 Patricia Arroyo-Parejo Drayer1
Patricia Arroyo-Parejo Drayer1 Wacharee Seeherunvong1*
Wacharee Seeherunvong1* Chryso P. Katsoufis1
Chryso P. Katsoufis1 Marissa J. DeFreitas1,2Tossaporn Seeherunvong3
Marissa J. DeFreitas1,2Tossaporn Seeherunvong3 Jayanthi Chandar1,2
Jayanthi Chandar1,2 Carolyn L. Abitbol1*
Carolyn L. Abitbol1*- 1Division of Pediatric Nephrology, Department of Pediatrics, Holtz Children's Hospital, University of Miami Miller School of Medicine, Miami, FL, United States
- 2Pediatric Renal Transplantation, Miami Transplant Institute, Jackson Health System, Miami, FL, United States
- 3Division of Pediatric Endocrinology, Department of Pediatrics, University of Miami Miller School of Medicine, Miami, FL, United States
Background: Mutations of the Wilms tumor suppressor-1 gene (WT1) are associated with life-threatening glomerulopathy, disorders of sexual development, Wilm's tumor, and gonadal malignancies. Our objectives were to describe the clinical presentations, age of progression, and onset of complications of WT1 mutation through a case series and literature review.
Methods: A retrospective study included all patients followed at the University of Miami/Holtz Children's Hospital from January 2000 to December 2020 with a diagnosis of WT1 mutation. A literature review of WT1 mutation cases was analyzed for clinical manifestations, karyotype, and long-term outcomes.
Results: The WT1 mutation was identified in 9 children, median age at presentation of 0.9 years (range 1 week to 7 years). A total of four had female phenotypes, and 5 had abnormalities of male external genitalia, while all had XY karyotypes. All progressed to end-stage kidney disease (ESKD) and received a kidney transplant at a median age of 5 years (1.5–15 years). During a median time of follow-up of 9 years (range 2–28 years), there were 2 allograft losses after 7 and 10 years and no evidence of post-transplant malignancy. From 333 cases identified from the literature review, the majority had female phenotype 66% (219/333), but the predominant karyotype was XY (55%, 183/333). Of the female phenotypes, 32% (69/219) had XY sex reversal. Wilm's tumor occurred in 24%, predominantly in males with gonadal anomalies.
Conclusions: Early recognition of WT1 mutation is essential for comprehensive surveillance of potential malignancy, avoidance of immunosuppressants for glomerulopathy, and establishing long-term multidisciplinary management.
Introduction
Wilms tumor suppressor gene-1 (WT1) mutations are associated with life-threatening nephropathy including congenital or/ infantile nephrotic syndrome (CNS) and/or childhood onset steroid-resistant nephrotic syndrome (SRNS) as well as a spectrum of disorders of sexual development (DSD), Wilm's tumor and gonadal malignancies. SRNS is one of the most common causes of end-stage kidney disease (ESKD) in children and adolescents with up to 25% of cases associated with a genetic abnormality preferentially expressed in the podocytes (1, 2). Traditional syndromic terminology includes the terms of Denys-Drash Syndrome, which represents early-onset nephrotic syndrome, Wilm's tumor, and male pseudo-hermaphroditism as well as Frasier Syndrome with late-onset SRNS and gonadal dysgenesis.
The WT1 gene is localized on chromosome 11p13 and encodes a transcriptional factor of the zinc-finger protein family. The advances in genetic diagnostic tests have increased the ability to identify genetic causes of childhood SRNS. The WT1 gene is a tumor suppressor gene necessary for the development of the kidney and gonads. In the fetal kidney, WT1 protein is highly expressed in the areas of active glomerulogenesis, which supports a major role of the gene in the development and maturation of the glomerular filtration barrier (2). Germline heterozygous WT1 mutations are known to manifest as the developmental genital abnormalities including 46XY sex reversal with complete female phenotype and various anomalies of the male genitalia such as cryptorchidism and hypogonadism (3, 4). In the recent years, the consensus has been to move away from these terms since the spectra of clinical and phenotypic diseases overlap (5). The risk of Wilm's tumor and gonadoblastoma in individuals with WT1 mutations is ominous and merits close surveillance for yet an undetermined period. Therefore, early recognition of WT1 mutation is critical for the early detection of neoplasia and counseling regarding sexual development and reproductive health. A number of international studies have described the incidence and prevalence of this rare disease and case reports, with comprehensive reviews on the clinical manifestations of WT1 gene mutations in the pediatric population (6–10).
Our objectives through this case series and contemporary literature review were to describe the clinical presentation, which includes extra-renal manifestations, age of diagnosis, progression, and complications of WT1 glomerulopathy. We also sought to develop a paradigm for earlier recognition and genetic diagnosis through the phenotypic correlations.
Methods
A retrospective chart review of patients followed at the University of Miami/Holtz Children's Hospital from January 2000 to December 2020 with a diagnosis of WT1 gene mutation was performed. Data collection that included age at presentation, sex, ethnicity, karyotype, initial presentations of kidney and extrarenal disease, age at diagnosis of WT1 mutation, at ESKD, and at transplantation as well as complications related to WT1 mutation that includes malignancy and gonadal disorders was recorded.
A comprehensive literature search and review for original articles and case reports with WT1 mutation and glomerulopathy published from 2000 to August 2021 was performed through PubMed. The core concept was to identify information on the spectrum of clinical manifestations and diagnosis of WT1 mutation. More specifically, sex, karyotype, and significant complications that include type of malignancy were extracted when available. The two authors independently performed the literature search using keywords: WT1 mutation, Denys-Drash, Frasier, genotype, CNS, SRNS, glomerulopathy, or nephropathy. The abstracts of all selected articles were reviewed, and references from retrieved articles were used to identify other relevant sources. To make data extraction accurate and consistent, the studies that did not specify clinical manifestations or karyotype were excluded. Only articles in the English language were reviewed.
This study was approved by the institutional review board of the University of Miami with the waiver of informed consent and protection of privacy in compliance with the Health Insurance Portability and Accountability Act (HIPAA).
Statistical Analyses
All data were analyzed with descriptive statistics with values reported as the median with range for non-parametric data. Normality was determined by the D'Agostino & Pearson test. Odds ratios for risk of malignancy were determined by Fisher's exact test with the 95% confidence intervals (95% CI) reported. Statistical analyses were performed using the GraphPad Prism version 9.12 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com. p-value < 0.05 was considered as statistically significant.
Results
Case Series of WT1 Glomerulopathy
Wilms tumor suppressor-1 gene mutation associated with glomerulopathy was identified in nine children (Table 1), of whom four were Hispanic, three Caucasian, one Asian, and one Middle Eastern. The median age at renal presentation was 0.9 years (range 1 week to 7 years). Of the 9 cases, 4 presented as CNS in the first year of life. Two phenotypic males presented with Wilms tumor at <2 years of age. The first (Patient #6) had proteinuria and hypertension early after birth and developed Wilm's tumor at 5 months of age. The second (Patient #8) developed Wilm's tumor at 2 years of age with proteinuria and CKD 3 years later. Two presented with SRNS at 3 and 5 years of age and had received multiple immunosuppressive treatments before diagnosis of WT1 mutation. Only one (Patient #4) presented with advanced kidney failure and hypertension at 7 years of age.
