 Hongping Li1†
Hongping Li1† Jingbo Jiang
Jingbo Jiang Chuan Nie
Chuan Nie Bin Xiao
Bin Xiao Jieyang Yu
Jieyang Yu- 1Department of Neonatology, Shenzhen Children's Hospital, Shenzhen, China
- 2Department of Neonatology, Guangdong Women and Children Hospital, Guangzhou, China
- 3Department of Critical Care Medicine, Baise Maternal and Child Hospital, Baise, China
Objectives: Vaginal microbiota is not only an important source of bacterial colonization for neonates, but also plays a crucial role in maternal and neonatal health. This study aimed to investigate the vaginal microbial community structure right after delivery and its impact on the neonatal oral microbiome.
Methods: In this study, 27 women were recruited from Bao'an Maternal and Child Health Hospital. Bacterial compositions of vaginal samples before and right after delivery and neonatal oral samples right after birth were investigated using 16S rRNA sequencing of V3–V4 hyperregions.
Results: Vaginal microbiome before delivery was dominated by the genus Lactobacillus. After delivery, the vaginal microbial community was altered, with significantly decreased proportion of Lactobacillus, increased alpha-diversity, and a more diverse ecological network. A large number subjects dominated by Lactobacillus species before delivery shifted to CST (community state type) IV after delivery. In addition, similar changes were observed in the neonatal oral microbiome, and its community profile was closer to vaginal samples after delivery than before delivery with principal coordinates analysis and microbial source tracking analysis.
Conclusion: The vaginal microbiome was altered right after delivery and impacted the colonization of the neonatal oral microbiome in China. Further, it is vital to understand the longitudinal influence on maternal and neonatal health of vaginal microbiome community changes after delivery.
Introduction
The vaginal microbiome is dynamic and influenced by many physiological changes such as sexual development, sexual intercourse, menstrual cycle, pregnancy, menopause, and hormone levels (1). Accumulating evidence proves that resident vaginal microbiota is related to bacterial vaginosis, menstruation, and sexually transmitted infections through interfering the proliferation of some organisms (1–4). Notably, dysbiosis of the vaginal microbiome community during pregnancy is associated with an increased risk of postpartum endometritis, preterm delivery, and spontaneous abortion (5–7). Not only as an important factor for women's reproductive health, vaginal microbiome in pregnancy also plays a key role in neonatal health (8). It is also understood that the bacterial community of the vagina of pregnant women is typically dominated by Lactobacillus species (9), many of which can produce biosurfactants and bacteriocins antagonistic to pathogens (10).
Understanding the community structure of the vaginal microbiome in pregnancy is important for maternal and neonatal health, which draws more and more attention. Several cross-sectional and longitudinal cohorts have been examined in studying the vaginal microbiome during pregnancy (9, 11, 12). Collectively, the results of these studies suggest that pregnancy leads to great stability, increases Lactobacillus proportional abundance, and reduces the richness and diversity of vaginal microbiome, although only a few studies investigate postpartum vaginal microbiome (13, 14). During the postpartum period, the vaginal microbiome is characterized by fewer Lactobacillus species and more diverse than that of pregnancy phase. However, all the vaginal samples included in the postpartum period are a few weeks after delivery; a comprehensive characterization of vaginal microbiome signature right after delivery has not yet been undertaken. The aim of this study was to characterize the composition of the vaginal microbiome right after delivery.
Neonates have been exposed to the maternal vaginal microbial ecosystem during the delivery process, and microbiota colonized at the vaginal birth canal is introduced to the neonate (15, 16). These pioneer microbial colonizers of the neonate, especially the gut and oral microbiome, play critical roles in neonatal health and development, including nutrient acquisition, immune programming, and protection from pathogens (16, 17), as the neonatal oral microbial structure is affected by multiple environmental factors, such as the maternal vaginal canal, skin-to-skin contact, maternal breast milk feeding, and so on (18, 19). As a valuable source of pioneer bacteria for neonatal oral microbiome, it is important to discern the relative potential contribution of the maternal vaginal community to the neonate oral microbiome. Herein, we tried to investigate the associations between the neonatal oral microbiome and vaginal microbiome before and right after delivery, respectively.
Methods
Study Subjects
Twenty-seven healthy and reproductive-aged women with gestational age >37 weeks were recruited in this study. They were asymptomatic and showed no clinical signs of vaginal disease upon examination by obstetricians. They were also with an uncomplicated singleton pregnancy and without medical problems or adverse outcomes during pregnancy, without known fetal anomalies or complications, and without antibiotics or other antimicrobial therapy during pregnancy. This study received ethics approval from Bao'an Maternal and Child Health Hospital (Shenzhen, China, no. LL2020012104). Written informed consent was obtained from all participants and parents/guardians of the recruited newborns. After obtaining written informed consent, all participants' demographic and clinical characteristics were collected via interview and by reviewing medical charts, including gestational age, height, weight, blood pressure, body mass index (BMI), ethnicity, age, and so on. Relevant clinical information was also obtained from neonates at birth.
Vaginal samples before delivery were collected at the first examination of hospital admissions of all participants (group BD, before delivery). Vaginal samples after delivery were collected before mothers left the delivery room with incision stitched (group AD, after delivery). The sterile swab was placed carefully on the vaginal sidewall about halfway between the introitus and the cervix, following the instructions reported previously (12), pressed firmly into the sidewall to a depth of roughly the diameter of the swab, rolled dorsally–ventrally back and forth four times to coat the swab. Neonatal oral samples were taken as soon as the newborns were delivered and before feeding by carefully swabbing the oral mucosa (group NO, neonatal oral) according to the previous study (20). Three sterile swabs were obtained for every sample by trained nurses to avoid insufficient DNA concentration. All samples were stored at 4°C and transferred to −80°C storage within 30 min after collection until DNA extraction.
