 Abirami Pararajasingam1
Abirami Pararajasingam1 Rachel E. Bradley2Jennifer Evans3Ashima Lowe1Richard Goodwin1
Rachel E. Bradley2Jennifer Evans3Ashima Lowe1Richard Goodwin1 Stephen Jolles2*
Stephen Jolles2*- 1Department of Dermatology, Aneurin Bevan University Health Board, Newport, United Kingdom
- 2Immunodeficiency Centre for Wales, University Hospital of Wales, Cardiff, United Kingdom
- 3Department of Paediatrics, University Hospital of Wales, Cardiff, United Kingdom
Aicardi-Goutières syndrome (AGS) is a rare hereditary early-onset encephalopathy. The syndrome was first described in 1984, and is characterised by upregulation of the type I interferon (IFN) pathway, which is involved in the host immune response against viral infections, including SARS-CoV-2. Whilst defects in type I IFN pathways have been described in association with severe coronavirus disease 2019 (COVID-19), less is known about the outcomes of upregulation. We describe an unusual case of generalised panniculitis as a post-COVID-19 phenomenon in a child with AGS. Our patient was initially managed with systemic steroid therapy, but due to relapse of symptoms on weaning, an alternative therapy was sought. In this case, a novel use of ruxolitinib, a JAK inhibitor, has resulted in lasting remission without complications. We discuss the probable protective role of IFN upregulation following COVID-19 infection in AGS and possible immunological mechanisms driving the panniculitis and therapeutic response in our case.
Introduction
Aicardi-Goutières syndrome (AGS) was first described by Aicardi and Goutières in their report of eight children from five families with an early-onset encephalopathy characterised by basal ganglia calcification, white matter abnormalities, and a chronic cerebrospinal lymphocytosis (1). There is a typical elevation of interferon (IFN) alpha levels in both cerebrospinal fluid (CSF) and serum of these patients (2). More recently, evidence of abnormal IFN activity was demonstrated by identifying an “interferon signature” in the peripheral blood. This measures the expression of IFN-stimulated genes (e.g., IFI27, IFI44L, IFIT1, ISG15, RSAD2, and SIGLEC1) and has been identified in patients with mutations in genes associated with AGS (3, 4).
Type I IFN was originally described as a soluble factor, produced by cells treated with inactivated non-replicating viruses, that subsequently successfully blocked or interfered on infection with the live virus (5, 6). The rapid induction and amplification of the type I IFN system is a key component of antiviral immunity (e.g., against the novel coronavirus) which activates the JAK-STAT signalling pathway and transcription of IFN-stimulated genes (7).
In AGS, the main extraneurological symptoms are chilblain-like cutaneous lesions in up to 40% of cases, usually on the fingers, toes, and ears (8). Some children with AGS also develop symptoms overlapping with systemic lupus erythematosus (SLE) (9). There appears to be a phenotypic overlap between AGS, in utero HIV-1 infection, and SLE, suggesting that these result from the common pathological feature of type I IFN upregulation (10). Mutations in nine genes, namely, TREX1 (AGS1), RNASEH2A (AGS2), RNASEH2B (AGS3), RNASEH2C (AGS4), SAMHD1 (AGS5), ADAR1 (AGS6), IFIH1 (AGS7), LSM11, and RNU7-1, have been identified as the causative agent of AGS (4, 11). These genes are involved in the DNA damage response and sensing free cytosolic DNA/RNA. Defects in these genes result in an inappropriate innate immune response, triggering an increased secretion of the antiviral cytokine type I IFN and upregulation of IFN-stimulated genes, ultimately responsible for the main features of the disease (12). At a cellular level, this resembles the type I IFN response following the exposure to viral DNA or RNA, thus explaining why the AGS phenotype may mimic that of viral infections. However, in contrast to viral infection, AGS is considered to represent an abnormal response to endogenous or self-derived nucleic acids (10).
Genotype-phenotype correlation has been observed, and there are certain core features that overlap across mutations in the nine genes with the vast majority of affected subjects displaying severe motor and cognitive impairment (13). In a large cohort of 252 patients with AGS, the most common clinical features were identified as cognitive impairment, dystonia, microcephaly, and chilblain lesions reported in 92, 75, 63, and 42% of cases, respectively (14). Chilblain lesions are the most well-documented extraneurological sign. These cutaneous manifestations are characterised by areas of inflammation and intermittent necrosis, found mainly on the fingers, toes, pressure points, and ears (13). Acrocyanosis is also very frequent, as is periungual erythema (15).
The course of AGS is variable; most affected individuals present around 4 months of age with a subacute onset encephalopathy followed by marked developmental delay/regression as well as signs of neurological impairment and a slowing of head growth over a few months (8). Early mortality is described in some, not long following symptom onset, whilst others go on to have a more stable clinical course, without significant further deterioration (16).