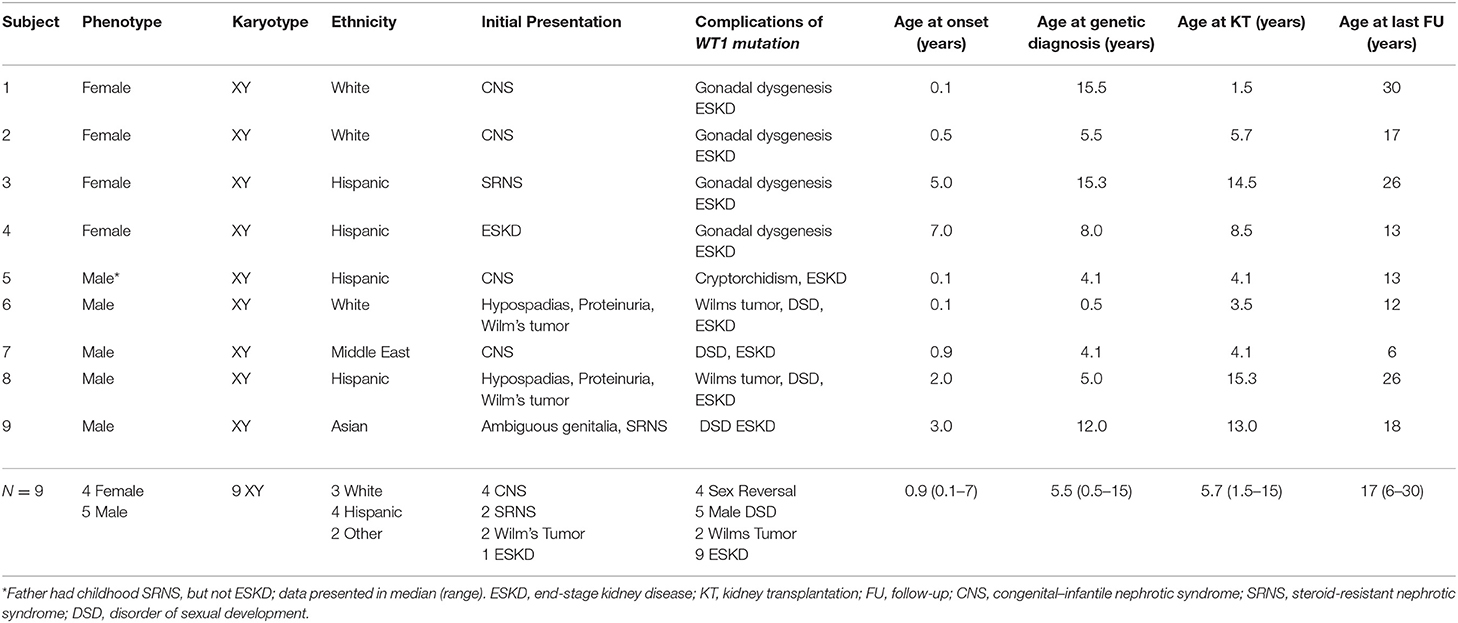
Table 1. Case series at university of Miami.
Of the nine subjects, all had 46XY karyotype, but only five had male phenotype with various degrees of abnormal male genital development with ambiguous genitalia (N = 1), cryptorchidism (N = 4), and hypospadias (N = 2). The other four subjects had complete sex reversal and presented as female phenotype. Among the four phenotypic females, two had delayed diagnosis of WT1 mutation at 15 years of age when XY karyotype was discovered as a component of the workup for delayed puberty. This was despite their presentation early in life with CNS and childhood onset SRNS, respectively. The other two phenotypic females had genetic diagnosis during the evaluation of SRNS with microarray analysis which revealed the XY karyotype at 2 and 7 years of age, respectively. All phenotypic female patients underwent prophylactic gonadectomy after diagnosis of WT1 mutation and have required long-term hormonal replacement. Each of the five phenotypic males has a disorder of sexual development (DSD), which ranges from hypospadias (2), cryptorchidism (2) to ambiguous genitalia with penoscrotal hypospadias, and bilateral cryptorchidism (1) requiring multiple urological surgeries. During the observation period, two males entered puberty, one had hypogonadism, and all continue to be monitored for gonadal tumor and sexual development and function. The median time from renal presentation to diagnosis of WT1 mutation was 7.6 years for the phenotypic female patients and 2.2 years for males. Although those of female phenotype tended to be older at genetic diagnosis, none of the age milestones comparing male vs. female phenotypes were significantly different in this small case series.
In our case series, eight of the nine subjects had bilateral nephrectomy before or at the time of transplantation. Those with CNS may have had bilateral nephrectomy because of heavy proteinuria. Those with known WT1 mutation likely had native nephrectomy at the time of transplantation as a prophylactic measure. There was no specific protocol regarding the recommendation for prophylactic nephrectomy. However, all phenotypic 46XY females underwent prophylactic gonadectomy after WT1 mutation was diagnosed while males with DSD did not.
All progressed to ESKD and received a kidney transplant at a median age of 5 years (range 1.5 to 15 years) with a median follow-up of 9 years (range 2–28 years). There were two allograft losses after 7 and 10 years and no evidence of post-transplant malignancy.
Results From Literature Review
Of 76 articles identified with keywords, 43 were excluded due to the lack of WT1 glomerulopathy cases, the lack of clinical data on sex/karyotype, age at presentation, or duplicated database (Figure 1). Thus, 33 articles were eligible for comprehensive review. A number of eight were cohort studies of subjects with WT1 glomerulopathy from various countries (6–8, 10–14) and 25 were from case series or case reports (15–39).
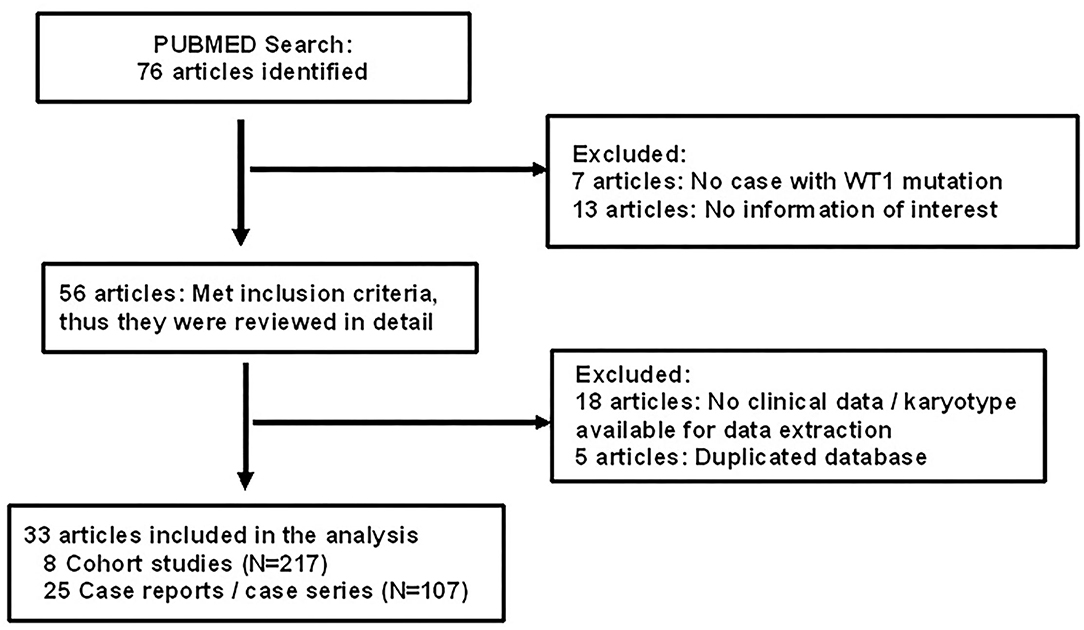
Figure 1. Flow chart showing identification and selection of cases having WT1 glomerulopathy with data on age, karyotype, and phenotype.