Microbiome Profiling
DNA was extracted from vaginal and oral swabs using QIAamp DNA Mini kit (Qiagen, Germany). The concentrations and purity were measured using the NanoDrop One (Thermo Fisher Scientific, Waltham, MA, USA). The average DNA concentration was 41.52 ng/μL per sample. The DNA concentration of vaginal samples before delivery (69.68 ± 85.19 ng/μL) was higher than the samples after delivery (33.15 ± 49.63 ng/μL) and higher than that of the neonatal samples (21.71 ± 45.50 ng/μL).
V3–V4 hypervariable regions of 16S rRNA genes were amplified with forward primer 338F (5′-ACTCCTACGGGAGGCAGCAG-3′), reverse primer 806R (5′-GGACTACHVGGGTWTCTAAT-3′). Polymerase chain reaction (PCR) reactions, containing 25 μL 2 × Premix Taq (Takara Biotechnology, Dalian Co. Ltd., China), 2 μL of each 10 mM primer and 3 μL DNA template in a volume of 50 μL, were amplified under the following thermal profile: 94°C for 5 min, then 30 cycles of 94°C for 30 s, 52°C for 30 s, 72°C for 30 s, followed by 72°C for 10 min. Amplification products were visualized with 1% agarose gel electrophoresis. These PCR products were then pooled in equimolar and paired-end sequences on an Illumina MiSeq machine (Illumina, Inc., San Diego, CA, USA) with V3 chemistry.
Sequencing reads were assigned to each sample according to barcode sequences and filtered reads containing ambiguous bases or mismatches in the primer regions using custom Perl scripts. Then, the 16S rRNA gene sequences generated were analyzed using the bioinformatics software package QIIME2 (version 2019.4) (21). Paired-end reads were first denoised via DADA2 (22) offered by QIIME2 with command “qiime dada2 denoise-paired,” to merge paired-end reads and quality filtering and to exclude chimeric and phiX sequences. Further sequences were classified against Greengenes (13_8 revision) database at the species level using command “qiime feature-classifier classify-sklearn.” Meanwhile, an array of alpha- and beta-diversity measures was generated using command “qiime phylogeny align-to-tree-mafft-fasttree” and “qiime diversity core-metrics-phylogenetic” at a sampling depth of 1,000. Alpha-diversity was calculated by Shannon's diversity index, observed feature number, and Pielou's measure of species evenness. Weighted and unweighted UniFrac distances were used to represent beta-diversity.
Moreover, sequences were separately analyzed using Mothur software against Greegenes (13_8 revision) database as described previously (20, 23), where the sequences were clustered into OTUs (operational taxonomic units) based on the similarity threshold of 0.97.
Statistical Analysis
The continuous demographic variables were presented as the mean ± standard deviation (SD), including alpha- and beta-diversity indexes. The categorical characteristics were reported as numbers (%). All comparisons of this study were performed in R software at 0.05 level of significance using χ2 and t-tests for categorical and continuous variables, respectively.
Principal coordinates analysis (PCoA) was performed based on unweighted and weighted UniFrac distances using QIIME2 software and accompanied with permutational multivariate analysis of variance (PERMANOVA, 999 permutations) with R package “vegan.” LEfSe (linear discriminant analysis effect size) software (24) was used to analyze the significant different microbes between two groups with linear discriminant analysis >3.0 and P < 0.05. The microbial ecological network was constructed using the SPIEC-EASI method (25) and visualized with Gephi 0.9.2 software. SourceTracker 1.0.1 software (26) was further used to investigate the transmission from vaginal microbiome to neonatal oral microbiome.
Assignments of vaginal microbial profiles to community state types (CSTs) were performed according to community composition, and the community clustering was performed using McQuitty linkage hierarchical clustering analysis with R software as previously reported (12).
Results
Maternal and Neonatal Clinical Data
Demographic and clinical characteristics of the women and newborns are provided in Table 1. A total of 27 healthy and asymptomatic women were recruited into this study. All of the subjects were Han ethnicity ranging from 19 to 34 years old (mean age, 28.1 years). The average BMI was 25.2 kg/m2 (range, 21.1–34.1 kg/m2). All women gave birth between the 37th and 42nd gestational week with 14.26-kg (range, 9.0–26.30 kg) average gestational weight gain. The average birth weight of 27 newborns was 3,205.9 g (range, 2,600–3,850 g), including 15 boys and 12 girls.
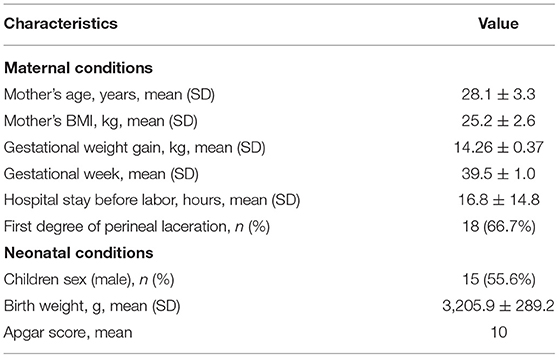
Table 1. Demographic and clinical data for study participants.