AGS is a rare genetic disease, and as such, treatment approaches are based on case series with limited clinical trial data. There is no cure for AGS, and treatments aim to limit the symptoms. As the pathogenesis of AGS is related to inappropriate activation of the innate immune response, generalised immunosuppression with high-dose corticosteroid therapy has been trialled and targetted therapies such as reverse transcriptase inhibitors and JAK inhibitors have been used.
Whereas genetic and acquired defects in the type I IFN pathway have been shown to predispose to more severe outcomes in coronavirus disease 2019 (COVID-19) (17–20), the consequences of this infection in the setting of upregulated type I IFN signalling are less well understood. We report a case of a child with AGS presenting acutely with generalised panniculitis as a post-COVID-19 phenomenon with a beneficial response to the ruxolitinib, a JAK 1/2 inhibitor.
Case Description
Our patient was born to consanguineous parents from Pakistan and had a confirmed diagnosis of AGS with previous genetic testing confirming homozygous SAMHD1 mutations (c. 427C > T). She had an older sibling who was confirmed to have the same genotype and who displayed an almost identical phenotype, manifesting in severe neurological deficit, spastic quadriplegia, and dystonia. Her regular medication consisted of sodium valproate (160 mg twice daily) and baclofen. Although the older sibling had developed cutaneous manifestations in the form of acral perniosis and periungual necrosis at the age of 8 years, there were no prior cutaneous changes in our patient.
At 5 years of age, the patient was admitted with a 3-week history of cutaneous changes affecting her face and upper limbs but was otherwise well. Her grandfather, who co-habited with the family, had developed a febrile illness and tested positive for COVID-19 by PCR a few weeks previously. Despite this, she had no history of fever, cough, or other systemic symptoms. No other family member was unwell, including the patient’s sibling, who remained well and completely asymptomatic.
