 Yitong Shen
Yitong Shen Anne P. R. Boulton
Anne P. R. Boulton Robert L. Yellon
Robert L. Yellon Matthew C. Cook
Matthew C. Cook
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 17 January 2023
Sec. Pediatric Immunology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1098426
This article is part of the Research Topic Dermatologic Manifestations of Primary Immune Deficiency Disorders in Children View all 6 articles
More than 400 single gene defects have been identified as inborn errors of immunity, including many arising from genes encoding proteins that affect NF-κB activity. We summarise the skin phenotypes in this subset of disorders and provide an overview of pathogenic mechanisms. NF-κB acts cell-intrinsically in basal epithelial cells during differentiation of skin appendages, influences keratinocyte proliferation and survival, and both responses to and amplification of inflammation, particularly TNF. Skin phenotypes include ectodermal dysplasia, reduction and hyperproliferation of keratinocytes, and aberrant recruitment of inflammatory cells, which often occur in combination. Phenotypes conferred by these rare monogenic syndromes often resemble those observed with more common defects. This includes oral and perineal ulceration and pustular skin disease as occurs with Behcet's disease, hyperkeratosis with microabscess formation similar to psoriasis, and atopic dermatitis. Thus, these genotype-phenotype relations provide diagnostic clues for this subset of IEIs, and also provide insights into mechanisms of more common forms of skin disease.
Mammalian nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) comprises a family of homo- and hetero-dimers made of five rel-homology domain-containing proteins (c-Rel, RelA, p105/50, p100/52, and RelB, encoded by REL, RELA, NFKB1, NFKB2, and RELB, respectively) (1, 2) (Table 1). Discovery of NF-κB provided the first evidence for tissue-specific transcription based on post-translational regulation of transcription factors in response to cellular stimulation or environmental cues (3). Nuclear translocation of NF-κB influences numerous transcriptional programs critical for activation, differentiation and survival of cells of innate and adaptive immunity, but also for organogenesis of primary and secondary lymphoid organs. Expression of NF-κB is not confined to the immune system. Many non-haemopoietic tissues also rely on NF-κB to maintain homeostasis. This includes the skin and other ectoderm-derived tissues, where NF-κB operates cell-intrinsically to maintain keratinocyte adhesion, homeostasis, and epidermal organisation.
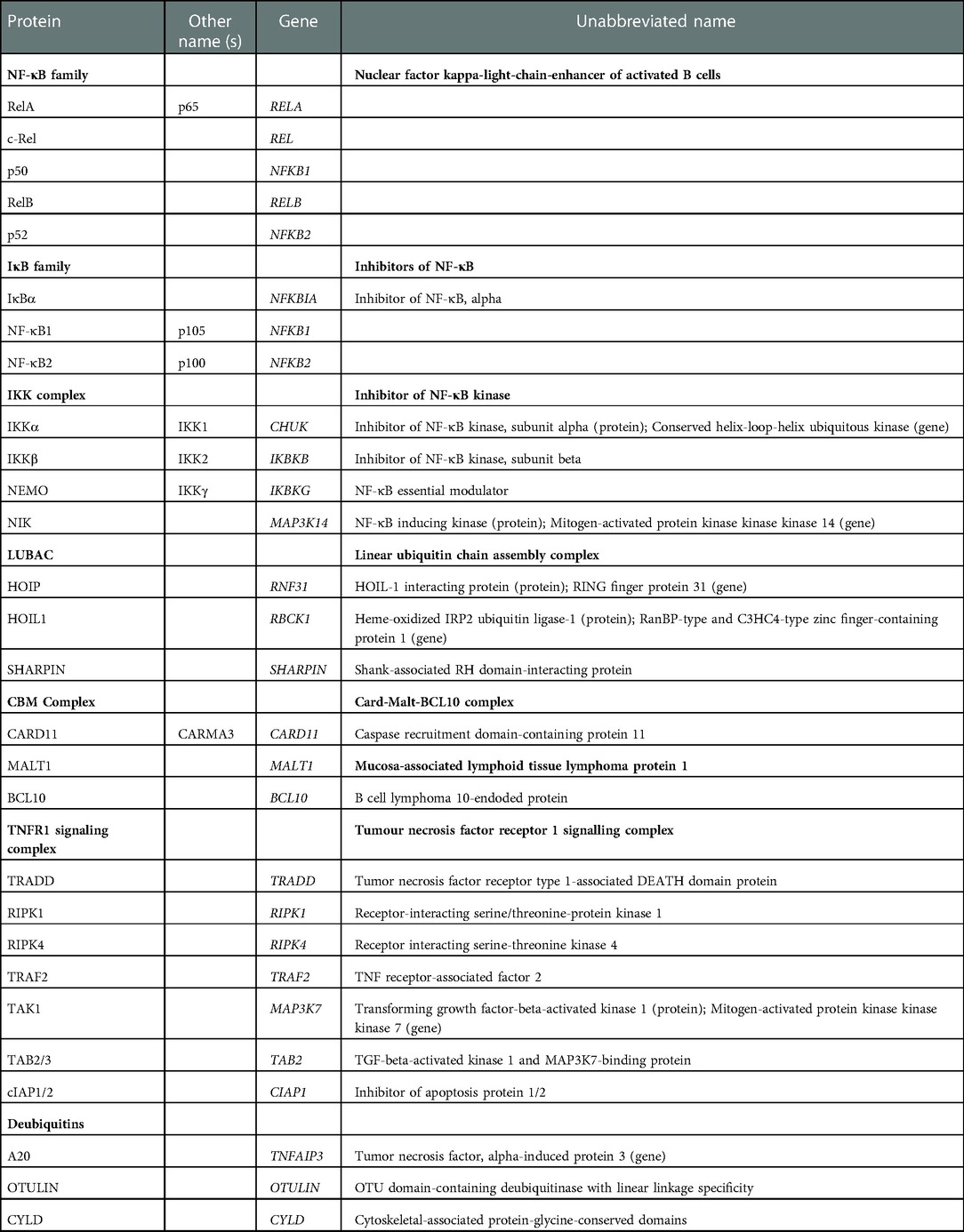
Table 1. Summary of NF-κB proteins and their regulators.
Genetic defects have been identified in NF-κB molecules, their upstream signalling pathways, and regulator components (4–7). Since NF-κB is expressed in both the immune system and cellular components of the skin, it is not surprising that many of the inborn errors of immunity (IEI) arising from defects in NF-κB result in dermatological and mucosal phenotypes. Indeed, these are sometimes the presenting features.
Transcriptionally active forms of NF-κB consist of dimers that recognize cis-acting κB sites in promoter and enhancer elements (e.g., the decameric sequence 5′-GGGACTTTCC-3′; consensus 5′-GGGPuNNPyPyCC-3) of many genes (8). A defining feature of NF-κB is its rapid response to cues from the cell-environment, which is possible because NF-κB dimers and higher order complexes exist pre-formed and maintained in a latent state within the cytoplasm by a family of inhibitors of NF-κB (IκB), including IκBα, IκBβ, IκBδ (p100, see below), IκBε, BCL3, and IκBNS, each containing approximately six ankyrin repeats (Table 1) (9–11) NF-κB becomes transcriptionally active when these IκB molecules undergo transient degradation, which is triggered by the IκB kinase (IKK) complex (Figure 1).