Changed Microbial Community After Delivery Accompanied With High Alpha-Diversity
To compare the overall vaginal community before and after delivery, PCoA was implemented on weighted and unweighted UniFrac distances separately (Figure 1). Both weighted and unweighted UniFrac distances showed that vaginal samples before delivery separated well from the subjects after delivery with PERMANOVA (R2 = 0.281, P = 0.001; R2 = 0.283, P = 0.001). Significant differences were also observed between vaginal samples before delivery and neonatal oral samples in unweighted (R2 = 0. 229, P = 0.001) and weighted UniFrac distances (R2 = 0. 261, P = 0.001), whereas a subtle difference was observed in unweighted UniFrac distances when comparing vaginal samples after delivery and neonatal oral samples (R2 = 0.027, P = 0.038), but no difference in weighted UniFrac distances (R2 = 0.026, P = 0.172). It indicated that the neonatal oral microbial community was closer to the vaginal microbial community after delivery than before delivery. Further, SourceTracker analysis demonstrated that an average of 30.11% neonatal oral microbiota potentially transmitted from vaginal microbiome before delivery, whereas 79.11% potentially transmitted from vaginal microbiome after delivery.
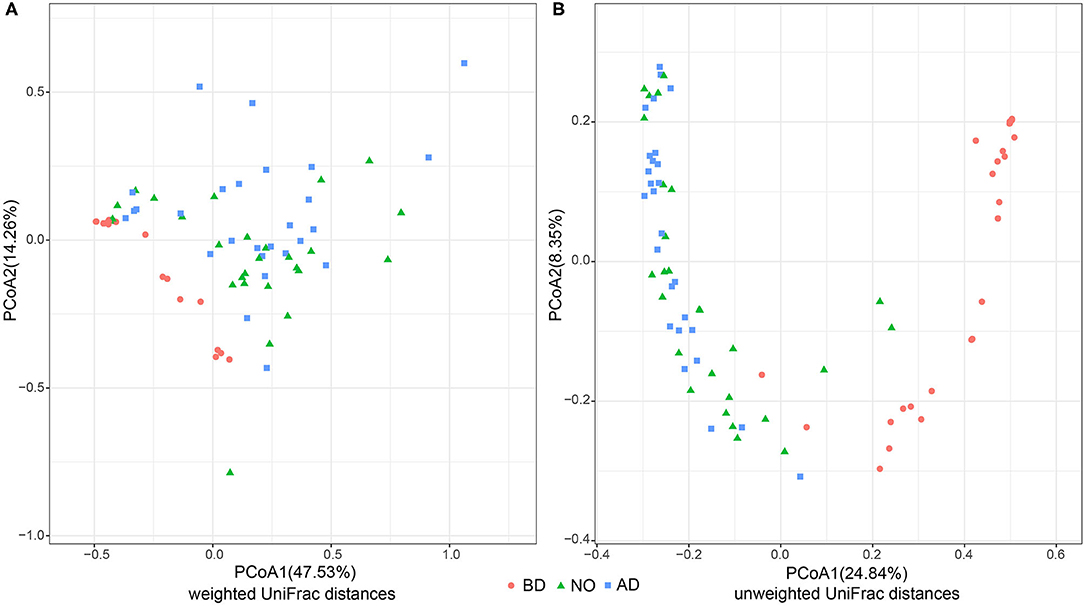
Figure 1. PCoA analysis of three groups based on weighted and unweighted UniFrac distances (group BD, before delivery; group NO, neonatal oral; group AD, after delivery). (A) PCoA analysis performed on weighted UniFrac distances. (B) PCoA analysis performed on unweighted UniFrac distances.
The vaginal microbiota after delivery and neonatal oral microbiota were associated with higher alpha-diversity, evenness, and richness (Figure 2). The mean Shannon index value was 0.92 ± 1.06 of the BD group, which was significantly lower than that of group AD (4.22 ± 1.50, P < 0.01) and NO (4.19 ± 2.22, P < 0.01) (Figure 2A). Increased mean Pielou index values were also observed in the AD (0.55 ± 0.16, P < 0.01) and NO (0.56 ± 0.24, P < 0.01) groups compared with group BD (0.25 ± 0.22) (Figure 2B). Meanwhile, the mean observed feature number increased significantly in neonatal oral samples (179.89 ± 113.37 vs. 13.30 ± 14.83, P < 0.01) and in vaginal samples after delivery (194.33 ± 85.69 vs. 13.30 ± 14.83, P < 0.01) (Figure 2C), compared with the vaginal samples before delivery.
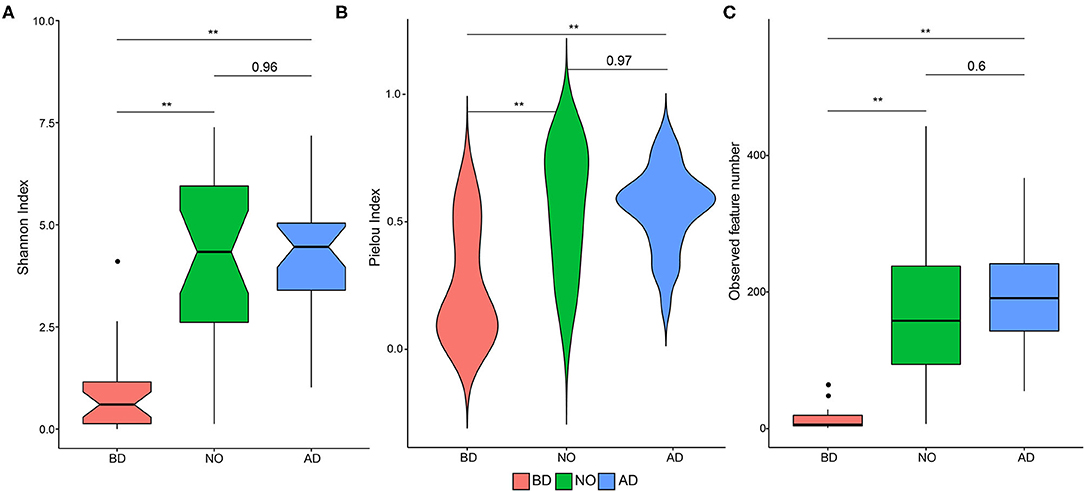
Figure 2. Alpha-diversity plots in microbial community and the comparisons among the three groups (group BD, before delivery; group NO, neonatal oral; group AD, after delivery). (A) Shannon index, (B) Pielou index, (C) observed feature number. Notes, **means P value < 0.01.