There were 324 subjects extracted from the literature review and 9 from our case series for a total of 333 cases of WT1 glomerulopathy. The details of phenotype/karyotype, age at presentation, renal and extrarenal presentations, and complications are summarized in Tables 2, 3.
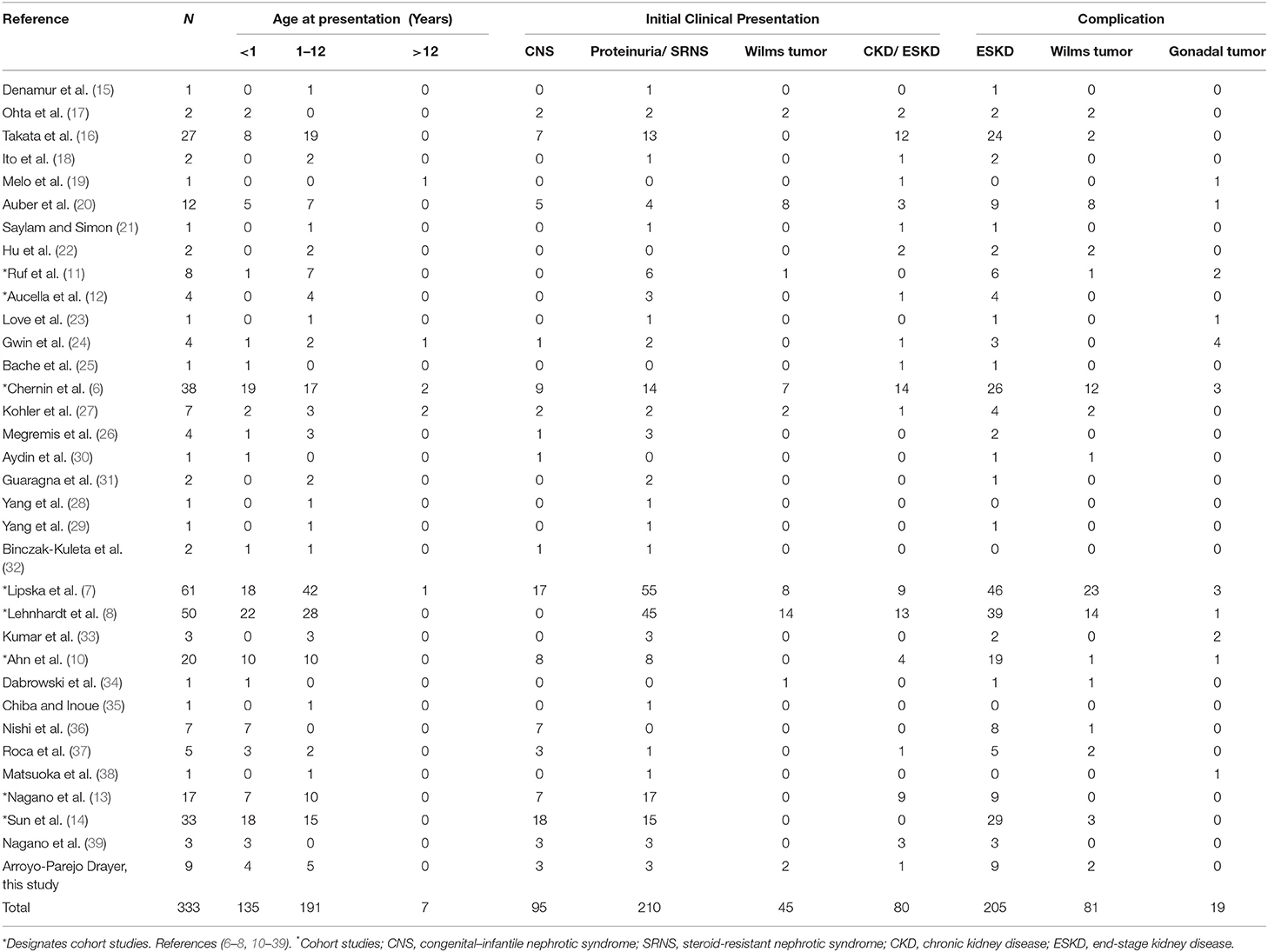
Table 2. Compiled data on age of onset, presentation, and complications in WT1 glomerulopathy.
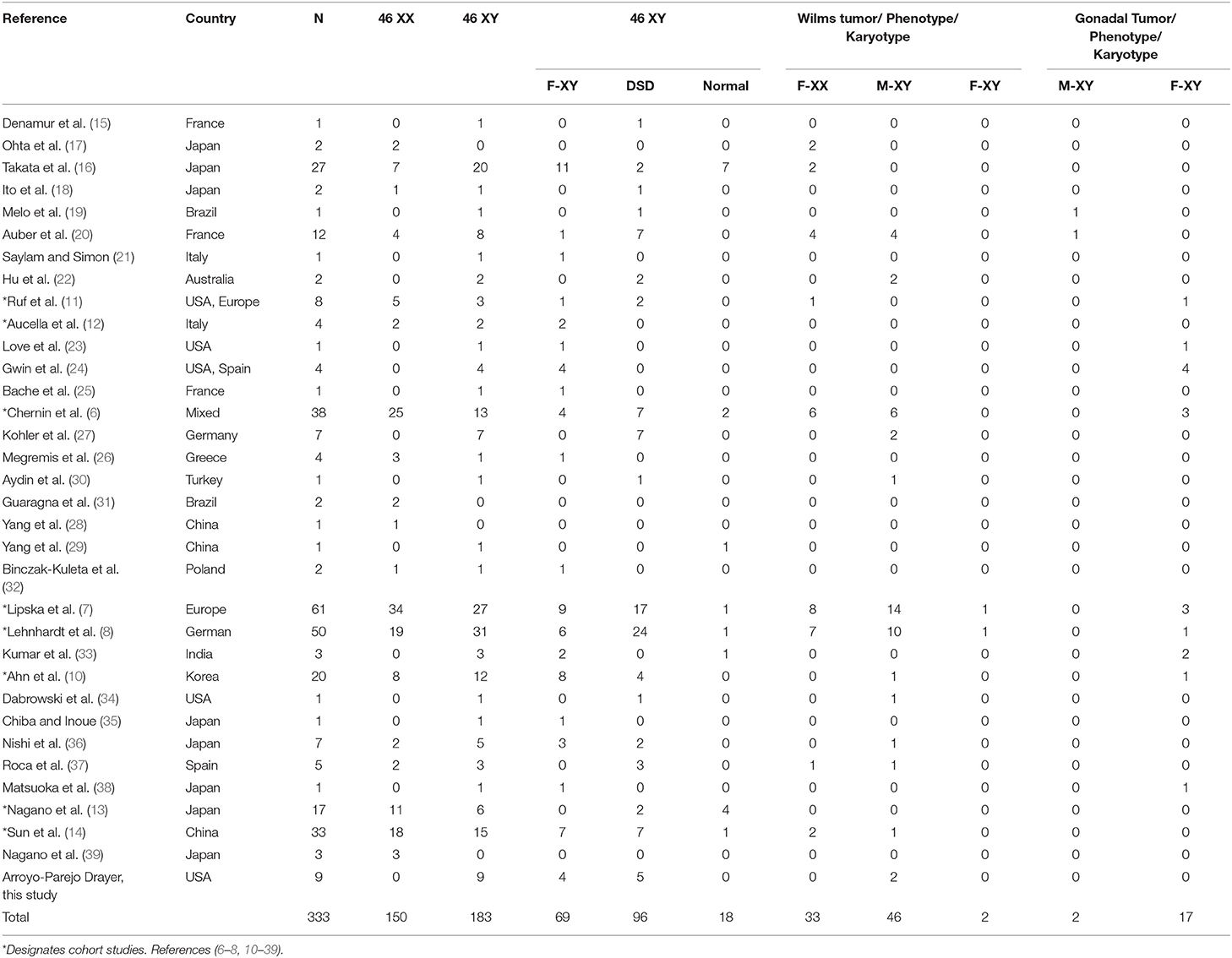
Table 3. Karyotype–phenotype and associated Wilms and gonadal tumors in WT1 glomerulopathy.