On physical examination of the new skin changes, there were dusky areas of induration involving the central and lower face, bilateral forearms, and extending onto her hands with associated oedema (Figure 1). There was notable sparing of dependent areas, including the back and posterior legs. Scattered petechial macules were also noted on the left forearm and foot. Aside from the cutaneous changes, no other extraneurological signs were found on examination. She was afebrile and the remaining observations were within the normal range. Investigations demonstrated a mild transaminitis with an ALT of 49 U/L (9–25), a raised amylase of 153 U/L (25–101), D-dimer of 1,785 μg/L (<500), and lactate dehydrogenase of 525 U/L (192–321). Throat swabs for SARS-CoV-2 RNA PCR were negative. Of note, chest radiography, renal function, autoimmune studies, and C-reactive protein levels were normal; however, there was a significant elevation in the terminal complement complex (TCC) at this stage at 244 ng/ml (0–80).
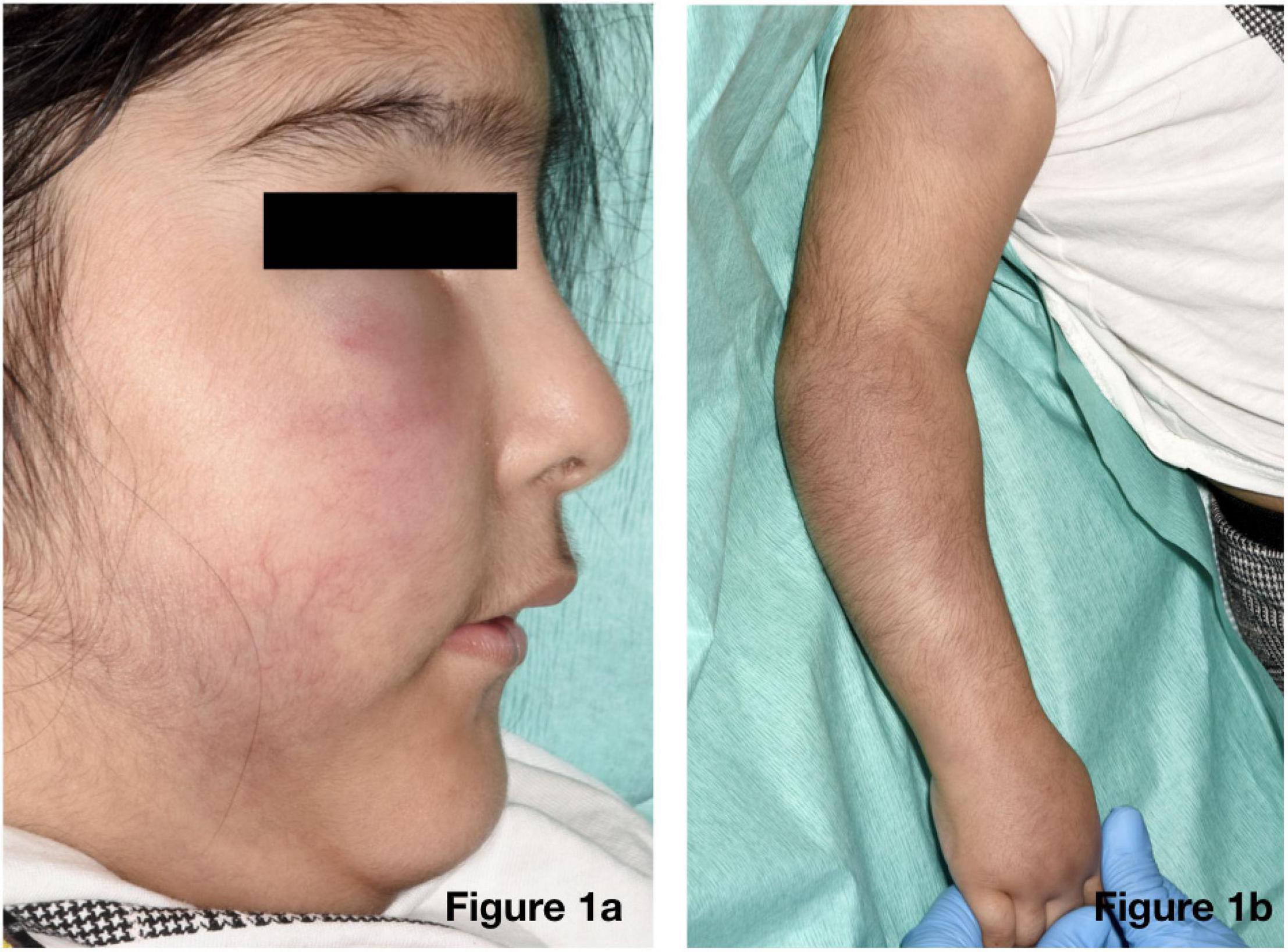
Figure 1. Physical examination revealed dusky indurated plaques on the central and lower face (a) and arms (b) with associated oedema.
Suspicion of a post-COVID-19 phenomenon was supported, with SARS-CoV-2 antibody testing demonstrating immunoglobulin G (IgG) positivity (in the absence of vaccination). Her cutaneous changes continued to progress, spreading across the chest, arms, and thighs, without any corresponding deterioration in general health. An incisional biopsy revealed a predominantly lymphocytic, nodular, perivascular, and periadnexal inflammatory infiltrate in the deep dermis and subcutis, with associated oedema, fat necrosis, and karyorrhexis in the subcutaneous fat, confirming a panniculitis (Figure 2). Viral particles were not identified on electron microscopy. Neither the patient nor her sister had previous evidence of panniculitis.
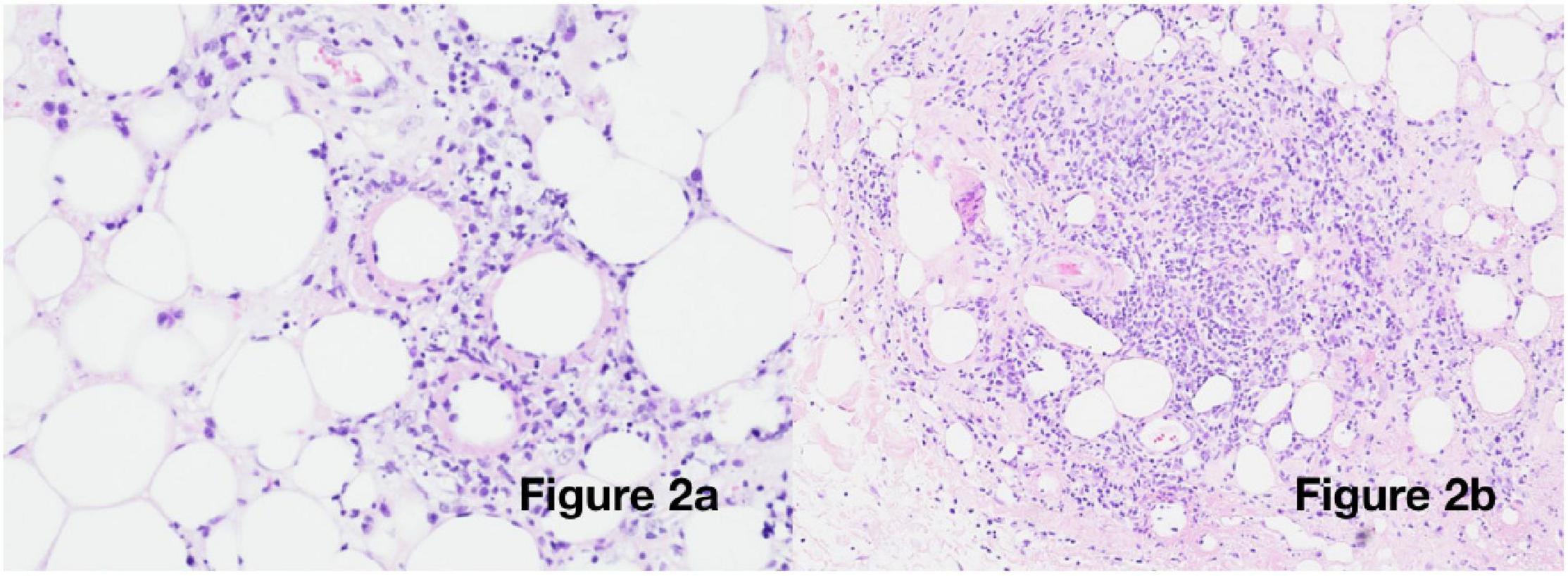
Figure 2. Histological examination: Incisional biopsy from the right upper arm [haematoxylin and eosin (H&E) staining] showing (a) 20× magnification: vasculitis and fibrinoid necrosis and (b) 10× magnification: deep subcutaneous infiltrate with oedema and karyorrhexis.