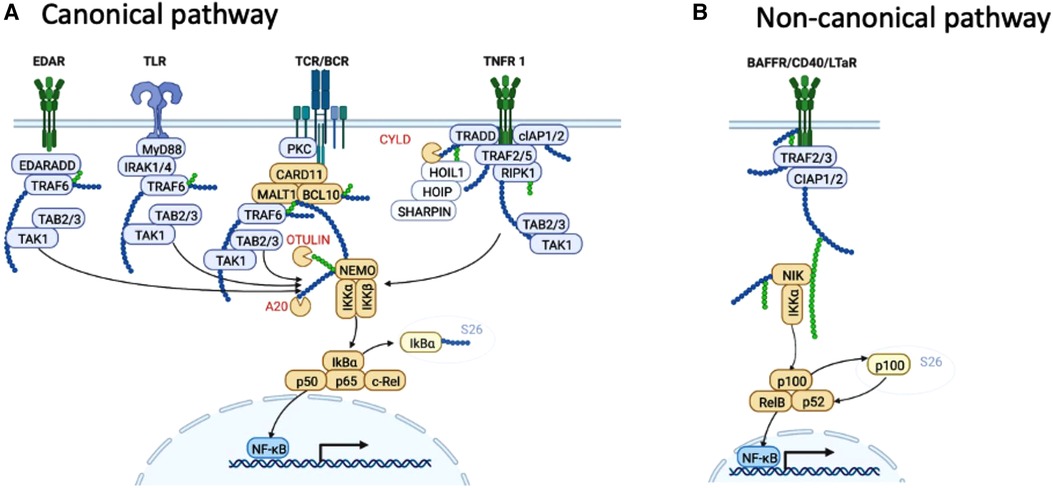
Figure 1. NF-κB pathways. (A) Summary of ligands and signalling events that lead to activation of the canonical (classical) NF-κB pathway. (B) Summary of ligands and signalling events that lead to activation of the non-canonical (alternative) NF-κB pathway. Proteins discussed in this review are shown in orange. Created with BioRender.com.
NF-κB activation can proceed through either of two pathways determined by specific kinases, referred to as canonical (or classical) and non-canonical (or alternative). Dimers comprising p50 (NF-κB1), RelA and c-Rel become transcriptionally active via the canonical pathway, while dimers comprising RelB and p52 become transcriptionally active via the non-canonical pathway (12). Tissue-specific regulation of NF-κB results from cell context-specific cues that activate NF-κB via various receptors. In the immune system, these include pattern recognition receptors such as toll-like receptors (TLR), NOD-like receptors, and pro-inflammatory cytokine receptors, especially IL-1 and TNF. The canonical pathway can also be activated downstream of B cell and T cell receptors, and in basal epithelial cells, from ligation of ectodysplasin associated receptor (EDAR) (Figure 1). Different membrane-proximal signalling pathways are responsible for delivering the signal after different types of receptor are liganded. Thus, a complex of TRAF6, MyD88 and IRAK1/4 signal from TLRs, protein kinase C (PKC) activates the CARD11-BCL10-MALT1 (CBM) complex downstream of T cell receptor (TCR) and B cell receptor (BCR), and the so-called complex 1, comprising RIPK1, TRADD, cIAP1 and 1, and TAB2 and 3 signal from TNFR1. These membrane proximal events converge on the IKK complex comprising IKKα, IKKβ, and IKKγ (aka NEMO) (13–16), which phosphorylate IκB molecules (17), tagging them for ubiquitination and 26S proteasomal degradation. As a result, NF-κB components p65, c-Rel and p50 are liberated to translocate to the nucleus.
While bearing similarities to canonical activation, the non-canonical pathway differs in kinetics and mechanism of activation, and hinges on p100 processing to p52, which unlike p105 processing, is regulated rather than constitutive (18). NF-κB-inducing kinase (NIK) undergoes constant turnover in resting cells but after appropriate stimulation, NIK turnover is retarded to permit formation of complexes with IKKα, which are responsible for phosphorylation of p100 (NF-κB2). Limited proteasomal degradation of phosphorylated p100 yields p52, which forms transactivating heterodimers with RelB (19).
NF-κB is subject to tight regulation mediated by many layers of feedback. Negative regulation is intrinsic to NF-κB signalling. For example, p100 and p105 which are precursors of active NF-κB transcription factors are themselves members of the IκB family (20), and p50 and p52 homodimers are inhibitory (21). Post-transcriptional regulation by phosphorylation and both K63 and M1 linear ubiquitination affects protein abundance and protein-protein interactions. Linear ubiquitination is mediated by the linear ubiquitin (LUBAC) complex, comprising HOIP, HOIL1 and SHARPIN (22). Deubiquitinases A20, CYLD, and OTULIN are critical regulators of NF-κB activity (23, 24). The deubiquitinating action of A20 has been attributed to the N-terminal OTU domain that removes K63 complexes from NEMO, RIPK1, TRAF6, and MALT1. The action of A20 is complex, however, as C-terminal zinc fingers have E3 ligase activity, and A20 also protects M1 chains (added by HOIP) from removal, which might limit apoptosis after TNF ligation (25). Otulin (OTU deubiquitinase with linear linkage specificity) removes linear polyubiquitin from proteins that had been modified by the LUBAC complex (26).
NF-κB mediates organisation and differentiation during development of the ectoderm into skin, its appendages (apocrine glands, hair, and nails), as well as mammary glands, nervous system, placodes, anterior hypophysis, lens, and olfactory epithelium (Table 2) (27, 28). Signals necessary for differentiation of ectodermal progenitors for formation of skin appendages depends on ligation of the TNFR superfamily member EDAR and its downstream adaptor protein EDARADD, which activates canonical NF-κB. Defects in EDAR and EDARADD result in ectodermal dysplasia but as they are skin-specific, no systemic features (27, 29–31). These findings provide important insights into the phenotypes conferred by defects in NF-κB but this review will focus on defects that are associated with concurrent immune defects.
Table 2. Ectodermal tissues.
In addition to ectodermal development, NF-κB is also important for regulation of keratinocyte growth and differentiation, skin inflammation, and cutaneous immunity (32) (Figure 2). Mouse studies have identified how the interplay of these signals regulates keratinocyte homeostasis. Dominant negative mutation of Nfkbia (IκBα) results in neonatal epidermal hyperplasia, which appears to be a cell-intrinsic action in keratinocytes (33, 34). Consistent with these observations, constitutive p65 activity has been found to act cell-intrinsically in basal keratinocytes to inhibit their proliferation (33).
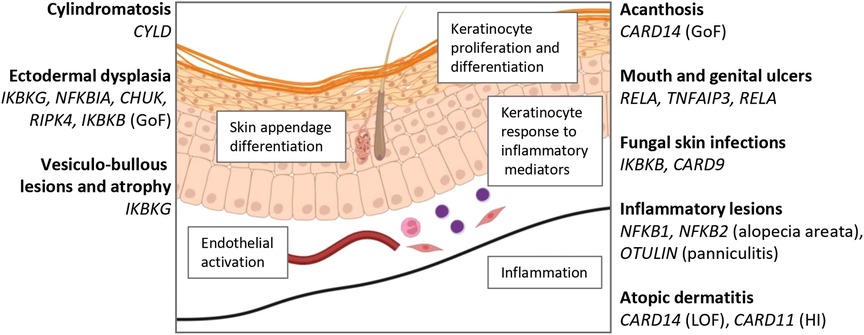
Figure 2. Summary of skin pathology associated with defects in NF-κB. Created with BioRender.com.
The hypothesis that NF-κB arrests keratinocyte proliferation permitting differentiation was challenged by a mouse model of epidermal keratinocyte-specific IKKβ deficiency, in which mice were born with macroscopically normal skin and developed a cutaneous inflammatory response before progressive dermatitis commencing on day 4–5 (35). In this model, epidermal proliferation was blocked by TNFR deficiency, suggesting that NF-κB regulates the proliferative response of keratinocytes in response to TNFR1 ligation (32, 35).
TNF-dependent inflammation is also a feature of both models in which there is reduction or enhanced NF-κB activity. Ikbkg (NEMO) deficiency was shown to arrest keratinocyte growth and trigger dependent inflammation, both of which were TNF-dependent (36). Similarly, Tak1 deletion, which reduces canonical NF-κB activation also results in TNF-mediated skin inflammation (37). On the other hand, Tnfaip3 deletion, which enhances NF-κB responses, resulted in marked TNF-dependent epidermal proliferation (38), and keratinocyte apoptosis and dermal TNF-dependent inflammation was observed with keratinocyte-conditional deficiency of Traf2 (39).
IKKα deficiency identified a further level of complexity. IKKα is critical for normal keratinocyte differentiation, including expression of filaggrin and loricin, as well as for tooth development (40–42), however, this action is mediated independently of its kinase activity and, indeed, NF-κB signalling (43). The action of IKKα nevertheless depends on its nuclear localisation (44). Defects in keratinocytes conferred by Chuk−/− (i.e., IKKα-deficient) can be overcome by a soluble factor originally named keratinocyte differentiation factor, but the mechanism of action of IKKα on keratinocyte differentiation remains unclear (43). A potential mechanism is via epidermal growth factor receptor (EGFR) signalling, as IKKα negatively regulates EGFR via ADAM (A disintegrin and metalloprotease) expression in keratinocytes (45).