Compositional Differences of Vaginal Microbiome Between Before and After Delivery
The microbiome of study participants were characterized using high-throughput sequencing of the 16S rRNA V3–V4 high-variable regions. A total of 3,788,402 reads were included in the analysis. The average sequence read count was 46,770 per sample, with a median of 46,158 (range, 13,434–71,182), and the mean and median read lengths were 420 and 423 base pairs, respectively. The average relative abundance of each group at the phylum and genus levels is shown in Figure 3. It showed that the top 10 phyla were Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Cyanobacteria, Firmicutes, OD1, Planctomycetes, Proteobacteria, and Tenericutes (Figure 3A). Meanwhile, the top 10 genera were Atopobium, Enterococcus, Gardnerella, Lactobacillus, Prevotella, Pseudomonas, Ralstonia, Staphylococcus, Streptococcus, and Thiobacillus (Figure 3B).
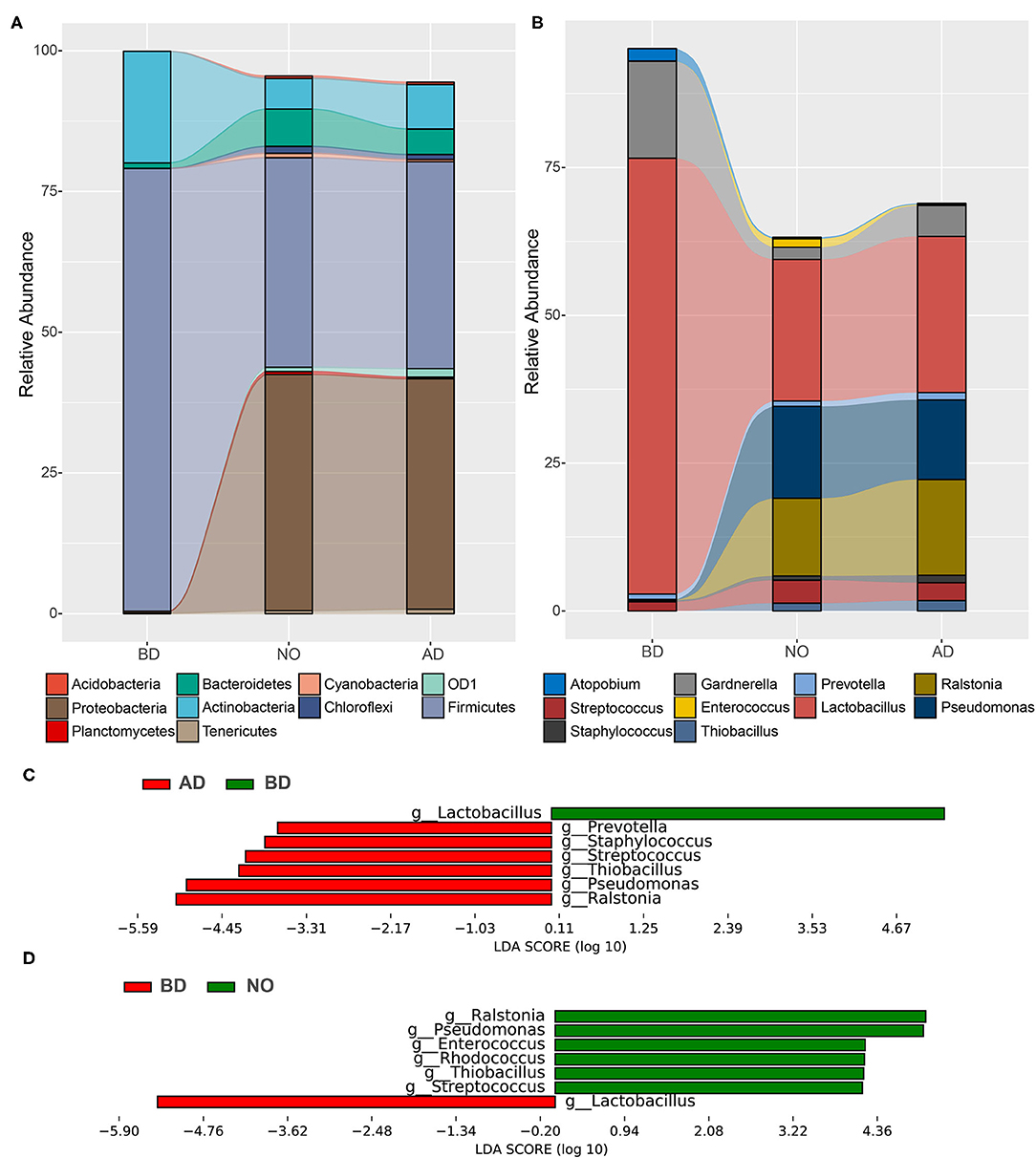
Figure 3. Relative proportions and differences of abundant microbes among the three groups. (A) Relative abundance at the phylum level. (B) Relative abundance at the genus level. (C) Significant different genera between BD and AD groups. (D) Significant different genera between the BD and NO groups.