She was commenced on prednisolone 20 mg (1 mg/kg) once daily, and within a few days, there was an improvement in the dusky discolouration across the face, limbs, and chest, with near-resolution and softening of the panniculitis observed after a few weeks. As the prednisolone dose was weaned to 0.25 mg/kg/day, the panniculitis relapsed and the higher dose was promptly reintroduced, with good effect. Further attempts at dose reduction were met with a similar deterioration, and although she remained systemically well, the lesions were associated with significant discomfort. Lymphocyte subsets taken 4 months following the onset of rash and 3 months following the trials of steroids showed CD3+ (T cells) 2,780 × 106 cells/L (900–4,500) and CD19+ (B cells) 3,690 × 106 cells/L (200–2,100).
Ruxolitinib, a JAK1/2 inhibitor, was commenced, building up to 5 mg twice daily. The rash improved and oedema resolved, and it was possible to wean off systemic steroids. Unfortunately, 10 days after stopping prednisolone, some oedema and discomfort reappeared, without any skin discolouration. Although this was a much less severe relapse than before, it necessitated the short-term re-introduction of prednisolone, alongside the ruxolitinib. A second attempt at steroid withdrawal was successful, and 4 months on, our patient remains stable on ruxolitinib alone. A timeline of events is shown in Figure 3.
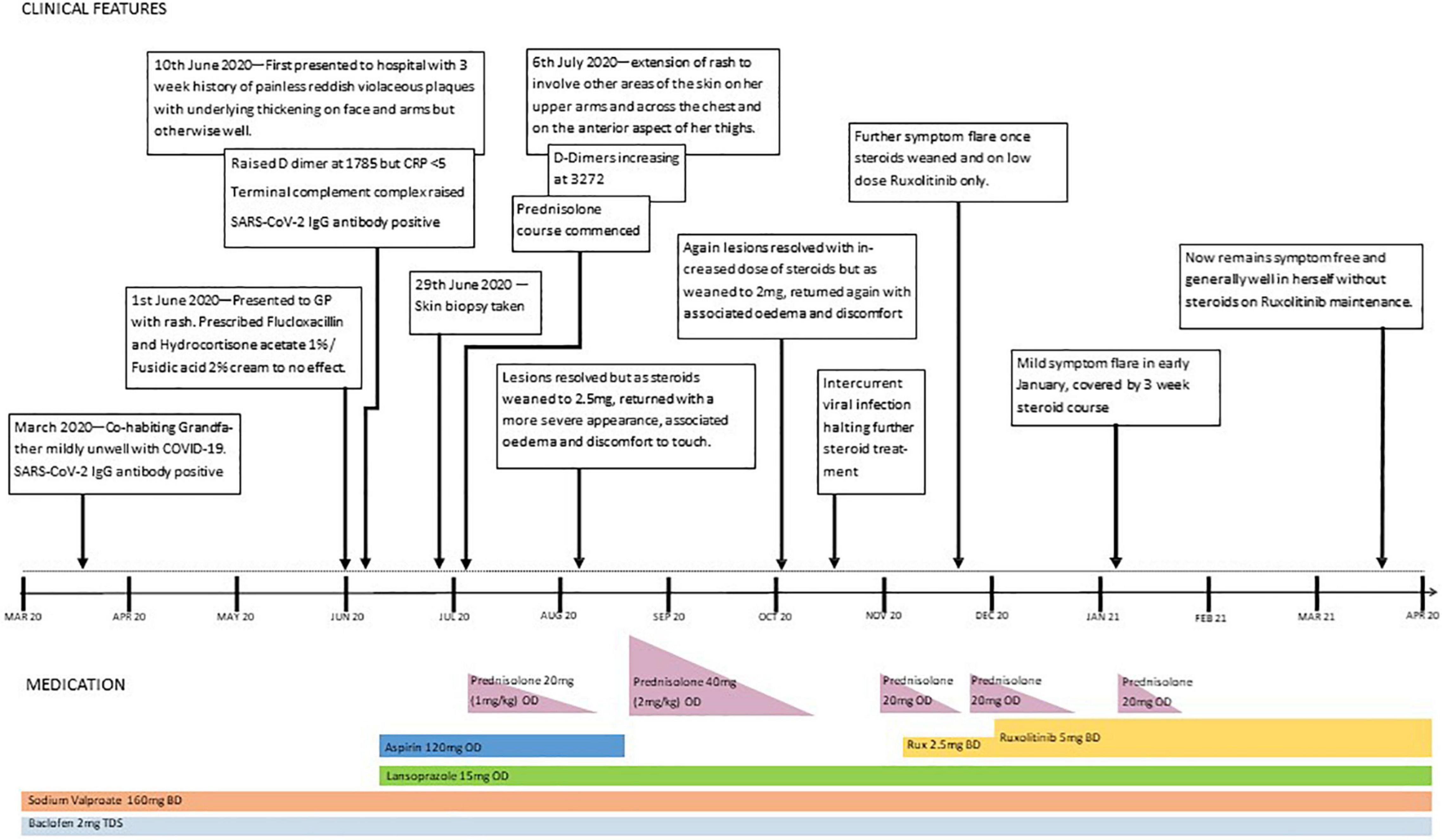
Figure 3. Timeline with the most relevant data of the clinical case.