As outlined below, the spectrum of skin pathology observed in patients with NF-κB defects reflects the requirement for tight regulation of NF-κB. For many genes in the NF-κB pathway, severe phenotypes have been reported for complete loss of function mutations. Components of the NF-κB pathway also appear to be susceptible to the actions of missense mutations, possibly because of the large number of protein-protein interactions required for NF-κB activation. In many instances, phenotypes are also conferred by hypo- and hypermorphic mutations, and these phenotypes often differ significantly from those conferred by loss-of-function (LoF) mutations.
It is important to note that as a consequence of the complex regulatory network for NF-κB, the outcome of a hypermorphic or hypomorphic mutation of a specific protein depends on whether the affected protein is a positive or negative regulator of NF-κB. For example, gain-of-function (GoF) mutation of a negative regulator such as IκBα will have a similar functional consequence as LoF mutation of a IKK complex component such as NEMO (46). Furthermore, as revealed by mutations in p100 (NFKB2), a protein with both IκB activity (via the p100 precursor) and transcriptional activity (via p52), both LoF and GoF can be conferred by the same mutation.
Loss-of-function (amorphic) mutations in IKBKG result in incontinentia pigmenti (IP) (47). IP usually begins shortly after birth. The phenotype is variable but in classic form is characterised by four distinct dermatological stages, beginning with vesiculobullous eruption (Stage I) then verrucae (Stage II), hyperpigmentation (Stage III), and finally atrophic hypopigmentation (Stage IV) (48, 49). Mucocutaneous ulcers of both mouth and perineum are also common, and have resulted in the concurrent diagnosis of Behcet's disease and IP (50, 51). Now that similar manifestations have been observed with TNFAIP3 haploinsufficiency (see below), it seems likely that these manifestations reflect the action of NEMO on A20 ubiquitination (52). In 30%–40% of cases, the molecular defect also occurs in vascular endothelium, which results in systemic inflammation and vascular occlusive complications, particularly in retinae and brain.
Murine Ikbkg deficiency is embryonic lethal on day d12 as a result of hepatic necrosis (53). In humans, hemizygosity for amorphic IKBKG alleles is usually lethal as well; IP is therefore almost always an XLD disorder of females, who are mosaic for cells lacking functional NEMO. In some cases, hyperpigmented whorls form that map to lines of Blaschko, reflecting migration of cells carrying the defect from the neural crest (54). Phenotypic heterogeneity is attributed to skewed X inactivation, and the propensity for selection against cells in which the normal X chromosome has been inactivated (55). Furthermore, evolution of the phenotype with age reflects selection against cells expressing the defective allele. Typically, these become less prominent during infancy (56). Similar mechanisms account for rare instances of in IP males, where it usually made compatible with survival by X-polyploidy (XXY) or somatic mosaicism (57, 58).
In stages I and II of IP there is inflammation with epidermal oedema (spongiosis), epidermal blisters, and dermal cellular infiltrates, which progresses to apoptosis of mutant keratinocytes and skin atrophy with loss of skin appendages in stages III and IV (59). NEMO-deficient cells are more susceptible to undergoing apoptosis under the influence of pro-inflammatory cytokines TNF, IL1β (60) and genotoxic stress (61). During apoptosis, they release mediators including HSPs that ligate TLRs on and nearby cells with intact IKK complex. This triggers an inflammatory response and infiltrate (62), including release of proteases that disrupt desmosomes to cause blisters (Figure 3). In other words, there is a cycle of inflammation and apoptosis that continues until IKK-deficient cells are eliminated. These mechanisms may provide clues to the pathophysiology of rarer and more recently described NF-κB genodermatoses.
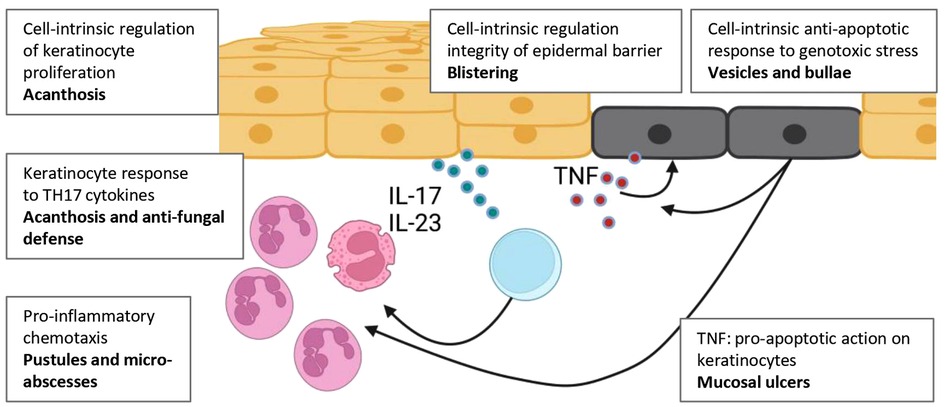
Figure 3. Overview of skin pathologies in which NF-κB is thought to contribute. NF-κB acts cell-intrinsically within keratinocytes, haemopoietic cells and other cells of the dermis, including fibroblasts (not shown). Cell-intrinsic defects in keratinocytes affect their proliferative capacity, propensity to undergo apoptosis, and capacity to contribute to the local antimicrobial response. NF-κB signalling also mediates (at least in part) keratinocytes responses to exogenous stimuli including pro-inflammatory cytokines (TNF, IL-1β) and Th17 cytokines. Keratinocyte activation and apoptosis can stimulate production of these cytokines. Created with BioRender.com.
IP is allelic with AED-ID, which results from hypomorphic IKBKG mutations. Features of AED-ID include hypohidrosis (sparse sweat glands), hypotrichosis (sparse hair), and periorbital wrinkling (63) (Table 3). Xerotic skin may take the form of severe ichthyotic like lesions. Various other cutaneous manifestations occur, including oral and perineal ulcers, palmoplantar keratoderma, hypopigmented papules seborrheic dermatitis, prurigo, and erythroderma, which may be severe (69).

Table 3. Links to images of conditions discussed.
Extracutaneous manifestations can include hypodontia, delayed tooth eruption and abnormal tooth shape (conical, accessory cusps, tulip-shaped teeth, and microdontia), low nasal bridge, frontal bossing and hypoplastic nasal alae (49, 69, 70). Severely affected individuals may show osteopetrosis and lymphedema (71). Isolated phenotypic features can phenotypes arise from defects in receptors responding to cues in cells that also signal though IKK (72, 73). At the other end of the spectrum, IKBKG mutations can present with mucocutaneous phenotypes of ichthyosis, eczematous dermatitis, palmoplantar keratoderma and recurrent mucocutaneous ulcers (69).
Immunodeficiency is a frequent but not invariable feature of hypomorphic IKBKG mutations. Indeed, some patients present with immunodeficiency in the absence of AED (74). Many patients are hypogammaglobulinaemic for IgG, sometimes with elevated IgM (63, 75–78). Most patients exhibit impaired polysaccharide-specific antibody responses (76) and some have NK cell defects (79). Pyogenic bacterial infections typically occur early in life, and later MSMD (80, 81). The reason for different immunological phenotypes remains to be resolved with certainty (8).
A large structural LoF variant in IKBKG (Δexons4–10) accounts for about 80% of IP (82). More than 100 hypomorphic mutations in IKBKG have been described with considerable phenotypic variation (83, 84). Mutations affecting leucine zipper domain or N-terminal coiled-coil domains have a less severe clinical phenotype and may have isolated hypotonia (Mild AED-ID) (84). Mutations causing decreased protein expression, folding or stability can cause immunodeficiency without AED (74). Genetic diagnosis is problematic because a non-functional partial second copy of the IKBKG gene (pseudogene) is located distal to exon 10 thus making traditional PCR based molecular diagnostics tests difficult (85).
