari, Lloyd-Puryear and Ghazal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hassan Ghazal, aGFzc2FuLmdoYXphbEBmdWxicmlnaHRtYWlsLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.