Discussion
Our patient had an asymptomatic COVID-19 illness with the development of detectable antibody response and the post-infection cutaneous manifestations of panniculitis in the setting of AGS. A recent report documenting the course of COVID-19 in 94 patients with inborn errors of immunity described three patients with AGS including our patient (21). While numbers are very limited, it is of interest that all three, including the two other patients with biallelic RNASEH2B variants, had completely asymptomatic “silent” COVID-19 infections possibly suggesting a protective effect of IFN upregulation in the setting of viral infection. Indeed, one recent case report shows increased IFN levels from baseline in a patient with AGS suffering from mild COVID-19 illness (22).
This theory concurs with studies demonstrating that disruption of type I IFN signalling is associated with severe COVID-19 illness. In a study, 23 of 659 (3.5%) patients with life-threatening COVID-19 pneumonia displayed genetic defects abolishing induction or amplification of type I IFN (23). A study of 50 adult patients with COVID-19 showed that IFN levels were significantly reduced in critically unwell patients compared to those with mild to moderate infection with the suggestion that this resulted in uncontrolled proliferation of the virus (24). Similarly, the examination of post-mortem COVID-19-infected lung tissue demonstrates that SARS-CoV-2 evokes a distinctive transcriptional response, which lacks type I and type III IFN expression (17–20). In addition, it has been noted that of 987 individuals with life-threatening COVID-19 infection, 13.7% had autoantibodies against type I IFNs detected (25).
It is well reported that children with COVID-19 generally present with milder disease. This may be due to a prompt innate immune response, where IFN signalling plays a key role, which is more frequently dealing with respiratory viruses and vaccinations that children are regularly exposed to in early life (26). However, few children go on to develop the post-infectious multi-system inflammatory syndrome (MIS-C), usually manifesting a number of weeks following the initial infection. One study found that 3/18 patients with MIS-C had genetic mutations affecting the regulation of IFN signalling (27).
Although our patient did not report any typical symptoms of COVID-19 infection, she subsequently manifested persistent skin lesions over her face, limbs, and chest at a time when the SARS-CoV-2 PCR was negative with positive SARS-CoV-2 serology. The sparing of dependent areas observed in our case is suggestive of a chilblain-like process, which is a recognised cutaneous feature of COVID-19 (19) and a hallmark feature of genetic type I interferonopathies like AGS (17–20, 28). This is likely an inflammatory process mediated by IFN in an individual that is genetically prone to such immune dysregulation that has been triggered by the initial viral infection.
From a physiological perspective, chilblains occur often due to cold-induced constriction of small arteries and veins, hence usually manifesting on distal extremities. In our patient, her immobility and supine positioning may function to protect the posterior aspect of her torso and limbs from temperature drops and partly explain the sparing of these sites. In the context of COVID-19, chilblain-like lesions (perniosis) characteristically occur a few weeks following the acute infection which is often mild or asymptomatic (29). Typical histopathological features, which were seen in our patient, include a perivascular lymphocytic infiltrate with dermal oedema. However, in addition to these classical histological features of perniosis, our patient also had more extensive involvement of the subcutaneous fat, in keeping with a panniculitis. Panniculitis with or without lipodystrophy has been reported in children with type I interferonopathies (30).
So far, generalised panniculitis is an unreported cutaneous feature of COVID-19, and it may therefore be possible that the combination of infection with COVID-19 on a background of underlying AGS with associated IFN upregulation resulted in the observed prolonged panniculitis. Indeed, a number of reports have suggested that the mechanism behind chilblain-like lesions in the setting of SARS-CoV-2 infection is IFN-driven (31–33). In addition, there are a number of case reports describing panniculitis at the administration site of IFN therapy and associations with systemic therapy (34–41), suggesting a direct role for IFN in the development of panniculitis. The delayed manifestation of the IFN-driven inflammatory process observed, whilst limited to the skin, may be reflective of delayed MIS-C seen in children with loss-of-function mutations associated with enhanced IFN signalling (42).