Rare cases of AED-ID have been observed in individuals with heterozygous defects in NFKBIA that confers defective phosphorylation and, therefore, resistance to IKK-mediated NF-κB activation (79, 86). The clinical phenotype of the index case was severe recurrent pyogenic bacterial infection, with low IgG and high IgM. T cells were refractory to activation. There were features of mild ED, with sparse hair, rough skin and conical teeth.
Homozygous LoF mutations in CHUK have been reported in patients with phenotypes on the popliteal-pterygia spectrum. Homozygous LoF mutations were reported in two fetuses with cocoon syndrome characterised by embryonic encasement with immobile limbs, omphalocele and craniofacial defects (29). Another child was reported with Bartocas-Papas syndrome, including alopecia totalis, microphthalmia, ankyloblepharon, cleft palate and popliteal webs (87) (Table 3). Another case arising from heterozygous de novo CHUK mutations was identified in a patient with ankyloblepharon-ectodermal defects-cleft lip/palate (AEC syndrome), as well as buccal synechiae, hypoplastic thumbs and 3rd-5th toe syndactyly (88). Ectodermal defects included sparse hair, conical teeth and nail defects. A patient with a similar phenotype was noted to by compound heterozygous mutations for a frameshift CHUK mutation transmitted from the unaffected mother and a de novo missense mutation (89). The latter two cases were also noted to have recurrent chest infections and hypogammaglobulinaemia.
It is interesting that immune deficiency has been observed in most of the cases that were live born. Bainter and colleagues reported a patient with homozygous missense CHUK mutation, and combined immune deficiency with total B-cell lymphopenia, panhypogammaglobulinaemia, and a progressive T-cell lymphopenia (90). Complications included recurrent pneumonia, failure to thrive, impetigo (S. aureus), mucocutaneous candidiasis, and epidermodysplasia verruciformis from HPV + . The child succumbed to overwhelming viraemia. As noted above, mouse models suggest that IKKα deficiency confers abnormalities in skin differentiation independently of NF-κB. In this case, the amino acid substitution was shown to prevent IKKα binding to NIK, and a defect in p100 processing was demonstrated in patient-derived fibroblasts but it remains unclear whether the skin manifestations were NF-κB-dependent. An engineered mouse model of this patient provided some insights into these manifestations, with a defect in cutaneous IL17A expression, absent lymph nodes and disorganised splenic architecture.
IKKβ deficiency results in severe combined immunodeficiency (SCID) with an inflammatory skin phenotype. The initial report described patients with a severe combined immune deficiency (SCID) phenotype complicated by disseminated mycobacteria (BCGosis, and M. avium), candidiasis, and recurrent gram negative infections (91). Dissemination of BCG to the skin resulting in a generalised rash has been described, and that case also featured ectodermal dysplasia (92).
Individuals have also been described with heterozygosity GoF missense IKBKB mutations (93, 94). The probands presented with recurrent respiratory infections/otitis media, cutaneous abscesses, bronchiectasis, ectodermal dysplasia, and lymphopenia, plus features of mild ED including abnormal dentition, nail defects, early onset cataracts and anhidrosis (93).
Heterozygous mutations in NFKB1 encoding the precursor p105 results in p50 haploinsufficiency which appears to be the most common cause of Mendelian common variable immunodeficiency (CVID) (95). The associated clinical phenotypes are heterogenous, including a variety of autoinflammatory and rheumatological features (95–99) Autoimmune skin manifestations include vitiligo, alopecia, psoriasis and mucosal ulceration. Indeed, the phenotypic spectrum of NFKB1 haploinsufficiency appears to be broad and also encompasses mouth and genital ulceration, pyoderma gangrenosum, and erythema nodosum. In addition, skin infections such as bacterial abscesses, necrotizing cellulitis, folliculitis as well as viral infections have been observed (96, 100). It is associated with increased rates of lymphoproliferative disease, autoimmune haematological cytopenia, and enteropathies.
Immune dysregulation is observed in Nfkb1−/− mouse models. Mice lacking p105 and p50 subunits display chronic inflammation, telomere shortening and cellular senescence associated with premature ageing (101, 102). Nfkb1−/− mice display multi-organ autoimmunity with increased IL-6 production and activation of autoreactive CD8+ T cells (103, 104). Nfkb1−/− mice are reported to display late onset al.opecia. A similar human phenotype was observed in New Zealand family with p50 haploinsufficiency (99).
Haploinsufficiency for RELA has been reported to present with mouth and genital ulcers, recurrent fever, colitis and mucocutaneous ulceration (105, 106). Ulceration was attributed to heightened sensitivity to the apoptotic effects of TNF. Consistent with this, Rela+/− mice demonstrated impaired NF-κB activation and develop cutaneous ulceration from TNF exposure.
Homozygous hypomorphic REL deficiency has been reported in a patient with combined immunodeficiency and susceptibility to intracellular pathogens, sclerosing cholangitis, but no features of ED were described (107).
Human pathogenic variants in NFKB2 were identified independently by several groups (108–110). Most patients were identified within cohorts of primary antibody deficiency patients, although they also suffer with organ-specific autoimmunity including alopecia areata, pneumonitis, autoimmune hepatitis, and arthritis (108, 111). Almost all NFKB2 mutations described to date in patients with autoimmunity are missense and frameshift mutations located in exons 22 and 23 and affect the C-terminal portion of the protein that is responsible for regulating p100 processing (111).
Analysis of engineered mouse models has revealed that autoimmunity is T-cell dependent, and arises largely due to T cell-extrinsic actions of non-processible p100, which as described above, has IκB activity (112). In this case, p100 accumulation confers defects in thymic negative selection of conventional autoreactive T cells, and also impairs thymic selection of Tregs. This dominant GoF action of p100 stands in contrast with NFKB1 variants, which mostly confer haploinsufficiency and therefore halve the abundance of p105/p50, and reveals that central T cell tolerance appears to be exquisitely sensitive to the IκB action of p100 (112). It remains to be determined with ED accounts for skin or pituitary defects.
Murine models of RelB deficiency have also been shown to develop an inflammatory skin phenotype similar to human atopic dermatitis (Barton et al., 2000). Autosomal recessive RelB deficiency have been found in family of patients presenting with combined immunodeficiency, autoimmune skin diseases and failure to thrive (Sharfe et al., 2015).
Rare NIK deficiency arising from biallelic mutations in MAP3K14 have been reported. Patients presented with recurrent viral, bacterial, and cryptosporidium infections (113). Immunological assessment identified B-cell lymphopenia, impaired class-switch recombination and somatic hypermutation, decreased marginal zone and memory B cells, and hypogammaglobulinaemia. No skin phenotype was described.
Four mammalian protein paralogs (CARD9, CARD10, CARD11 and CARD14) are characterized by presence of both CARD and coiled-coil domains operate in different tissues though a similar mechanism of recruiting BCL10 and MALT into a so-called CBM complex. The CBM complex regulates NF-κB, as well as other biochemical processes including mTORC1 activation (114, 115). CARD10 and CARD14 are expressed in keratinocytes, while CARD11 is expressed in lymphocytes and masts cells, and CARD9 is expressed in dendritic cells and neutrophils (116).
Human deficiencies of different CARD9, 11 and 14 have been described, and confer different skin phenotypes, although with some interesting shared features (117–122). Loss of function variants of CARD11 have been identified in young children with SCID-phenotype. Complete deficiency of BCL10 and MALT1 have also been associated with combined immunodeficiency (123–128). MALT1 deficiency has also been associated with vitiligo, eczema, and erythroderma resembling Omenn syndrome (124–126, 129, 130). Mouse models suggest this is due to reduced Tregs (131).
Interestingly, hypomorphic mutations (dominant negative or haploinsufficiency) result in a complex phenotype of immune dysregulation that encompasses atopy, including atopic dermatitis and food allergy, and sometimes features of immune deficiency and autoimmunity (119–121, 132–134). Consistent with the proposition that hypomorphic rather than LoF mutations confer a skin phenotype, partial rescue of CARD11 function via somatic reversion of a LoF mutation has been reported to result in acquisition of an Omenn-like syndrome including hyper IgE and eczema (135). The mutations exhibit incomplete penetrance for AD. Similarly, atopic dermatitis (AD) plus hyper-IgE was observed with incomplete penetrance in one hypomorphic Card11 hypomorphic mouse strain, while another manifested only late-onset hyper IgE (136–138). Neither phenotype was observed with complete Card11 deficiency (139). In both humans and their mouse models, reduced CARD11 activity results in Th2-skewed immune responses, although there is evidence for both cell-intrinsic mechanisms, and cell-extrinsic effects via deficiency of Tregs (137, 140).