Phenotype–Karyotype and Genital Abnormalities
Figure 2 shows the phenotype–karyotype distribution of the 333 cases of the WT1 mutation compiled in the literature review and case series. Of the 333 subjects, the predominant karyotype was XY (N = 183; 55%). In contrast, the majority phenotype was female with 219/333 (66%) including the 69/219 (32%) having XY sex reversal. Of note, the male phenotypes were in the minority at 114/333 (34%). Of those with the male phenotype, 84% (96/114) had a DSD including hypospadias, cryptorchidism, and/or ambiguous genitalia.
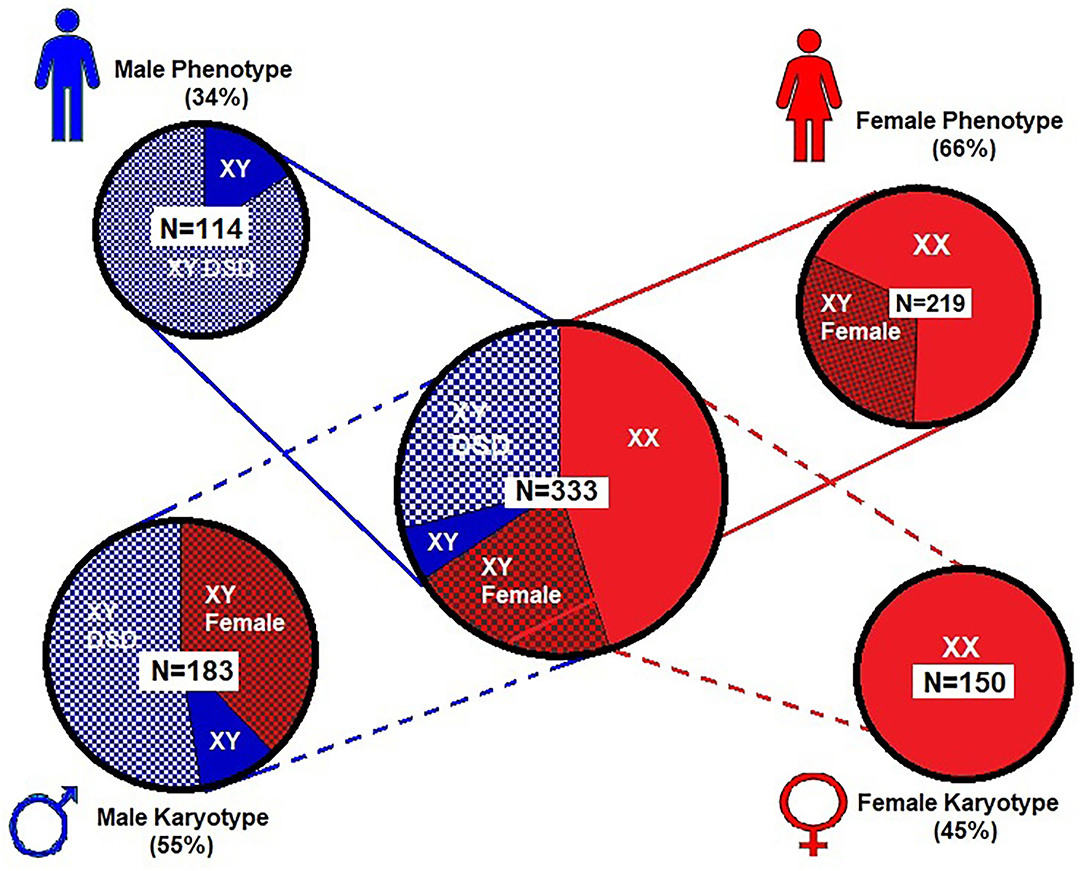
Figure 2. Proportional distribution of phenotype–karyotype for 333 subjects from literature review and our case series. Female (XX) karyotype is only 45% of the entire series while the male (XY) karyotype predominates (55%) and includes predominantly males with disorders of sexual development (DSD). However, the female phenotype was nearly two times that of the male phenotype and includes those with 46 XY sex reversal. For graphic clarity, the phenotype is represented by the male and female iconic figures in blue and red, respectively  . The karyotypes are graphically represented by the male and female gender icons in blue and red, respectively
. The karyotypes are graphically represented by the male and female gender icons in blue and red, respectively 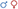 .
.
Renal Presentation and Progression to ESKD
Most cases of WT1 mutation (98%) had clinical presentation before 12 years of age. Renal presentation that includes CNS, SRNS, isolated proteinuria, chronic kidney disease (CKD), or ESKD occurred across all age groups. Up to 41% presented in the first year of life with CNS, proteinuria, ESKD, and/or Wilm's tumor. Males often received early recognition with abnormal genitalia at birth but subsequently developed proteinuria and/or Wilm's tumor. Females from 46 XY sex reversal were more likely to have late diagnosis due to delayed puberty.
Malignancy
The risk of malignancy relative to phenotype–karyotype was analyzed among the entire cohort. Of the 333 subjects, 29% (98/333) developed a malignancy which included 81 (24%) with Wilm's tumor and 98 (29%) with any tumor including Wilm's or gonadal tumor. Wilm's tumor usually presented early before 2 years, and rarely after 5 years of age.
Figure 3 and Table 4 demonstrate the proportion and odds ratios of subjects who developed malignancy relative to phenotype and karyotype. The male karyotype (XY) was predominant with 183/333 = 55% of the cohort. This included the females with 46 XY sex reversal who comprised 69/183 = 38% of the male karyotype. Those with male karyotype, regardless of phenotype (male-or-female XY), had an odds ratio for developing any malignancy including Wilm's or gonadal tumor of 1.95, 95% CI: 1.2→ 3.2; p < 0.01. The major risk of malignancy was with the male phenotype, the majority of whom (96/114 = 84%) had a DSD while only 18 had normal male genitalia (18/114 = 16%). None of the normal males developed a malignancy. Those with male karyotype had a higher but non-significant risk of developing Wilm's tumor as compared to the female karyotype. The XY karyotype odds ratio was 1.26 (95% CI: 0.8→ 2.1) while those with female karyotype had a reciprocal odds ratio of 0.79 (95% CI: 0.5→ 1.3; p = 0.44) for the development of Wilm's tumor which was not significant. Overall, those with male phenotype including largely those with DSD had an odds ratio of 3.56 (95% CI 2.1→ 5.9; p < 0.0001) of developing Wilm's tumor. Those with female phenotype including the 46 XY sex reversal females had a reciprocal odds ratio of 0.28 (95% CI 0.2→ 0.5; p < 0.0001) for developing Wilm's tumor. Gonadal tumors occurred only in XY males with DSD (n = 2) and 46 XY females with sex reversal (n = 17).