Treatment of the cutaneous manifestations in our patient was initially responsive to high-dose corticosteroid therapy. A number of reports of treatment of AGS with steroids confirm a reduction in IFN-alpha levels in the CSF but no clinical improvement (43–46). More recently described is a case where neuroradiological changes on MRI showed an improvement after high-dose steroids and IV immunoglobulin (47). When steroid weaning failed to maintain remission, therapy targetted against IFN and its signalling pathways directly was initiated in the form of a JAK inhibitor which proved effective for the patients cutaneous symptoms. There are encouraging results from a number of publications using the JAK inhibitors, ruxolitinib (48–52) and baricitinib (53), in type I interferonopathies and in AGS specifically with some neurological and developmental improvement reported with ruxolitinib. In addition, reports of the first trial of baricitinib in 35 patients with AGS showed similar improvements. A phase 2 trial of baricitinib in AGS is now currently underway (ClinicalTrials.gov Identifier: NCT03921554).
Conclusion
We suggest that our patient developed COVID-19-triggered, prolonged auto-inflammatory, type I IFN-driven panniculitis, resulting from the exceedingly rare combination of AGS and COVID-19 infection. Therapy was challenging as the panniculitis was responsive only to very high doses of steroids and relapsed on tapering the dose. The novel use of ruxolitinib, used to both control the panniculitis and spare steroids, was effective and well tolerated, and it will be important to determine if ongoing therapy is needed.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the Immunodeficiency Centre for Wales Research Budget.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to acknowledge Kristina Robinson for the careful review of histology and electron microscopy.
References
1. Aicardi J, Goutières F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. (1984) 15:49–54. doi: 10.1002/ana.410150109
2. Lebon P, Badoual J, Ponsot G, Goutières F, Hémeury-Cukier F, Aicardi J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J Neurol Sci. (1988) 84:201–8. doi: 10.1016/0022-510x(88)90125-6
3. Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. (2013) 12:1159–69. doi: 10.1016/S1474-4422(13)70258-8
4. Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet. (2020) 52:1364–72. doi: 10.1038/s41588-020-00737-3
5. Lindenmann J, Burke DC, Isaacs A. Studies on the production, mode of action and properties of interferon. Br J Exp Pathol. (1957) 38:551–62.
6. Isaacs A, Lindenmann J. Virus interference. I. the interferon. Proc R Soc Lond B Biol Sci. (1957) 147:258–67.
7. Picard C, Belot A. Does type-I interferon drive systemic autoimmunity? Autoimmun Rev. (2017) 16:897–902. doi: 10.1016/j.autrev.2017.07.001
8. Orcesi S, La Piana R, Fazzi E. Aicardi–Goutières syndrome. Brit Med Bull. (2009) 89:183–201. doi: 10.1093/bmb/ldn049
9. Crow YJ, Black DN, Ali M, Bond J, Jackson AP, Lefson M, et al. Cree encephalitis is allelic with Aicardi-Goutiéres syndrome: implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet. (2003) 40:183–7. doi: 10.1136/jmg.40.3.183
10. Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol (2015) 15:429–40. doi: 10.1038/nri3850
11. Livingston JH, Crow YJ. Neurologic phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutieres syndrome and beyond. Neuropediatrics. (2016) 47:355–60. doi: 10.1055/s-0036-1592307
12. Coquel F, Neumayer C, Lin YL, Pasero P. SAMHD1 and the innate immune response to cytosolic DNA during DNA replication. Curr Opin Immunol. (2019) 56:24–30. doi: 10.1016/j.coi.2018.09.017
13. Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. (2007) 81:713–25. doi: 10.1086/521373
14. Ji T, Wang J, Li H, Zhao L, Sang Y, Wu Y. [Clinical and genetic analysis of a family with Aicardi-Goutières syndrome and literature review]. Zhonghua Er Ke Za Zhi. (2014) 52:822–7.