CARD14 expression is greatest in epithelia, particularly in the skin and respiratory tract. In both humans and mice, GoF CARD14 mutations have been associated with psoriasis, or psoriatic-like syndromes of pityriasis rubra pilaris and palmoplantar pustulosis (66, 141, 142). Like dominant negative CARD11 mutations, CARD14 mutations that attenuate NF-κB activity are associated with AD and hyper-IgE (122), whereas complete absence of CARD14 is not associated with AD (141).
Intact CBM complexes are required for normal host defence, particularly at epithelial surfaces. Loss of function mutations in CARD9 have been linked to autosomal recessive forms of susceptibility to chronic mucocutaneous candidiasis, deep dermatophytosis, and other infections involving yeast-like fungi (117), which appears to result from defective neutrophil recruitment and Th17 induction after ligation of dectin1 and dectin2 by fungal antigens.
RIPK4 (receptor interacting serine-threonine kinase) interacts with PKCδ and mediates activation of NF-κB (143). RIPK4 and is critical for regulation of keratinocyte differentiation as well as craniofacial and limb development (144). The kinase domain of RIP4 activates NF-κB signalling during epidermal inflammatory responses (145). Loss of function mutations in RIPK4 have been identified in patients with a popliteal-pterygium syndrome (Bartsocas-Papas syndrome) characterised by numerous craniofacial, musculoskeletal, genitourinary, gastrointestinal, cardiac and neurodevelopmental defects, resembling IKKα deficiency (68, 146, 147). Similar severe morphological defects and skin abnormalities of ectodermal origin can be seen in CHUK (IKKα) deficiency, as described above.
Haploinsufficiency for TNFAIP3 results in an auto-inflammatory phenotype known also as Familial Behçet-like Autoinflammatory Syndrome or HA20 (65, 148). Reduction in A20 results in increased activity of canonical NF-κB, c-Jun N-terminal kinase, and p38. HA20 usually presents in children <10 years of age with recurrent oral and genital ulcers. Skin involvement is noted in approximately 50% of cases, with pathergy, pustules, folliculitis and acneiform eruptions (Table 3). Additional manifestations include periodic fever, recurrent infections, ocular inflammation, gastrointestinal symptoms ranging from pain to inflammatory disease with perforation, and polyarthropathy. Some patients exhibit frank immune deficiency or organ-specific autoimmunity (149, 150).
Autosomal dominant mutations in CYLD cause familial cylindromatosis characterised by multiple benign skin tumours (cylindromas) (151, 152). CYLD is expressed at highest levels in brain, testis, skin and thymus (153). CYLD is a deubiquitinase that targets several proteins, including NEMO, TRAF2, TRAF6, BCL3, and TRAF2 (154). CYLD is therefore a negative regulator of NF-κB-mediated pro-survival genes, and an inhibitor of necroptosis mediated by RIPK1 after ligation of TLRs or TNFR1 (155–160). Mutations in CYLD account for what had previously been three skin appendage tumour syndromes, familial cylindromatosis, Spiegler-Brooke syndrome (BSS), and multiple familial trichoepithelioma. Cylindromas arising from hair follicles or sweat glands usually on the scalp or face. BSS consists of a triad of cylindromas, trichoepitheliomas arising from hair follicles usually affecting nose and nasolabial folds and spiradenomas arising from sweat glands typically on the head, neck and trunk (161) (Table 3). CYLD deficiency is not associated with overt immune deficiency. Although a Cyld−/− mouse model exhibited a T cell developmental defect this was not observed in other models (162). These models also feature susceptibility to chemically-induced skin tumours induced colonic inflammation and increased incidence of tumors and enhanced NF-κB activity in lymphocytes and myeloid cells (162).
(OTU Deubiquitinase With Linear Linkage Specificity) is a negative regulator of NF-κB. Homozygous deficiency of OTULIN results in OTULIN-related Autoinflammatory Syndrome (ORAS), which presents in infancy with neutrophlic nodular panniculitis, lipodystrophy and cutaneous vasculitis, as well as recurrent fevers, diarrhoea, and arthritis, and is characterised by prominent acute phase responses and hypergammaglobulinaemia (163–165). More recently, a different syndrome characterised by skin necrosis and necrotising pneumonia due to S. aureus has been identified in individuals with OTULIN haploinsufficiency (166). Patients often present in adolescence. The phenotype is thought to result in a cell-intrinsic susceptibility of fibroblasts to cytotoxic effects of staphylococcal α-toxin and appears to be mediated by accumulation of caveolin-1 rather than heightened NF-κB activity in response to TNF.
Cutaneous manifestations provide important clues to IEI arising from defects in NF-κB. The manifestations are diverse (Table 4; Figure 2). Disorders associated with prominent defects in ectodermal development include those arising from IKBKG and NFKBIA variants, manifesting as ED with loss of skin appendages, and popliteal-pterygium spectrum (Bartsocas-Papas syndrome), from defects in CHUK and RIPK4. By contrast, defects of regulatory proteins (A20, OTULIN) result in predominantly inflammatory manifestations of mucocutaneous ulceration and neutrophil accumulation in the skin, features that make these syndromes an important differential diagnosis for Behcet's disease. CYLD defects result in excessive expansion of keratinocytes and skin appendages (Figure 3).
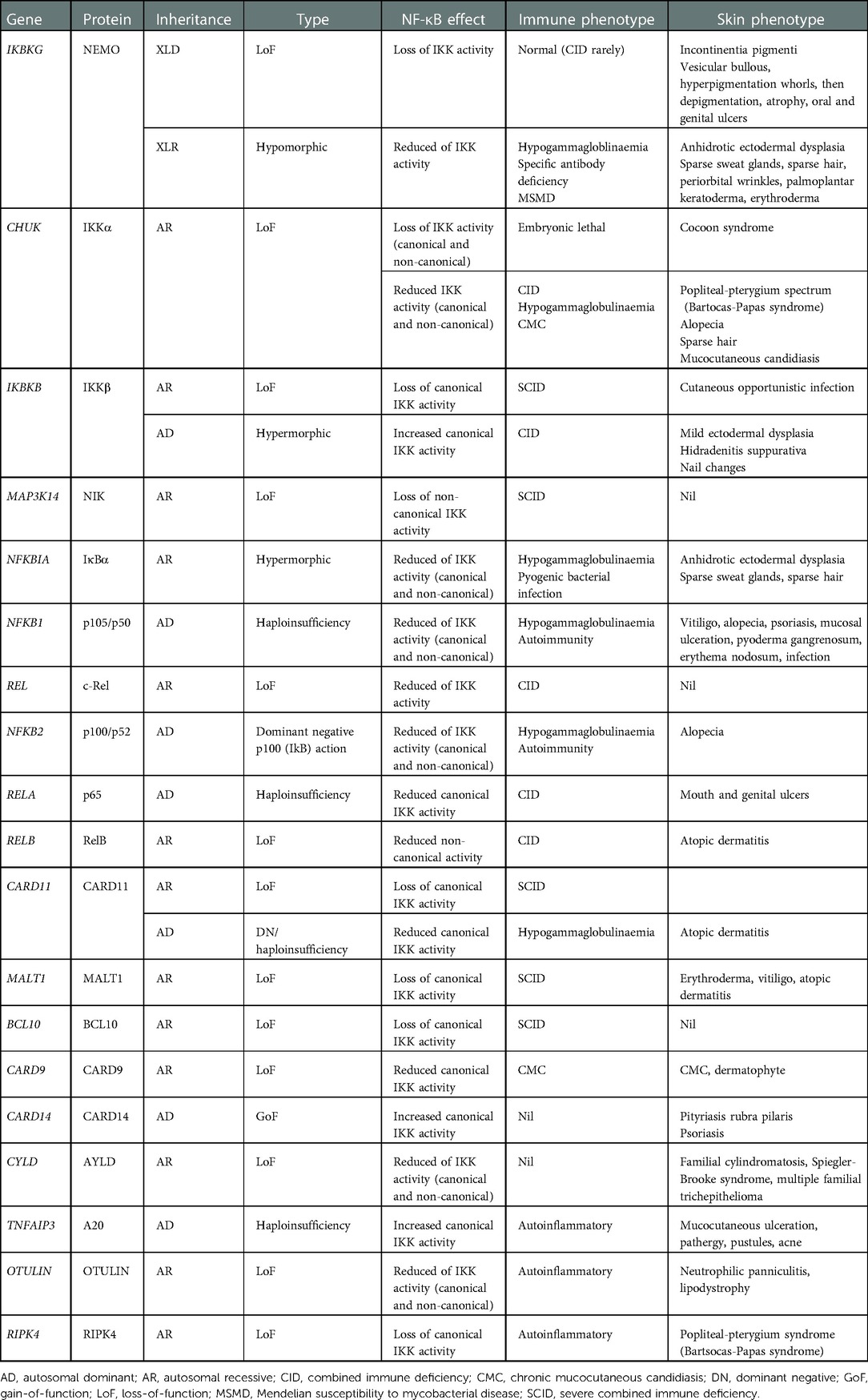
Table 4. Summary of skin and immune phenotypes with NF-κB defects.