The results also demonstrated that pregnancy was associated with a microbiome largely dominated by phyla Firmicutes (average, 78.65%) and Actinobacteria (average, 19.83%), which accounted for more than 98% of the microbial population, while substantial shifts in bacterial phylum community were observed at neonatal mucous and vaginal samples after delivery (Figure 3A), with dramatically decreased proportions of Firmicutes (37.20 and 36.75%) and Actinobacteria (5.44 and 7.92%), increased abundances of Proteobacteria (41.94% and 40.93% vs. 0.35%) and Bacteroidetes (6.64% and 4.52% vs. 0.97%). As the important genus in phylum Firmicutes, the average proportion of Lactobacillus decreased from 73.71% before delivery to 26.42% after delivery and 23.94% in neonatal oral, accompanied with increases in genera Streptococcus (3.04% vs. 1.56%), Ralstonia (16.19% vs. 0.04%), Pseudomonas (13.45% vs. 0.09%), and Thiobacillus (1.74% vs. 0%) (Figure 3B).
Next, the LEfSe software was used to detect significant differences in relative abundances of genera, accounting for >1% on average in at least one group. Figure 3C included a list of genera that are significantly different between vaginal samples before and after delivery. Among them, the genera Prevotella, Staphylococcus, Streptococcus, Thiobacillus, Pseudomonas, and Ralstonia increased significantly after delivery, whereas genus Lactobacillus decreased significantly. In addition, the significantly different genera between vaginal samples before delivery and neonatal oral samples are presented in Figure 3D. Compared with vaginal samples before delivery, the relative abundances of genera Ralstonia, Pseudomonas, Enterococcus, Rhodococcus, Thiobacillus, and Streptococcus were higher, whereas genus Lactobacillus was dramatically lower in neonatal oral samples. Notably, there was no difference between vaginal samples after delivery and neonatal oral samples. These observations collectively suggested that the vaginal microbial composition after delivery was significantly different from before delivery.
Different Ecological Network of Vaginal Microbiome Between Before and After Delivery
SpiecEasi software was used to construct ecological association networks at the family level of the vaginal microbiome before and after delivery, respectively. The results demonstrated that the ecological network of group BD included 55 nodes and 59 edges (Figure 4A). Modularity analysis indicated that family Lactobacillaceae coexisted with Bifidobacteriaceae. However, 293 nodes and 850 edges were presented in the AD group (Figure 4B), and the family Lactobacillaceae coexisted with other 40 families with modularity analysis. This was in accordance with the results observed by PCoA, indicating an obviously different vaginal microbial community right after delivery.
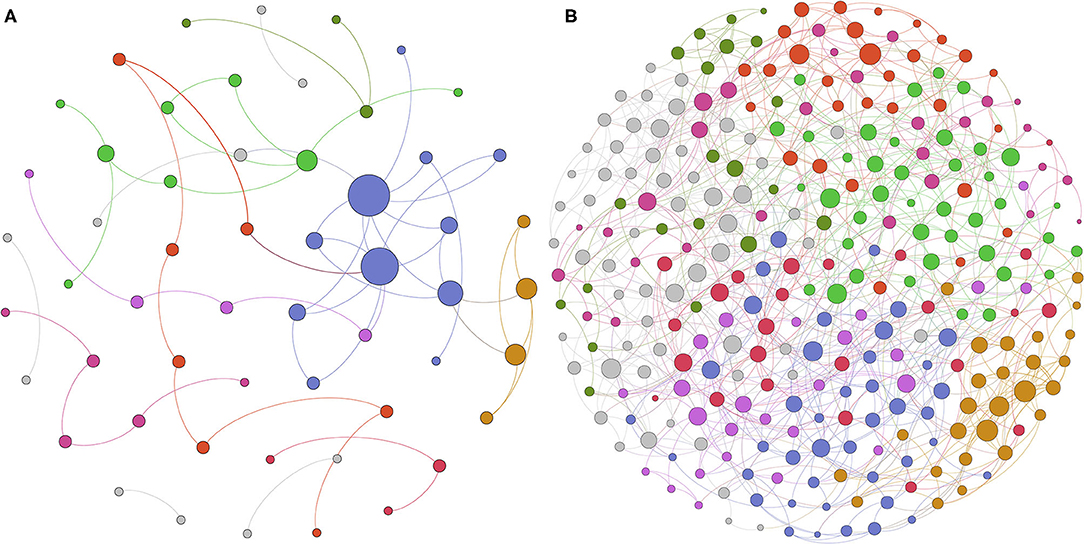
Figure 4. Different microbioal ecological network between before and after delivery with SpiecEasi analysis. (A) Microbial ecological network at the family level in the BD group. (B) Microbial ecological network at the family level in the AD group.
Community State Types Analysis and Its Changes After Delivery
Categorizing microbiome profiles based on the taxon with the largest proportion of reads and hierarchical clustering analysis of bacterial species from the pregnant vaginal microbiome profiles revealed three major CSTs (Figure 5A). Among all samples, eight samples were dominated by the species Lactobacillus iners (CST III). Six samples were assigned to CST IV, which were dominated by the genus Gardnerella, and also typified by higher proportions of Aerococcus, Atopobium, Bifidobacterium, Corynebacterium, Dialister, Finegoldia, Megasphaera, Mobiluncus, Peptoniphilus, Prevotella, Ralstonia, Staphylococcus, Streptococcus, and Sneathia than other CSTs. The rest of the 13 samples were dominated by the species Lactobacillus helveticus or Lactobacillus delbrueckii, which were clustered into a new observed community type and named CST VI in this study.