15. Goutières F. Aicardi–Goutières syndrome. Brain Dev. (2005) 27:201–6. doi: 10.1016/j.braindev.2003.12.011
16. Lanzi G, Fazzi E, D’Arrigo S, Orcesi S, Maraucci I, Uggetti C, et al. The natural history of Aicardi-Goutières syndrome: follow-up of 11 Italian patients. Neurology. (2005) 64:1621–4. doi: 10.1212/01.WNL.0000159864.05826.08
17. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. (2020) 181:1036–45.e. doi: 10.1016/j.cell.2020.04.026
18. Chen S, Bonifati S, Qin Z, St Gelais C, Kodigepalli KM, Barrett BS, et al. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-kappaB and interferon pathways. Proc Natl Acad Sci USA. (2018) 115:E3798–807. doi: 10.1073/pnas.1801213115
19. Galván Casas C, Català A, Carretero Hernández G, Rodríguez-Jiménez P, Fernández-Nieto D, Rodríguez-Villa Lario A. Classification of the cutaneous manifestations of COVID-19: a rapid prospective nationwide consensus study in Spain with 375 cases. Br J Dermatol. (2020) 183:71–7. doi: 10.1111/bjd.19163
20. Eleftheriou D, Brogan PA. Genetic interferonopathies: an overview. Best Pract Res Clin Rheumatol. (2017) 31:441–59. doi: 10.1016/j.berh.2017.12.002
21. Meyts I, Bucciol G, Quinti I, Neven B, Fischer A, Seoane E, et al. Coronavirus disease 2019 in patients with inborn errors of immunity: an international study. J Allergy Clin Immunol. (2020) 147:520–31. doi: 10.1016/j.jaci.2020.09.010
22. Badolato R, Cattalini M, Scaduto R, Roversi S, Galli J, Ferraro RM, et al. IFN-α levels in ruxolitinib-treatead Aicardi-Goutières patient during SARS-CoV-2 infection: a case report. Clin Immnol (Orlando Fla). (2021) 227:108743. doi: 10.1016/j.clim.2021.108743
23. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. (2020) 370:eabd4570. doi: 10.1126/science.abd4570
24. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. (2020) 369:718–24. doi: 10.1126/science.abc6027
25. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science (New York N.Y). (2020) 370:eabd4585. doi: 10.1126/science.abd4585
26. ostagliola G, Spada E, Consolini R. Age-related differences in the immune response could contribute to determine the spectrum of severity of COVID-19. Immun Inflamm Dis. (2021) 9:331–9. doi: 10.1002/iid3.404
27. Chou J, Platt CD, Habiballah S, Nguyen AA, Elkins M, Weeks S, et al. Mechanisms underlying genetic susceptibility to multisystem inflammatory syndrome in children (MIS-C). J Allergy Clin Immunol. (2021) 148:732.e–8.e. doi: 10.1016/j.jaci.2021.06.024
28. Volpi S, Picco P, Caorsi R, Candotti F, Gattorno M. Type I interferonopathies in pediatric rheumatology. Pediatr Rheumatol Online J. (2016) 14:35. doi: 10.1186/s12969-016-0094-4
29. Kanitakis J, Lesort C, Danset M, Jullien D. Chilblain-like acral lesions during the COVID-19 pandemic (”COVID toes”): Histologic, immunofluorescence, and immunohistochemical study of 17 cases. J Am Acad Dermatol. (2020) 83:870–5. doi: 10.1016/j.jaad.2020.05.145
30. Garg A, Hernandez MD, Sousa AB, Subramanyam L, Martinez de Villarreal L, dos Santos HG, et al. An autosomal recessive syndrome of joint contractures, muscular atrophy, microcytic anemia, and panniculitis-associated lipodystrophy. J Clin Endocrinol Metab. (2010) 95:58–63. doi: 10.1210/jc.2010-0488
31. Hubiche T, Cardot-Leccia N, Le Duff F, Seitz-Polski B, Giordana P, Chiaverini C, et al. Clinical, laboratory, and interferon-alpha response characteristics of patients with chilblain-like lesions during the COVID-19 pandemic. JAMA Dermatol. (2021) 157:202–6. doi: 10.1001/jamadermatol.2020.4324
32. Battesti G, Descamps V. Negative tests for SARS-CoV-2 infection do not rule out its responsibility for chilblains. Br J Dermatol. (2020) 183:1151. doi: 10.1111/bjd.19483
33. Battesti G, El Khalifa J, Abdelhedi N, Ferre V, Bouscarat F, Picard-Dahan C, et al. New insights in COVID-19-associated chilblains: a comparative study with chilblain lupus erythematosus. J Am Acad Dermatol. (2020) 83:1219–22. doi: 10.1016/j.jaad.2020.06.1018
34. Oeda E, Shinohara K. Cutaneous necrosis caused by injection of alpha-interferon in a patient with chronic myelogenous leukemia. Am J Hematol. (1993) 44:213–4. doi: 10.1002/ajh.2830440320
35. Bachmeyer C, Farge D, Gluckman E, Miclea JM, Aractingi S. Raynaud’s phenomenon and digital necrosis induced by interferon-alpha. Br J Dermatol. (1996) 135:481–3. doi: 10.1046/j.1365-2133.1996.d01-1028.x
36. Heinzerling L, Dummer R, Burg G, Schmid-Grendelmeier P. Panniculitis after subcutaneous injection of interferon beta in a multiple sclerosis patient. Eur J Dermatol. (2002) 12:194–7.
37. O’Sullivan SS, Cronin EM, Sweeney BJ, Bourke JF, Fitzgibbon J. Panniculitis and lipoatrophy after subcutaneous injection of interferon beta-1b in a patient with multiple sclerosis. J Neurol Neurosurg Psychiatry. (2006) 77:1382–3. doi: 10.1136/jnnp.2006.094813
38. Poulin F, Rico P, Côté J, Bégin LR. Interferon beta-induced panniculitis mimicking acute appendicitis. Arch Dermatol. (2009) 145:916–7. doi: 10.1001/archdermatol.2009.106
39. Baek YS, Kim JY, Choi JE, Ahn HH, Kye YC, Seo SH. Pegylated interferon Alpha2a induced panniculitis in patient with chronic hepatitis C. Korean J Dermatol. (2012) 50:640–3.