It is still too early to arrive at a coherent account of all the genotype-phenotype relations for each of the IEIs described in this review. They nevertheless provide insights into pathogenic pathways likely to be relevant to more common disorders where it can be assumed that pathology arises from complex interactions of more than one gene defect (Figure 4). These include psoriasis, skin cancers, and syndromes associated with mucocutaneous ulceration such as Behcet's disease and inflammatory bowel disease.
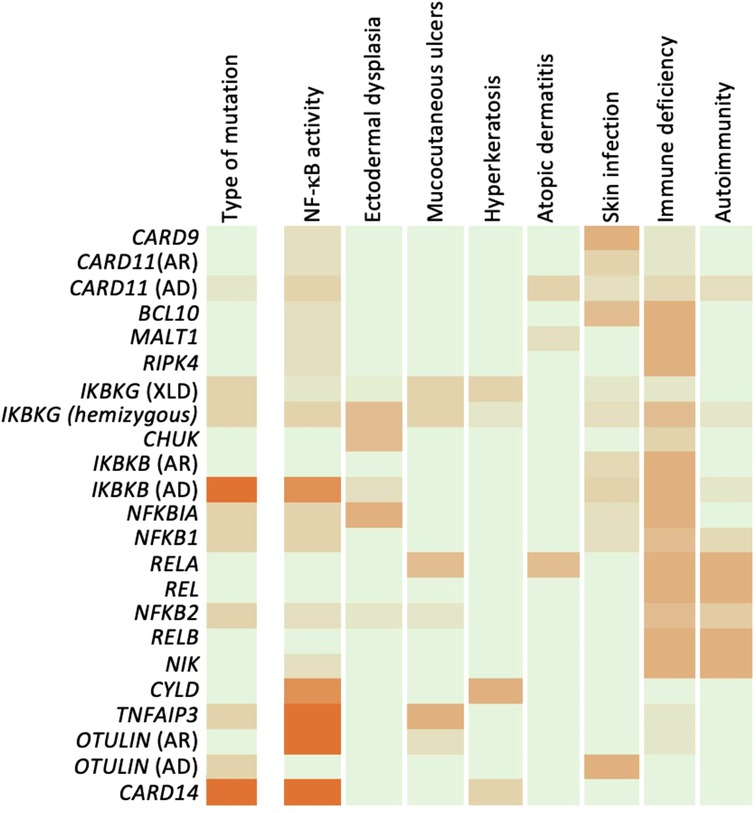
Figure 4. Genotype-phenotype relation of human NF-κB defects. Summary of mutations and phenotypes discussed, expressed as a heatmap. For genotype and NF-κB effect, reduced activity appears green and increased activity in orange. For phenotypes, absence of phenotype is indicated in white, while severity of phenotype is shown in orange.
Psoriasis is characterised by acanthosis, increased dermal vascularity, and neutrophilic microabscesses in the epidermis. IEIs provide evidence for the importance of NF-κB in regulating keratinocyte proliferation. Of the IEIs discussed here, GoF variants in CARD14 account for segregation of psoriasis with the Psors2 (17q25-ter) locus on genome-wide linkage scans. Disease-associated alleles of CARD14 result in increased NF-κB activity in keratinocytes in response to IL-17A (167). Furthermore, while some models have suggested pathogenic pathways common to psoriasis and atopic dermatitis, it is noteworthy that CARD14 defects with the opposite polarity to those responsible for psoriasis (i.e., LoF alleles) cause severe atopic dermatitis (122). Furthermore, while excessive NF-κB signals triggered by Th17 cytokines appears to contribute to psoriatic changes, underactivity of this pathway may explain predilection to cutaneous yeast and fungal infections, as observed with hypomorphic defects in CARD9.
Both CARD14 GoF and HA20 result in increased NF-κB activity. CARD14 GoF results in both plaque and pustular psoriasis. The most consistent clinical feature of HA20 appears to be severe and early onset mouth and genital ulcers, nevertheless, palmoplantar pustules are also observed frequently (65), whereas plaque psoriasis does not appear to be a feature of HA20. HA20 is also a profoundly inflammatory disorder, and these differences probably reflect consequences of A20 deficiency within cells of the immune system. Further complexity, however, is revealed by the Behcet's disease-like presentations observed in association with NFKB1 haploinsufficiency. Thus, attributing the aphthosis to NF-κB alone might be an oversimplification.
A number of variables are likely to account for the phenotypic variation from Mendelian defects in NF-κB, including the severity of the biochemical defect. As illustrated for CARD11, OTULIN, and IKBKB, autosomal dominant defects that confer quantitative changes in NF-κB signalling result in phenotypes that differ significantly from LoF mutations. Other explanations for phenotypic variation include the consequences of feed-forward loops, particularly for the autoinflammatory disorders (exemplified by HA20). In the interests of concision, we have concentrated on changes in NF-κB, but many of the molecules discussed also regulate other pathways. Whether or not this is the case, background genetic variation will of course also contribute to phenotypic variation.
Finally, as noted above, there are differences in cell-specific expression. Pathology in the skin is common with NF-κB disorders because it acts intrinsically in keratinocytes, as well as in inflammatory cells that infiltrate the epidermis. Expression of some of the regulators described in this review, however, are restricted to one compartment or another. Dissecting these mechanisms is likely to require accurate models of the defects in question, in which the genetic defects can be isolated to either keratinocytes or components of the immune system. The phenotypes observed with IEIs provide evidence that this line of enquiry is likely to be informative for more common disease.
YS, APRB and RLY wrote the manuscript. MCC conceived the review and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Royal Society Wolfson Fellowship (MCC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. (2008) 132:344–62. doi: 10.1016/j.cell.2008.01.020
3. Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein nf-kappa B by a posttranslational mechanism. Cell. (1986) 47:921–8. doi: 10.1016/0092-8674(86)90807-X
4. Puel A, Picard C, Ku C-L, Smahi A, Casanova J-L. Inherited disorders of NF-κB-mediated immunity in man. Curr Opin Immunol. (2004) 16:34–41. doi: 10.1016/j.coi.2003.11.013
5. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: a blossoming of relevance to human Pathobiology. Cell. (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
6. Miraghazadeh B, Cook MC. Nuclear factor-kappaB in autoimmunity: man and mouse. Front Immunol. (2018) 9:613. doi: 10.3389/fimmu.2018.00613
7. Schnappauf O, Aksentijevich I. Mendelian Diseases of dysregulated canonical NF-κB signaling: from immunodeficiency to inflammation. J Leukoc Biol. (2020) 108:1–35. doi: 10.1002/JLB.2MR0520-166R
8. Baeuerle PA, Baltimore D. NF-kappa B: Ten years after. South San Francisco, California 94080, USA: Tularik Inc. (1996). 13–20 p.