Figure 3. This composite depicts the risks of malignancy in WT1 mutation according to phenotype and karyotype. (A) Distribution of karyotype and Wilm's tumor and gonadal tumors in 333 cases of WT1 mutation. Note that the true male XY (n = 18) without DSD developed no malignancy. Only those with XY karyotype and gonadal dysgenesis (male XY-DSD and female-XY) developed gonadal tumors. (B) Risks by odds ratios for developing Wilm's tumor and/or gonadal tumor by karyotype and phenotype in 333 cases of WT1 mutation. For graphic clarity, the phenotype is represented by the male and female iconic figures in blue and red, respectively . The karyotypes are graphically represented by the male and female gender icons in blue and red, respectively .
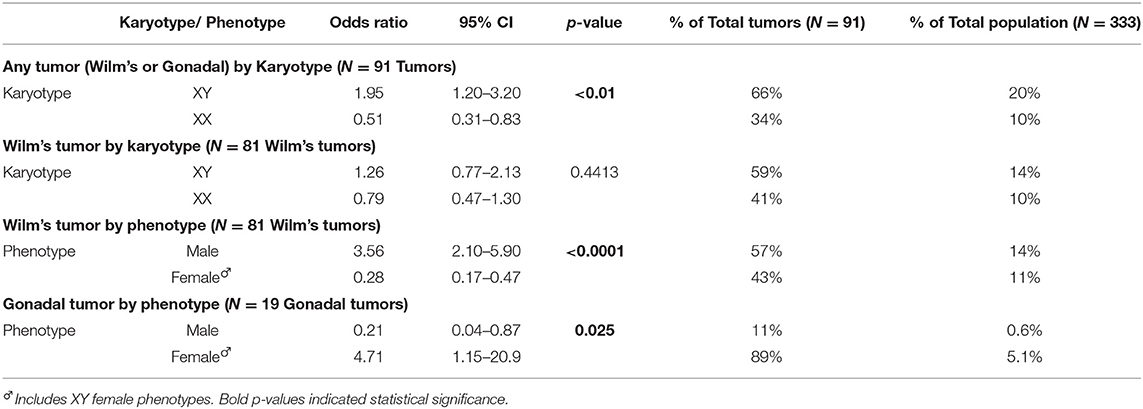
Table 4. Online: Odds ratios for risks of malignancy in WT1 mutation according to phenotype–karyotype in a case series of 333 subjects.
Discussion
With this case series and extensive literature review, we have compiled important clinical, sex, and karyotype characteristics and projected long-term outcomes of 333 cases with an estimated 500 cases reported worldwide since it was first described in 1991 (5, 40). Although considered an extremely rare disease (41), the broad spectrum of clinical manifestations has become more apparent, and it is suspected that many cases may go unrecognized (8, 27, 42, 43).
Proteinuria is the most frequent and pervasive manifestation of the WT1 mutation from birth through childhood to adulthood (5, 8, 42, 43). It occurred in 92% of the 333 cases at presentation in our current review, which is consistent with the previous reviews (8, 42, 43). The progression to ESKD is also synchronous with proteinuria. The concurrence of CNS with renal failure is a frequent clinical presentation in infancy, whereas SRNS with slower progression to ESKD occurs later in childhood.
In this case series and literature review, we were unable to refine the clinical phenotypes with the specific WT1 mutation analyses which have become more available in the recent years. These studies have been able to categorize the various mutations with broader clinicopathologic correlations (39). Over 30 mutations or missense variants of the WT1 protein have been analyzed, reported, and correlated with phenotypes that include proteinuria, ESKD, genital anomalies, and Wilm's tumor (13). Subjects with exon 6-9 WT1 missense mutations display partial gonadal dysgenesis and late-onset nephropathy. The intron 9 splice-site mutations with the 3 amino acid residues–lysine (K), threonine (T), and serine (S)–referred to as the +KTS is an important element in gonadal development and the WT1 variant that affects the +KTS/-KTS ratio of the isoforms to <2.0 results in abnormal gonadal development with sexual reversal (female XY-DSD) phenotype and late-onset nephropathy. Importantly, the XY sex reversal with female phenotype appears to be protective against the development of Wilm's tumor while it is associated with high risk for gonadoblastoma (19, 44, 45). Although rare, there were 2 cases of Wilm's tumor with female XY sex reversal in this case series (7, 8).
Malignancy is the most ominous complication of the WT1 mutation and its occurrence relative to the clinical phenotypic–genotypic profile was delineated by the analysis of the 333 cases in our review. Importantly, Wilm's tumor with WT1 mutation tends to occur early with a median age of 15–19 months vs. a median age of 36 months in children without a pathogenic WT1 variant (5). Any early-onset Wilm's tumor or bilateral Wilm's tumor should have WT1 mutation analysis. The 20-year registry of Wilm's tumor has shown that 1.3% of those with unilateral and 15% with bilateral Wilm's tumor progressed to ESKD (46). Hence, this mutation should be considered in toddlers, as well as in cases of bilateral Wilm's tumor (5). A recent single center retrospective analysis of 25 long-term Wilm's tumor survivors with WT1 pathogenic variants confirmed male predominance (60%), early age occurrence (median 14 months), multiple genetic variants, high incidence of bilateral disease (52%), and genitourinary malformations (44%) (47). This recent report emphasized the importance of early genetic diagnosis to better inform surgical management toward the preservation of renal mass when oncologic management is not compromised. Importantly, our report is the first to describe long-term outcomes of those with WT1 mutations post-transplantation. It is notable that we report a cumulative of over 170 patient years with no evidence of post-transplant malignancy.
The complete sex reversal with 46 XY female phenotype is associated with late-onset SRNS, and CKD has been typically categorized as “Frasier syndrome.” These patients are often not diagnosed until adolescence when they fail to achieve puberty. Importantly, this unique phenotype is associated with the KTS gene variant that seems to impart “protection” against the development of Wilm's tumor. It does not prevent gonadoblastoma because the gonads are dysplastic. As in our series, 2 phenotypic females presented with CNS and childhood SRNS, but they were not diagnosed with WT1 mutation until adolescence when a karyotype was obtained as workup for delayed puberty. These are examples of the wide spectrum of clinical presentations and progression of disease that make the designations of Denys-Drash and Frasier syndromes indistinct. This contributes to the paradigm that calls for the genetic investigation including karyotype of any phenotypic female who presents with CNS, SRNS or proteinuria, or ESKD concerning for possible 46XY sex reversal associated with WT1 glomerulopathy. The fact that the XY karyotype predominates in most series suggests that we are probably missing the diagnosis in a number of females, especially those with familial proteinuria (5, 8, 42, 43).