40. Urosevic-Maiwald M, Nobbe S, Kerl K, Benz R. Disseminated ulcerating lupus panniculitis emerging under interferon therapy of hairy cell leukemia: treatment- or disease-related? J Dermatol. (2014) 41:329–33. doi: 10.1111/1346-8138.12389
41. Song JS, Sohn JH, Jeong JY, Min JH, Choi WS, Kim OZ, et al. Repeated Panniculitis Induced by Pegylated Interferon Alpha 2a in a Patient with Chronic Hepatitis C. Korean J Gastroenterol. (2016) 67(5):272–6. doi: 10.4166/kjg.2016.67.5.272
42. Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K, et al. Immune dysregulation and multisystem inflammatory syndrome in children (MIS-C) in individuals with haploinsufficiency of SOCS1. J Allergy Clin Immunol. (2020) 146:1194.e–200.e. doi: 10.1016/j.jaci.2020.07.033
43. Kuijpers TW. Aicardi–Goutie‘res syndrome: immunophenotyping in relation to interferon-alpha. Eur J Paediatr Neurol. (2002) 6(Suppl. A):A59–64 Discussion A65-6. doi: 10.1053/ejpn.2002.0576.
44. Orcesi S, Pessagno A, Biancheri R, La Piana R, Mascaretti M, Rossi A, et al. Aicardi-Goutières syndrome presenting atypically as a sub-acute leukoencephalopathy. Eur J Paediatr Neurol. (2008) 12(5):408–11. doi: 10.1016/j.ejpn.2007.10.005
45. D’Arrigo S, Riva D, Bulgheroni S, Chiapparini L, Lebon P, Rice G, et al. Aicardi-Goutières syndrome: description of a late onset case. Dev Med Child Neurol. (2008) 50:631–4. doi: 10.1111/j.1469-8749.2008.03033.x
46. De Laet C, Goyens P, Christophe C, Ferster A, Mascart F, Dan B. Phenotypic overlap between infantile systemic lupus erythematosus and Aicardi–Goutie‘res syndrome. Neuropediatrics. (2005) 36(6):399–402. doi: 10.1055/s-2005-873058
47. Tonduti D, Izzo G, D’Arrigo S, Riva D, Moroni I, Zorzi G, et al. Spontaneous MRI improvement and absence of cerebral calcification in Aicardi-Goutières syndrome: diagnostic and disease-monitoring implications. Mol Gen Met. (2019) 126:489–94. doi: 10.1016/j.ymgme.2019.02.006
48. Frémond ML, Rodero MP, Jeremiah N, Belot A, Jeziorski E, Duffy D, et al. Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J Allergy Clin Immunol. (2016) 138:1752–5. doi: 10.1016/j.jaci.2016.07.015
49. McLellan KE, Martin N, Davidson JE, Cordeiro N, Oates BD, Neven B, et al. JAK 1/2 Blockade in MDA5 Gain-of-Function. J Clin Immunol. (2018) 38:844–6. doi: 10.1007/s10875-018-0563-2
50. Kothur K, Bandodkar S, Chu S, Wienholt L, Johnson A, Barclay P, et al. An open-label trial of JAK 1/2 blockade in progressive IFIH1-associated neuroinflammation. Neurology. (2018) 90:289–91. doi: 10.1212/WNL.0000000000004921
51. Briand C, Frémond ML, Bessis D, Carbasse A, Rice GI, Bondet V, et al. Efficacy of JAK1/2 inhibition in the treatment of chilblain lupus due to TREX1 deficiency. Ann Rheum Dis. (2019) 78:431–3. doi: 10.1136/annrheumdis-2018-214037
52. Alsohime F, Martin-Fernandez M, Temsah MH, Alabdulhafid M, Le Voyer T, Alghamdi M, et al. JAK inhibitor therapy in a child with inherited USP18 deficiency. N Engl J Med. (2020) 382:256–65. doi: 10.1056/NEJMoa1905633
Keywords: Aicardi-Goutières syndrome, COVID-19, ruxolitinib, panniculitis, SAMHD1 mutation
Citation: Pararajasingam A, Bradley RE, Evans J, Lowe A, Goodwin R and Jolles S (2022) Case Report: Generalised Panniculitis as a Post-COVID-19 Presentation in Aicardi-Goutières Syndrome Treated With Ruxolitinib. Front. Pediatr. 10:837568. doi: 10.3389/fped.2022.837568
Received: 16 December 2021; Accepted: 01 March 2022;
Published: 25 April 2022.
Edited by:
Rita Consolini, University of Pisa, ItalyReviewed by:
Giorgio Costagliola, University of Pisa, ItalyAnnarosa Soresina, Azienda Socio Sanitaria Territoriale degli Spedali Civili di Brescia, Italy
Copyright © 2022 Pararajasingam, Bradley, Evans, Lowe, Goodwin and Jolles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen Jolles, am9sbGVzc3JAY2FyZGlmZi5hYy51aw==; orcid.org/0000-0002-7394-6804