9. Ghosh S, Baltimore D. Activation in vitro of NF-kappa B by phosphorylation of its inhibitor I kappa B. Nature. (1990) 344:678–82. doi: 10.1038/344678a0
10. May MJ, Kopp EB. NF-kappa B and rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. (1998) 16:225–60. doi: 10.1146/annurev.immunol.16.1.225
11. Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. (2002) 298:1241–5. doi: 10.1126/science.1071914
12. Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. (2006) 25:6706–16. doi: 10.1038/sj.onc.1209933
13. Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. (1997) 91:243–52. doi: 10.1016/S0092-8674(00)80406-7
14. Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. (1997) 278:860–6. doi: 10.1126/science.278.5339.860
15. Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV. Ikappab kinase-beta: nF-kappaB activation and complex formation with IkappaB kinase-alpha and NIK. Science. (1997) 278:866–9. doi: 10.1126/science.278.5339.866
16. Scheidereit C. Ikappab kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. (2006) 25:6685–705. doi: 10.1038/sj.onc.1209934
17. Didonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. (1997) 388:548–54. doi: 10.1038/41493
18. Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. (2001) 7:401–9. doi: 10.1016/S1097-2765(01)00187-3
19. Maier HJ, Marienfeld R, Wirth T, Baumann B. Critical role of RelB serine 368 for dimerization and p100 stabilization*. J Biol Chem. (2003) 278:39242–50. doi: 10.1074/jbc.m301521200
20. Basak S, Kim H, Kearns JD, Tergaonkar V, O’Dea E, Werner SL, et al. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. (2007) 128:369–81. doi: 10.1016/j.cell.2006.12.033
21. Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. (2002) 9:625–36. doi: 10.1016/S1097-2765(02)00477-X
22. Heger K, Wickliffe KE, Ndoja A, Zhang J, Murthy A, Dugger DL, et al. OTULIN Limits cell death and inflammation by deubiquitinating LUBAC. Nature. (2018) 559:120–4. doi: 10.1038/s41586-018-0256-2
23. Lork M, Verhelst K, Beyaert RCYLD. A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: so similar, yet so different. Cell Death Differ. (2017) 24:1172–83. doi: 10.1038/cdd.2017.46
24. Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol. (2012) 12:774–85. doi: 10.1038/nri3313
25. Draber P, Kupka S, Reichert M, Draberova H, Lafont E, de Miguel D, et al. LUBAC-Recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep. (2015) 13:2258–72. doi: 10.1016/j.celrep.2015.11.009
26. Keusekotten K, Elliott PR, Glockner L, Fiil BK, Damgaard RB, Kulathu Y, et al. OTULIN Antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell. (2013) 153:1312–26. doi: 10.1016/j.cell.2013.05.014
27. Cui C-Y, Schlessinger D. EDA Signaling and skin appendage development. Cell Cycle (Georgetown, Tex). (2006) 5:2477–83. doi: 10.4161/cc.5.21.3403
28. Wright JT, Fete M, Schneider H, Zinser M, Koster MI, Clarke AJ, et al. Ectodermal dysplasias: classification and organization by phenotype, genotype and molecular pathway. Am J Med Genet A. (2019) 179:442–7. doi: 10.1002/ajmg.a.61045
29. Kere J, Srivastava AK, Montonen O, Zonana J, Thomas N, Ferguson B, et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet. (1996) 13:409–16. doi: 10.1038/ng0895-409
30. Monreal AW, Ferguson BM, Headon DJ, Street SL, Overbeek PA, Zonana J. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet. (1999) 22:366–9. doi: 10.1038/11937
31. Headon DJ, Emmal SA, Ferguson BM, Tucker AS, Justice MJ, Sharpe PT, et al. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature. (2001) 414:913–6. doi: 10.1038/414913a
32. Pasparakis M. Role of NF-κB in epithelial biology. Immunol Rev. (2012) 246:346–58. doi: 10.1111/j.1600-065x.2012.01109.x
33. Seitz CS, Lin Q, Deng H, Khavari PA. Alterations in NF-κB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-κB. Proc National Acad Sci. (1998) 95:2307–12. doi: 10.1073/pnas.95.5.2307
34. van Hogerlinden M, Rozell BL, Toftgård R, Sundberg JP. Characterization of the progressive skin disease and inflammatory cell infiltrate in mice with inhibited NF-κB signaling. J Invest Dermatol. (2004) 123:101–8. doi: 10.1111/j.0022-202x.2004.22706.x
35. Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. (2002) 417:861–6. doi: 10.1038/nature00820
36. Nenci A, Huth M, Funteh A, Schmidt-Supprian M, Bloch W, Metzger D, et al. Skin lesion development in a mouse model of incontinentia pigmenti is triggered by NEMO deficiency in epidermal keratinocytes and requires TNF signaling. Hum Mol Genet. (2006) 15:531–42. doi: 10.1093/hmg/ddi470
37. Omori E, Matsumoto K, Sanjo H, Sato S, Akira S, Smart RC, et al. TAK1 Is a master regulator of epidermal homeostasis involving skin inflammation and apoptosis*. J Biol Chem. (2006) 281:19610–7. doi: 10.1074/jbc.m603384200
38. Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. (2000) 289:2350–4. doi: 10.1126/science.289.5488.2350
39. Etemadi N, Chopin M, Anderton H, Tanzer MC, Rickard JA, Abeysekera W, et al. TRAF2 Regulates TNF and NF-κB signalling to suppress apoptosis and skin inflammation independently of sphingosine kinase 1. eLife. (2015) 4:e10592. doi: 10.7554/elife.10592
40. Ohazama A, Hu Y, Schmidt-Ullrich R, Cao Y, Scheidereit C, Karin M, et al. A dual role for ikkα in tooth development. Dev Cell. (2004) 6:219–27. doi: 10.1016/s1534-5807(04)00024-3
41. Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, et al. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. (1999) 284:316–20. doi: 10.1126/science.284.5412.316
42. Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, et al. Limb and skin abnormalities in mice lacking IKKalpha. Science. (1999) 284:313–6. doi: 10.1126/science.284.5412.313
43. Hu Y, Baud V, Oga T, Kim KI, Yoshida K, Karin M. IKKalpha controls formation of the epidermis independently of NF-kappaB. Nature. (2001) 410:710–4. doi: 10.1038/35070605
44. Sil AK, Maeda S, Sano Y, Roop DR, Karin M. Iκb kinase-α acts in the epidermis to control skeletal and craniofacial morphogenesis. Nature. (2004) 428:660–4. doi: 10.1038/nature02421
45. Liu B, Xia X, Zhu F, Park E, Carbajal S, Kiguchi K, et al. IKKα is required to maintain skin homeostasis and prevent skin cancer. Cancer Cell. (2008) 14:212–25. doi: 10.1016/j.ccr.2008.07.017
46. Boisson B, Quartier P, Casanova J-L. Immunological loss-of-function due to genetic gain-of-function in humans: autosomal dominance of the third kind. Curr Opin Immunol. (2015) 32:90–105. doi: 10.1016/j.coi.2015.01.005
47. Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, et al. Genomic rearrangement in NEMO impairs NF-κB activation and is a cause of incontinentia pigmenti. Nature. (2000) 405:466–72. doi: 10.1038/35013114
48. Carney R, Carney R. Inconinentia pigmenti. Arch Dermatol. (1970) 102:157–62. doi: 10.1001/archderm.1970.04000080029006
49. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-sulzberger syndrome). J Med Genet. (1993) 30:53. doi: 10.1136/jmg.30.1.53
50. Cinar OK, Romano M, Guzel F, Brogan PA, Demirkaya E. Paediatric behçet’s disease: a comprehensive review with an emphasis on monogenic mimics. J Clin Medicine. (2022) 11:1278. doi: 10.3390/jcm11051278
51. Endoh M, Yokozeki H, Maruyama R, Matsunaga T, Katayama I, Nishioka K. Incontinentia pigmenti and behçet’s disease: a case of impaired neutrophil chemotaxis. Dermatology. (1996) 192:285–7. doi: 10.1159/000246388