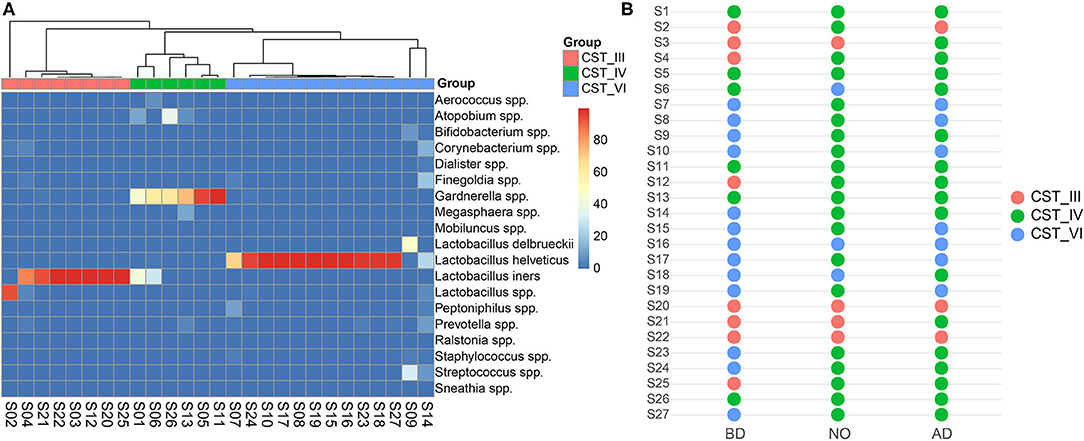
Figure 5. Vaginal community state types presented throughout before and after delivery. (A) Hierarchical clustering analysis using McQuitty linkage of microbial species abundance showed that these subjects were clustered into three major groups. (B) The switch of community state types among the three groups.
The dominance of CST IV was observed in neonatal samples and postpartum vaginal samples (n = 20 and n = 17 in groups NO and AD, respectively) (Figure 5B). Most subjects of group BD belonged to CST III and CST VI (15/21) switched to CST IV of group NO. Similarly, they (11/21) shifted to CST IV in the vaginal microbiome after delivery.
Discussion
In this study, the composition of the vaginal microbiota of women after delivery was characterized and compared with the profile before delivery. Increased microbial richness and diversity were observed in the vaginal samples right after delivery in this study, which was consistent with previous studies with 6 weeks' postpartum samples (13, 27). In addition, dramatic changes in community membership of the vaginal microbiome were observed right after delivery based on PCoA results. In concordance with previous observations (8, 9, 12), Lactobacillus was the predominant genus in most of the samples before delivery in this study, along with low proportions of Atopobium, Gardnerella, Prevotella, and Streptococcus. Whereas, after delivery, Lactobacillus significantly decreased, the genera Prevotella, Staphylococcus, Streptococcus, Thiobacillus, Pseudomonas, and Ralstonia increased significantly. Furthermore, a more diverse ecological network with more nodes and edges was presented in vaginal microbiota after delivery. Taken together, these results demonstrated that the vaginal microbial community right after delivery was altered.
At labor onset, the vaginal microbiome is still dominated by family Lactobacillaceae and Bifidobacteriaceae (28). Moreover, the microbiota of cesarean-born infants partially restore after being exposed to maternal vaginal fluids at birth with sterile gauze and become more and more similar to the microbiota of vaginal delivery infants (29). The previously published results indicate that lactic acid–producing bacteria are still abundant at labor or right after delivery. However, significant declines in abundances of Lactobacillus species and an increase in alpha-diversity right after delivery were observed in this study, potentially resulting from the disinfecting operation at labor of the study cohorts. In China, as vaginal examination of standard delivery in the textbook Obstetrics and Gynecology (eighth edition) required, soapy water and povidone-iodine are used to clean and disinfect the vulva during delivery in clinic (30). In clinical practice, all gynecologists use cotton ball stained or hand impregnated with povidone-iodine to sterilize the vulva (20). Povidone-iodine is a broad-spectrum antiseptic for preventing skin infections, and its efficacy, particularly on resistant microorganisms, has been shown (31). Its clinical use could alter the vaginal microbial community to a large extent. However, for the deficiency of samples without disinfection in this study, the exact effect of povidone-iodine use on vaginal microbiome was not clear. Follow-up investigation will be taken in our next study.
The vaginal microbiota of women can be generally clustered into five CSTs (9). Four of the CSTs are characterized by high levels of Lactobacillus crispatus (CST I), Lactobacillus gasseri (CST II), L. iners (CST III), and Lactobacillus jensenii (CST V). On the other hand, CST IV with reduced Lactobacillus species is enriched in a combination of various anaerobic bacteria (9, 32). In this study, the vaginal microbiome was clustered into three major groups by using hierarchical cluster analysis. CST III and CST IV are previously described in North America (33) and European (13, 34), whereas a new CST (named CST VI) dominated by L. helveticus was shown in this study. In order to avoid the taxonomic bias caused by the bioinformatics method, the sequences were aligned to the 16S rRNA database in the National Center for Biotechnology Information using BLAST. Meanwhile, the sequence reads were reanalyzed using Mothur software (23). Both of them confirmed that a number of samples were dominated by L. helveticus. Previous studies demonstrated that CSTs seem to depend on ethic and biogeographical background (13, 33), and these studies mainly focus on North America and Europe. The new CST dominated by L. helveticus might be closely related to our study samples as they all were recruited from Han ethnicity in this study. In further research, large cohort studies should be taken into account to confirm this observation. Beyond this, the V3–V4 regions of 16S rRNA were used to profile the vaginal microbiome community in this study, which was different with a previous study based on cpn60 gene (32). To cull out the discrepancy caused by the amplified region, lower taxonomic levels should be investigated with whole-genome sequencing in the next study. In addition, the results showed that most samples during pregnancy belonged to CST III and CST VI, with high abundance of Lactobacillus species, whereas after delivery the majority of CST III and CST VI before delivery switched to CST IV, in concordance with previous studies (9, 13, 35). Increased rate of CST IV was also observed in neonatal oral samples in this study. CST IV is associated with high Nugent scores and increased risk of bacterial vaginosis, which are linked to preterm birth and chorioamnionitis (13, 36). Further, it has been reported that Lactobacillus is the most prevalent and dominant bacterium in female reproductive tract, which appears to ensure normal vaginal microbiota and effectively inhibits the colonization of pathogens through producing lactic acid, H2O2, and so on (1). Thus, more attention should be paid on the maternal and neonatal health after delivery with Lactobacillus decrease induced by using povidone-iodine.