The risk of malignancy associated with WT1 mutation compels the discussion of whether prophylactic nephrectomy and/or gonadectomy should be considered once the diagnosis is confirmed. In the context of this study and others in the literature review, the characterization of the risks of malignancy according to phenotype–karyotype is extremely pertinent (14, 48, 49). Importantly, the current analysis of 333 cases shows that normal male XY phenotype–karyotype is at low risk for any malignancy. However, normal female XX phenotype–karyotype has very low risk for gonadoblastoma but remains at risk for Wilm's tumor, especially in early life. Historically, there has been an unsubstantiated concession to perform prophylactic nephrectomies in young children with WT1 mutation, especially in the setting of prior Wilm's tumor or in preparation for transplantation (48). Currently, there is no consensus for prophylactic nephrectomies. In the literature review, there were no specific recommendations in favor of prophylactic bilateral nephrectomy, and most recently, the recommendation has been for renal sparing surgery in the case of unilateral Wilm's tumor (14, 47–49). In marked contrast, there is a strong consensus for prophylactic gonadectomy in all phenotypic females with 46 XY sex reversal. Moreover, our results show the increased risk of malignancy in phenotypic males with DSD and emphasize the need for close surveillance and a low threshold for prophylactic gonadectomy (47–50).
Comprehensive data on the surgical and medical management of sexual health in children with WT1 mutations are lacking. There is a need to better understand hormonal replacement, erectile dysfunction, cosmesis for urogenital abnormalities, and reproductive capacity. Endocrine data are scarce in subjects with WT1 mutations. Long-term follow-up with the development of novel markers to evaluate and predict gonadal malignancies is imperative.
We propose a paradigm designed to initiate early comprehensive genetic testing followed by clinical surveillance (Figure 4). The goals are for (1) early phenotypic recognition leading to genomic diagnosis; (2) appropriate surveillance for progression to CKD, malignancy, and sexual health in patients with WT1 mutations; (3) genetic analysis to precede kidney biopsy in children with SRNS; (4) establish early medical management to modulate proteinuria and preserve long-term kidney function; (5) genetic counseling for the patient and family related to fertility and future pregnancies; and (6) expansion of the knowledge database by collaboration and reporting of new cases. Current recommendations include early genomic analyses by next generation sequencing (NGS) and karyotyping of any infants (< 1 year of age) with proteinuria including CNS, SRNS, and/or male DSD (51).
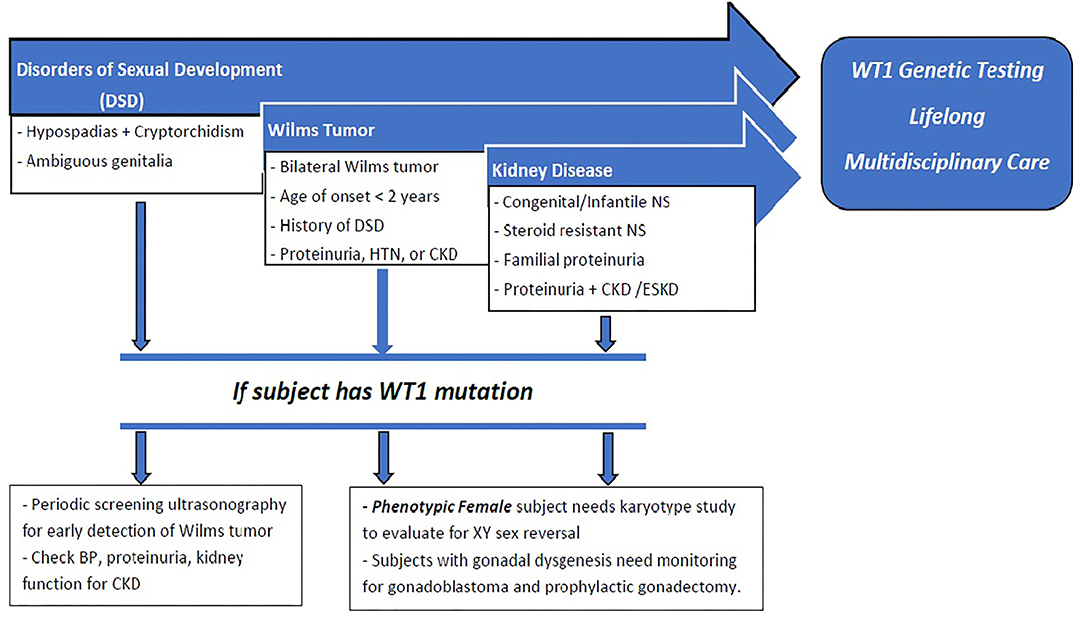
Figure 4. : Proposed paradigm for early recognition, comprehensive genetic testing followed by clinical surveillance of patients with WT1 mutation. Early presentation with DSD, CNS, SRNS, or Wilm's tumor should raise suspicion for WT1 mutation. Those with DSD should undergo periodic screening for Wilm's tumor, proteinuria, and development of CKD. All phenotypic females should have karyotype testing for possible 46XY sex reversal. All patients with WT1 mutation should receive long-term multidisciplinary medical, surgical, and psychosocial management. Phenotypic females with XY sex reversal should be closely surveilled for gonadoblastoma and recommended for prophylactic gonadectomy.
This study has several limitations including those inherent in extracting accurate detailed case characteristics from an in-depth literature review. There may have been publication bias from distinct case reports, particularly involving malignancy or karyotype–phenotype correlations. This is especially true in the case of XY-karyotype with severe gonadal dysmorphia and ambiguous phenotype. Nevertheless, the impact of WT1 mutations and the long-term consequences on patient's health merits comprehensive and contemporary review to promote early recognition and genetic screening.
Conclusions
Early recognition of WT1 gene mutation is essential for appropriate medical and surgical management. This includes the use of renal preservation therapy to modulate proteinuria and hyperfiltration with angiotensin blockade, avoidance of immunosuppressive treatment before kidney transplantation, surveillance, and early detection of malignancy. Further, these patients would benefit from multi-disciplinary care from primary care providers, nephrologists, endocrinologists, urologists, and psychologists to establish appropriate long-term management of psychosocial and sexual health. In this case series, kidney transplantation has provided long-term patient and allograft survival with no increased incidence of malignancy post-transplant.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by University of Miami Institution Review Board. Written informed consent from the participants' legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author Contributions
PA-P, WS, and CA wrote the first draft of the manuscript. WS, CK, MD, and CA participated in the critical analysis of the data and the major revisions of the manuscript. All authors contributed to the conception, design of the study, contributed to manuscript revision, read, and approved the final version of the manuscript.
Funding
This work was supported in part by the Lilian Barbarosh Pediatric Renal Research Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
A portion of the work was presented at the 2021 Florida Society of Nephrology Fellow's Awards. The authors would like to acknowledge Claudia Serrano, APRN, Teresa Canal Cano, RN, and the Pediatric Dialysis and Transplant nurses and physicians for their assistance in caring for these patients throughout the years.
References
1. McTaggart SJ, Algar E, Chow CW, Powell HR, Jones CL. Clinical spectrum of denys-drash and frasier syndrome. Pediatr Nephrol. (2001) 16:335–9. doi: 10.1007/s004670000541
2. Heathcott RW, Morison IM, Gubler MC, Corbett R, Reeve AE. A review of the phenotypic variation due to the denys-drash syndrome-associated germline WT1 mutation R362X. Hum Mutat. (2002) 19:462. doi: 10.1002/humu.9031
3. Kaltenis P, Schumacher V, Jankauskiene A, Laurinavicius A, Royer-Pokora B. Slow progressive FSGS associated with an F392L WT1 mutation. Pediatr Nephrol. (2004) 19:353–6. doi: 10.1007/s00467-003-1372-1
4. Little S, Hanks S, King-Underwood L, Picton S, Cullinane C, Rapley E, et al. A WT1 exon 1 mutation in a child diagnosed with denys-drash syndrome. Pediatr Nephrol. (2005) 20:81–5. doi: 10.1007/s00467-004-1649-z
5. Lipska-Zietkiewicz B.WT1 Disorder. In: Adam, MP Ardinger, HH Pagon, RA editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (1993-2021).
6. Chernin G, Vega-Warner V, Schoeb DS, Heeringa SF, Ovunc B, Saisawat P, et al. Genotype/phenotype correlation in nephrotic syndrome caused by WT1 mutations. Clin J Am Soc Nephrol. (2010) 5:1655–62. doi: 10.2215/CJN.09351209
7. Lipska BS, Ranchin B, Iatropoulos P, Gellermann J, Melk A, Ozaltin F, et al. Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int. (2014) 85:1169–78. doi: 10.1038/ki.2013.519
8. Lehnhardt A, Karnatz C, Ahlenstiel-Grunow T, Benz K, Benz MR, Budde K, et al. Clinical and molecular characterization of patients with heterozygous mutations in wilms tumor suppressor gene 1. Clin J Am Soc Nephrol. (2015) 10:825–31. doi: 10.2215/CJN.10141014
9. Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol. (2015) 10:592–600. doi: 10.2215/CJN.06260614
10. Ahn YH, Park EJ, Kang HG, Kim SH, Cho HY, Shin JI, et al. Genotype-phenotype analysis of pediatric patients with WT1 glomerulopathy. Pediatr Nephrol. (2017) 32:81–9. doi: 10.1007/s00467-016-3395-4
11. Ruf RG, Schultheiss M, Lichtenberger A, Karle SM, Zalewski I, Mucha B, et al. Prevalence of WT1 mutations in a large cohort of patients with steroid-resistant and steroid-sensitive nephrotic syndrome. Kidney Int. (2004) 66:564–70. doi: 10.1111/j.1523-1755.2004.00775.x
12. Aucella F, Bisceglia L, De Bonis P, Gigante M, Caridi G, Barbano G, et al. WT1 mutations in nephrotic syndrome revisited. high prevalence in young girls, associations and renal phenotypes. Pediatr Nephrol. (2006) 21:1393–8. doi: 10.1007/s00467-006-0225-0
13. Nagano C, Yamamura T, Horinouchi T, Aoto Y, Ishiko S, Sakakibara N, et al. Comprehensive genetic diagnosis of Japanese patients with severe proteinuria. Sci Rep. (2020) 10:270. doi: 10.1038/s41598-019-57149-5
14. Sun S, Xu L, Bi Y, Wang J, Zhang Z, Tang X, et al. Early diagnosis of WT1 nephropathy and follow up in a Chinese multicenter cohort. Eur J Med Genet. (2020) 63:104047. doi: 10.1016/j.ejmg.2020.104047
15. Denamur E, Bocquet N, Baudouin V, Da Silva F, Veitia R, Peuchmaur M, et al. WT1 splice-site mutations are rarely associated with primary steroid-resistant focal and segmental glomerulosclerosis. Kidney Int. (2000) 57:1868–72. doi: 10.1046/j.1523-1755.2000.00036.x
16. Takata A, Kikuchi H, Fukuzawa R, Ito S, Honda M, Hata J. Constitutional WT1 correlate with clinical features in children with progressive nephropathy. J Med Genet. (2000) 37:698–701. doi: 10.1136/jmg.37.9.698
17. Ohta S, Ozawa T, Shiraga H, Fuse H. Genetic analysis of two female patients with incomplete denys-drash syndrome. Endocr J. (2000) 47:683–7. doi: 10.1507/endocrj.47.683
18. Ito S, Takata A, Hataya H, Ikeda M, Kikuchi H, Hata J, et al. Isolated diffuse mesangial sclerosis and Wilms tumor suppressor gene. J Pediatr. (2001) 138:425–7. doi: 10.1067/mpd.2001.111317
19. Melo KF, Martin RM, Costa EM, Carvalho FM, Jorge AA, Arnhold IJ, et al. An unusual phenotype of Frasier syndrome due to IVS9 +4C>T mutation in the WT1 gene: predominantly male ambiguous genitalia and absence of gonadal dysgenesis. J Clin Endocrinol Metab. (2002) 87:2500–5. doi: 10.1210/jcem.87.6.8521
20. Auber F, Lortat-Jacob S, Sarnacki S, Jaubert F, Salomon R, Thibaud E, et al. Surgical management and genotype/phenotype correlations in WT1 gene-related diseases. J Pediatr Surg. (2003) 38:124–9. doi: 10.1053/jpsu.2003.50025
21. Saylam K, Simon P. WT1 gene mutation responsible for male sex reversal and renal failure: the Frasier syndrome. Eur J Obstet Gynecol Reprod Biol. (2003) 110:111–3. doi: 10.1016/S0301-2115(03)00088-5
22. Hu M, Craig J, Howard N, Kan A, Chaitow J, Little D, et al. A novel mutation of WT1 exon 9 in a patient with Denys-Drash syndrome and pyloric stenosis. Pediatr Nephrol. (2004) 19:1160–3. doi: 10.1007/s00467-004-1564-3
23. Love JD, DeMartini SD, Coppola CP. Prophylactic bilateral salpingo-oopherectomy in a 17-year-old with Frasier syndrome reveals gonadoblastoma and seminoma: a case report. J Pediatr Surg. (2006) 41:e1–4. doi: 10.1016/j.jpedsurg.2006.07.012
24. Gwin K, Cajaiba MM, Caminoa-Lizarralde A, Picazo ML, Nistal M, Reyes-Mugica M. Expanding the clinical spectrum of Frasier syndrome. Pediatr Dev Pathol. (2008) 11:122–7. doi: 10.2350/07-01-0209.1
25. Bache M, Dheu C, Doray B, Fothergill H, Soskin S, Paris F, et al. Frasier syndrome, a potential cause of end-stage renal failure in childhood. Pediatr Nephrol. (2010) 25:549–52. doi: 10.1007/s00467-009-1343-2
26. Megremis S, Mitsioni A, Fylaktou I, Tzeli SK, Komianou F, Stefanidis CJ, et al. Broad and unexpected phenotypic expression in Greek children with steroid-resistant nephrotic syndrome due to mutations in the Wilms' tumor 1 (WT1) gene. Eur J Pediatr. (2011) 170:1529–34. doi: 10.1007/s00431-011-1450-5
27. Kohler B, Biebermann H, Friedsam V, Gellermann J, Maier RF, Pohl M, et al. Analysis of the Wilms' tumor suppressor gene (WT1) in patients 46, XY disorders of sex development. J Clin Endocrinol Metab. (2011) 96:E1131–6. doi: 10.1210/jc.2010-2804
28. Yang YH, Zhao F, Feng DN, Wang JJ, Wang CF, Huang J, et al. Wilms' tumor suppressor gene mutations in girls with sporadic isolated steroid-resistant nephrotic syndrome. Genet Mol Res. (2013) 12:6184–91. doi: 10.4238/2013.December.4.5
29. Yang Y, Feng D, Huang J, Nie X, Yu Z. A child with isolated nephrotic syndrome and WT1 mutation presenting as a 46, XY phenotypic male. Eur J Pediat. (2013) 172:127–9. doi: 10.1007/s00431-012-1770-0
30. Aydin M, Hakan N, Zenciroglu A, Aydog O, Okumus N. Different clinical presentations of WT1 gene mutations. Eur J Pediatr. (2013) 172:1705–6. doi: 10.1007/s00431-013-2085-5
31. Guaragna MS, Lutaif AC, Piveta CS, Belangero VM, Maciel-Guerra AT, Guerra G, et al. Two distinct WT1 mutations identified in patients and relatives with isolated nephrotic proteinuria. Biochem Biophys Res Commun. (2013) 441:371–6. doi: 10.1016/j.bbrc.2013.10.064