By the time of delivery, the neonate is introduced as the bacteria colonized at the vaginal birth canal (37). Both the PCoA and SourceTracker analysis results consistently indicated that the neonatal oral microbiome was more closely related to the vaginal microbiome after delivery, whereas they were both notably different to the vaginal microbiome before delivery in this study. It indicated that the microbes first colonized at neonatal oral primarily transmitted from the vaginal microbiome right after delivery. That would be helpful to understand the establishment process of the first microbial colonizers of newborns, and it is essential for gaining a comprehensive understanding of neonatal development.
The absence of a longitudinal data set of maternal and neonatal health revealed some insights that need to be determined in further studies. First, during pregnancy, some vaginal microbiota profiles belonged to CST III, some belonged to CST IV, and some were clustered into CST VI. Their long-term impact on maternal and neonatal health should be conducted in the following study. Second, after delivery, some vaginal microbiome communities were still dominated by Lactobacillus, whereas others with a small proportion of Lactobacillus and similar phenomena were observed in the neonatal oral microbiome. However, the effects of these changes on maternal and neonatal health were not clear. Third, some pregnant women might not need to be disinfected at labor, and some neonates might need to be supplemented with probiotics during the growth and developmental process according to microbial profiles. The clinical operations should be changed according to the result of a large cohort study.
In summary, it was observed in this study that the vaginal microbiome right after delivery was altered, potentially due to perineal disinfection, which affected the colonization of the neonatal oral microbiome. The obvious changes of vaginal microbiome after delivery were the decreased relative abundance of Lactobacillus and changed ecological network. These observations will be helpful to understand the community profile of the vaginal microbiome right after delivery in Chinese pregnant women.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: PRJNA596821.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethical Committee of Shenzhen Bao'an Maternity and Child Health Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
JY and HL conceived of the presented idea and planned the experiments. JJ and QL carried out the experiments. BX and CN designed the computational framework and analyzed the data. HL, JJ, BX, and JY wrote the manuscript. All authors discussed the results and contributed to the final manuscript.
Funding
This work was funded by Health and Wellness Commission of Guangxi Zhuang Autonomous Region (20211814), Guangdong Natural Science Fund Project (2018A030313605), Guangzhou Science and Technology Commission Project (201804010090).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Greenbaum S, Greenbaum G, Moran-Gilad J, Weintraub AY. Ecological dynamics of the vaginal microbiome in relation to health and disease. Am J Obstet Gynecol. (2019) 220:324–35. doi: 10.1016/j.ajog.2018.11.1089
2. Gupta S, Kakkar V, Bhushan I. Crosstalk between vaginal microbiome and female health: a review. Microb Pathog. (2019) 136:103696. doi: 10.1016/j.micpath.2019.103696
3. Ravel J, Brotman RM, Gajer P, Ma B, Nandy M, Fadrosh DW, et al. Daily temporal dynamics of vaginal microbiota before, during and after episodes of bacterial vaginosis. Microbiome. (2013) 1:29. doi: 10.1186/2049-2618-1-29
4. Brotman RM. Vaginal microbiome and sexually transmitted infections: an epidemiologic perspective. J Clin Invest. (2011) 121:4610–7. doi: 10.1172/JCI57172
5. Zhao F, Chen Y, Gao J, Wu M, Li C, Wang Z, et al. Characterization of vaginal microbiota in women with recurrent spontaneous abortion that can be modified by drug treatment. Front Cell Infect Microbiol. (2021) 11:680643. doi: 10.3389/fcimb.2021.680643
6. Nasioudis D, Forney LJ, Schneider GM, Gliniewicz K, France M, Boester A, et al. Influence of pregnancy history on the vaginal microbiome of pregnant women in their first trimester. Sci Rep. (2017) 7:10201. doi: 10.1038/s41598-017-09857-z
7. Jacobsson B, Pernevi P, Chidekel L, Jörgen Platz-Christensen J. Bacterial vaginosis in early pregnancy may predispose for preterm birth and postpartum endometritis. Acta Obstet Gynecol Scand. (2002) 81:1006–10. doi: 10.1034/j.1600-0412.2002.811103.x
8. Fettweis JM, Serrano MG, Brooks JP, Edwards DJ, Girerd PH, Parikh HI, et al. The vaginal microbiome and preterm birth. Nat Med. (2019) 25:1012–21. doi: 10.1038/s41591-019-0450-2
9. DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci USA. (2015) 112:11060–5. doi: 10.1073/pnas.1502875112
10. O'Hanlon DE, Moench TR, Cone RA. In vaginal fluid, bacteria associated with bacterial vaginosis can be suppressed with lactic acid but not hydrogen peroxide. BMC Infect Dis. (2011) 11:200. doi: 10.1186/1471-2334-11-200
11. Walther-António MRS, Jeraldo P, Berg Miller ME, Yeoman CJ, Nelson KE, Wilson BA, et al. Pregnancy's stronghold on the vaginal microbiome. PLoS ONE. (2014) 9:e98514. doi: 10.1371/journal.pone.0098514
12. Serrano MG, Parikh HI, Brooks JP, Edwards DJ, Arodz TJ, Edupuganti L, et al. Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy. Nat Med. (2019) 25:1001–11. doi: 10.1038/s41591-019-0465-8
13. MacIntyre DA, Chandiramani M, Lee YS, Kindinger L, Smith A, Angelopoulos N, et al. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci Rep. (2015) 5:8988. doi: 10.1038/srep08988
14. Zhang X, Zhai Q, Wang J, Ma X, Xing B, Fan H, et al. Variation of the Vaginal Microbiome During and after Pregnancy in Chinese Women. Genomics Proteomics Bioinformatics. (2022). doi: 10.1016/j.gpb.2021.08.013. [Epub ahead of print].