32. Binczak-Kuleta A, Rubik J, Litwin M, Ryder M, Lewandowska K, Taryma-Lesniak O, et al. Retrospective mutational analysis of NPHS1, NPHS2, WT1 and LAMB2 in children with steroid-resistant focal segmental glomerulosclerosis - a single-centre experience. Bosn J Basic Med Sci. (2014) 14:89–93. doi: 10.17305/bjbms.2014.2270
33. Kumar AS, Srilakshmi R, Karthickeyan S, Balakrishnan K, Padmaraj R, Senguttuvan P. Wilms' tumour 1 gene mutations in south Indian children with steroid-resistant nephrotic syndrome. Indian J Med Res. (2016) 144:276–80. doi: 10.4103/0971-5916.195044
34. Dabrowski E, Armstrong AE, Leeth E, Johnson E, Cheng E, Gosiengfiao Y, et al. Proximal hypospadias and a novel WT1 variant: when should genetic testing be considered? Pediatrics. (2018) 141:S491–S5. doi: 10.1542/peds.2017-0230
35. Chiba Y, Inoue CN. Once-daily low-dose cyclosporine a treatment with angiotensin blockade for long-term remission of nephropathy in frasier syndrome. Tohoku J Exp Med. (2019) 247:35–40. doi: 10.1620/tjem.247.35
36. Nishi K, Inoguchi T, Kamei K, Hamada R, Hataya H, Ogura M, et al. Detailed clinical manifestations at onset and prognosis of neonatal-onset Denys-Drash syndrome and congenital nephrotic syndrome of the Finnish type. Clin Exp Nephrol. (2019) 23:1058–65. doi: 10.1007/s10157-019-01732-7
37. Roca N, Munoz M, Cruz A, Vilalta R, Lara E, Ariceta G. Long-term outcome in a case series of Denys-Drash syndrome. Clin Kidney J. (2019) 12:836–9. doi: 10.1093/ckj/sfz022
38. Matsuoka D, Noda S, Kamiya M, Hidaka Y, Shimojo H, Yamada Y, et al. Immune-complex glomerulonephritis with a membranoproliferative pattern in Frasier syndrome: a case report and review of the literature. BMC Nephrol. (2020) 21:362. doi: 10.1186/s12882-020-02007-0
39. Nagano C, Takaoka Y, Kamei K, Hamada R, Ichikawa D, Tanaka K, et al. Genotype-phenotype correlation in WT1 Exon 8 to 9 missense variants. Kidney Int Rep. (2021) 6:2114–21. doi: 10.1016/j.ekir.2021.05.009
40. Pelletier J, Bruening W, Li FP, Haber DA, Glaser T, Housman DE. WT1 mutations contribute to abnormal genital system development and hereditary Wilms' tumour. Nature. (1991) 353:431–4. doi: 10.1038/353431a0
41. Coppes MJ, Huff V, Pelletier J. Denys-Drash syndrome: relating a clinical disorder to genetic alterations in the tumor suppressor gene WT1. J Pediatr. (1993) 123:673–8. doi: 10.1016/S0022-3476(05)80839-X
42. Hall G, Gbadegesin RA, Lavin P, Wu G, Liu Y, Oh EC, et al. A novel missense mutation of Wilms' Tumor 1 causes autosomal dominant FSGS. J Am Soc Nephrol. (2015) 26:831–43. doi: 10.1681/ASN.2013101053
43. Benetti E, Caridi G, Malaventura C, Dagnino M, Leonardi E, Artifoni L, et al. A novel WT1 gene mutation in a three-generation family with progressive isolated focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. (2010) 5:698–702. doi: 10.2215/CJN.05670809
44. Kohler B, Schumacher V., l'Allemand D, Royer-Pokora B, Gruters A. Germline wilms tumor suppressor gene (WT1) mutation leading to isolated genital malformation without Wilms tumor or nephropathy. J Pediatr. (2001) 138:421–4. doi: 10.1067/mpd.2001.112512
45. Barbaux S, Niaudet P, Gubler MC, Grunfeld JP, Jaubert F, Kuttenn F, et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet. (1997) 17:467–70. doi: 10.1038/ng1297-467
46. Breslow NE, Collins AJ, Ritchey ML, Grigoriev YA, Peterson SM, Green DM. End stage renal disease in patients with Wilms tumor: results from the national wilms tumor study group and the United States renal data system. J Urol. (2005) 174:1972–5. doi: 10.1097/01.ju.0000176800.00994.3a
47. Falcone MP, Pritchard-Jones K, Brok J, Mifsud W, Williams RD, Nakata K, et al. Long-term kidney function in children with Wilms tumour and constitutional WT1 pathogenic variant. Pediatr Nephrol. (2021) 37:821–32. doi: 10.1007/s00467-021-05125-5
48. Hu M, Fletcher J, McCahon E, Catchpoole D, Zhang GY, Wang YM, et al. Bilateral Wilms tumor and early presentation in pediatric patients is associated with the truncation of the Wilms tumor 1 protein. J Pediatr. (2013) 163:224–9. doi: 10.1016/j.jpeds.2012.12.080
49. Auber F, Jeanpierre C, Denamur E, Jaubert F, Schleiermacher G, Patte C, et al. Management of wilms tumors in drash and frasier syndromes. Pediatr Blood Cancer. (2009) 52:55–9. doi: 10.1002/pbc.21759
50. Hol JA, Jewell R, Chowdhury T, Duncan C, Nakata K, Oue T, et al. Wilms tumour surveillance in at-risk children: Literature review and recommendations from the SIOP-Europe host genome working group and SIOP renal tumour study group. Eur J Cancer. (2021) 153:51–63. doi: 10.1016/j.ejca.2021.05.014
Keywords: Wilms tumor, congenital nephrotic syndrome, steroid-resistant nephrotic syndrome, 46XY sex reversal, disorders of sexual development
Citation: Arroyo-Parejo Drayer P, Seeherunvong W, Katsoufis CP, DeFreitas MJ, Seeherunvong T, Chandar J and Abitbol CL (2022) Spectrum of Clinical Manifestations in Children With WT1 Mutation: Case Series and Literature Review. Front. Pediatr. 10:847295. doi: 10.3389/fped.2022.847295
Received: 01 January 2022; Accepted: 14 March 2022;
Published: 15 April 2022.
Edited by:
Asha Moudgil, Children's National Hospital, United StatesReviewed by:
Gema Ariceta, Vall d'Hebron University Hospital, SpainWilliam Morello, Fondazione IRCCS Ca' Granda Ospedale Maggiore, Policlinico di Milano, Italy
Copyright © 2022 Arroyo-Parejo Drayer, Seeherunvong, Katsoufis, DeFreitas, Seeherunvong, Chandar and Abitbol. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolyn L. Abitbol, Y2FiaXRib2xAbWVkLm1pYW1pLmVkdQ==; Wacharee Seeherunvong, d3NlZWhlcnVudm9uZ0BtZWQubWlhbWkuZWR1