15. Dogra S, Sakwinska O, Soh SE, Ngom-Bru C, Brück WM, Berger B, et al. Dynamics of infant gut microbiota are influenced by delivery mode and gestational duration and are associated with subsequent adiposity. mBio. (2015) 6:e02419-14. doi: 10.1128/mBio.02419-14
16. Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, Aagaard KM. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med. (2017) 23:314–26. doi: 10.1038/nm.4272
17. Huurre A, Kalliomäki M, Rautava S, Rinne M, Salminen S, Isolauri E. Mode of delivery—effects on gut microbiota and humoral immunity. Neonatology. (2008) 93:236–40. doi: 10.1159/000111102
18. Lif Holgerson P, Harnevik L, Hernell O, Tanner AC, Johansson I. Mode of birth delivery affects oral microbiota in infants. J Dent Res. (2011) 90:1183–8. doi: 10.1177/0022034511418973
19. He J, Li Y, Cao Y, Xue J, Zhou X. The oral microbiome diversity and its relation to human diseases. Folia Microbiol (Praha). (2015) 60:69–80. doi: 10.1007/s12223-014-0342-2
20. Li H, Chen S, Wu L, Wang H, Xiao K, Gao Y, et al. The effects of perineal disinfection on infant's oral microflora after transvaginal examination during delivery. BMC Pregnancy Childbirth. (2019) 19:213. doi: 10.1186/s12884-019-2350-3
21. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. (2019) 37:852–7. doi: 10.1038/s41587-019-0209-9
22. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. (2016) 13:581–3. doi: 10.1038/nmeth.3869
23. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. (2009) 75:7537–41. doi: 10.1128/AEM.01541-09
24. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. (2011) 12:R60. doi: 10.1186/gb-2011-12-6-r60
25. Kurtz ZD, Müller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. Sparse and compositionally robust inference of microbial ecological networks. PLOS Comput Biol. (2015) 11:e1004226. doi: 10.1371/journal.pcbi.1004226
26. Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, et al. Bayesian community-wide culture-independent microbial source tracking. Nat Meth. (2011) 8:761–3. doi: 10.1038/nmeth.1650
27. Huang YE, Wang Y, He Y, Ji Y, Wang LP, Sheng HF, et al. Homogeneity of the vaginal microbiome at the cervix, posterior fornix, and vaginal canal in pregnant Chinese women. Microb Ecol. (2015) 69:407–14. doi: 10.1007/s00248-014-0487-1
28. Avershina E, Slangsvold S, Simpson MR, StorrØ O, Johnsen R, Øien T, et al. Diversity of vaginal microbiota increases by the time of labor onset. Sci Rep. (2017) 7:17558. doi: 10.1038/s41598-017-17972-0
29. Dominguez-Bello MG, De Jesus-Laboy KM, Shen N, Cox LM, Amir A, Gonzalez A, et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat Med. (2016) 22:250–3. doi: 10.1038/nm.4039
31. Durani P, Leaper D. Povidone–iodine: use in hand disinfection, skin preparation and antiseptic irrigation. Int Wound J. (2008) 5:376–87. doi: 10.1111/j.1742-481X.2007.00405.x
32. Albert AYK, Chaban B, Wagner EC, chellenberg JJ, Links MG, van Schalkwyk J, et al. A study of the vaginal microbiome in healthy Canadian women utilizing cpn60- based molecular profiling reveals distinct gardnerella subgroup community state types. PLoS ONE. (2015) 10:e0135620. doi: 10.1371/journal.pone.0135620
33. Zhou X, Brown CJ, Abdo Z, Davis CC, Hansmann MA, Joyce P, et al. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J. (2007) 1:121–33. doi: 10.1038/ismej.2007.12
34. Kiss H, Kögler B, Petricevic L, Sauerzapf I, Klayraung S, Domin K, et al. Vaginal Lactobacillus microbiota of healthy women in the late first trimester of pregnancy. BJOG. (2007) 114:1402–7. doi: 10.1111/j.1471-0528.2007.01412.x
35. Doyle R, Gondwe A, Fan YM, Maleta K, Ashorn P, Klein N, et al. Lactobacillus-deficient vaginal microbiota dominates postpartum women in rural Malawi. Appl Environ Microbiol. (2018) 84:e02150–17. doi: 10.1128/AEM.02150-17
36. Onderdonk AB, Delaney ML, Fichorova RN. The human microbiome during bacterial vaginosis. Clin Microbiol Rev. (2016) 29:223–38. doi: 10.1128/CMR.00075-15
Keywords: vaginal microbiota, neonatal oral microbiome, community state type, 16S rRNA sequencing, ecological network
Citation: Li H, Jiang J, Nie C, Xiao B, Li Q and Yu J (2022) Community Structure and Ecological Network's Changes of Vaginal Microbiome in Women Right After Delivery. Front. Pediatr. 10:750860. doi: 10.3389/fped.2022.750860
Received: 31 July 2021; Accepted: 11 February 2022;
Published: 28 March 2022.
Edited by:
Fiammetta Piersigilli, Cliniques Universitaires Saint-Luc, BelgiumReviewed by:
Yuriy Fofanov, University of Texas Medical Branch at Galveston, United StatesSara De Carolis, Agostino Gemelli University Polyclinic (IRCCS), Italy
Copyright © 2022 Li, Jiang, Nie, Xiao, Li and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jieyang Yu, eXVqaWV5YW5nNTQ4MUBzaW5hLmNvbQ==
†These authors have contributed equally to this work