52. Zilberman-Rudenko J, Shawver LM, Wessel AW, Luo Y, Pelletier M, Tsai WL, et al. Recruitment of A20 by the C-terminal domain of NEMO suppresses NF-κB activation and autoinflammatory disease. Proc National Acad Sci. (2016) 113:1612–7. doi: 10.1073/pnas.1518163113
53. Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, et al. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. (2000) 14:854–62. papers3://publication/uuid/46F0DE32-3521-4BEE-9C7B-4C9CC4A71CDC. doi: 10.1101/gad.14.7.854
54. Happle R. Lionization and the lines of blaschko. Hum Genet. (1985) 70:200–6. doi: 10.1007/BF00273442
55. Parrish JE, Scheuerle AE, Lewis RA, Levy ML, Nelson DL. Selection against mutant alleles in blood leukocytes is a consistent feature in incontinentia Pigmenti type 2. Hum Mol Genet. (1996) 5:1777–83. doi: 10.1093/hmg/5.11.1777
56. Courtois G, Smahi A. NF-κB-related genetic diseases. Cell Death Differ. (2006) 13:843–51. doi: 10.1038/sj.cdd.4401841
57. Scheuerle AE. American Journal of medical genetics. Am J Med Genet. (1998) 77:201–18. doi: 10.1002/(SICI)1096-8628(19980518)77:3%3C201::AID-AJMG5%3E3.0.CO;2-S
58. Kenwrick S, Woffendin H, Jakins T, Shuttleworth SG, Mayer E, Greenhalgh L, et al. Survival of male patients with incontinentia Pigmenti carrying a lethal mutation can be explained by somatic mosaicism or klinefelter syndrome. Am J Hum Genetics. (2001) 69:1210–7. doi: 10.1086/324591
59. Nelson DL. NEMO, NFκB signaling and incontinentia pigmenti. Curr Opin Genet Dev. (2006) 16:282–8. doi: 10.1016/j.gde.2006.04.013
60. Makris C, Godfrey VL, Krähn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, et al. Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol Cell. (2000) 5:969–79. doi: 10.1016/s1097-2765(00)80262-2
61. Huang TT, Wuerzberger-Davis SM, Wu Z-H, Miyamoto S. Sequential modification of NEMO/IKKγ by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell. (2003) 115:565–76. doi: 10.1016/s0092-8674(03)00895-x
62. Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. NEJM. (1997) 336:1066–71. doi: 10.1056/nejm199704103361506
63. Döffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-κB signaling. Nat Genet. (2001) 27:277–85. doi: 10.1038/85837
64. Hübner S, Schwieger-Briel A, Technau-Hafsi K, Danescu S, Baican A, Theiler M, et al. Phenotypic and genetic spectrum of incontinentia pigmenti – a large case series. JDDG J Der Deutschen Dermatologischen Gesellschaft. (2022) 20:35–43. doi: 10.1111/ddg.14638
65. Aeschlimann FA, Batu ED, Canna SW, Go E, Gül A, Hoffmann P, et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-κB-mediated autoinflammatory disease. Ann Rheum Dis. (2018) 77:728. doi: 10.1136/annrheumdis-2017-212403
66. Craiglow BG, Boyden LM, Hu R, Virtanen M, Su J, Rodriguez G, et al. CARD14-associated Papulosquamous eruption: a spectrum including features of psoriasis and pityriasis rubra pilaris. J Am Acad Dermatol. (2018) 79:487–94. doi: 10.1016/j.jaad.2018.02.034
67. Lakhani SR. Putting the brakes on cylindromatosis? New Engl J Medicine. (2004) 350:187–8. doi: 10.1056/nejmcibr032650
68. Kalay E, Sezgin O, Chellappa V, Mutlu M, Morsy H, Kayserili H, et al. Mutations in RIPK4 cause the autosomal-recessive form of popliteal pterygium syndrome. Am J Hum Genetics. (2012) 90:76–85. doi: 10.1016/j.ajhg.2011.11.014
69. Kohn LL, Braun M, Cordoro KM, McCalmont TH, Shah SD, Frieden IJ, et al. Skin and mucosal manifestations in NEMO syndrome: a case series and literature review. Pediatr Dermatol. (2022) 39:84–90. doi: 10.1111/pde.14905
70. Hanson EP, Monaco-Shawver L, Solt LA, Madge LA, Banerjee PP, May MJ, et al. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. (2008) 122:1169–1177.e16. doi: 10.1016/j.jaci.2008.08.018
71. Roberts CML, Angus JE, Leach IH, McDermott EM, Walker DA, Ravenscroft JC. A novel NEMO gene mutation causing osteopetrosis, lymphoedema, hypohidrotic ectodermal dysplasia and immunodeficiency (OL-HED-ID). Eur J Pediatr. (2010) 169:1403–7. doi: 10.1007/s00431-010-1206-7
72. Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RGH, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. (2000) 24:45–8. doi: 10.1038/71667
73. Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, McTigue MA, et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet. (2000) 25:153–9. doi: 10.1038/75997
74. Niehues T, Reichenbach J, Neubert J, Gudowius S, Puel A, Horneff G, et al. Nuclear factor κB essential modulator–deficient child with immunodeficiency yet without anhidrotic ectodermal dysplasia. J Allergy Clin Immun. (2004) 114:1456–62. doi: 10.1016/j.jaci.2004.08.047
75. Jain A, Ma CA, Liu S, Brown M, Cohen J, Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. (2001) 2:223–8. doi: 10.1038/85277
76. Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet. (2000) 67:1555–62. doi: 10.1086/316914
77. Aradhya S, Courtois G, Rajkovic A, Lewis RA, Levy M, Israël A, et al. Atypical forms of incontinentia Pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-γ). Am J Hum Genetics. (2001) 68:765–71. doi: 10.1086/318806
78. Miot C, Imai K, Imai C, Mancini AJ, Kucuk ZY, Kawai T, et al. Hematopoietic stem cell transplantation in 29 patients hemizygous for hypomorphic IKBKG/NEMO mutations. Blood. (2017) 130:1456–67. doi: 10.1182/blood-2017-03-771600
79. Orange JS, Brodeur SR, Jain A, Bonilla FA, Schneider LC, Kretschmer R, et al. Deficient natural killer cell cytotoxicity in patients with IKK-γ/NEMO mutations. J Clin Invest. (2002) 109:1501–9. doi: 10.1172/jci14858
80. Martinez-Pomar N, Munoz-Saa I, Heine-Suner D, Martin A, Smahi A, Matamoros N. A new mutation in exon 7 of NEMO gene: late skewed X-chromosome inactivation in an incontinentia pigmenti female patient with immunodeficiency. Hum Genet. (2005) 118:458. doi: 10.1007/s00439-005-0068-y
81. Orange JS, Jain A, Ballas ZK, Schneider LC, Geha RS, Bonilla FA. The presentation and natural history of immunodeficiency caused by nuclear factor κB essential modulator mutation. J Allergy Clin Immun. (2004) 113:725–33. doi: 10.1016/j.jaci.2004.01.762
82. Aradhya S, Woffendin H, Jakins T, Bardaro T, Esposito T, Smahi A, et al. A recurrent deletion in the ubiquitously expressed NEMO (IKK-γ) gene accounts for the vast majority of incontinentia pigmenti mutations. Hum Mol Genet. (2001) 10:2171–9. doi: 10.1093/hmg/10.19.2171
83. Hubeau M, Ngadjeua F, Puel A, Israel L, Feinberg J, Chrabieh M, et al. New mechanism of X-linked anhidrotic ectodermal dysplasia with immunodeficiency: impairment of ubiquitin binding despite normal folding of NEMO protein. Blood. (2011) 118:926–35. doi: 10.1182/blood-2010-10-315234
84. Heller S, Kölsch U, Magg T, Krüger R, Scheuern A, Schneider H, et al. T cell impairment is predictive for a severe clinical course in NEMO deficiency. J Clin Immunol. (2020) 40:421–34. doi: 10.1007/s10875-019-00728-y
85. Frans G, Meert W, Bosch JVDWT, Meyts I, Bossuyt X, Vermeesch JR, et al. Conventional and single-molecule targeted sequencing method for specific variant detection in IKBKG while bypassing the IKBKGP1 pseudogene. J Mol Diagnostics. (2018) 20:195–202. doi: 10.1016/j.jmoldx.2017.10.005
86. Courtois G, Smahi A, Reichenbach J, Doffinger R, Cancrini C, Bonnet M, et al. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest. (2003) 112:1108–15. doi: 10.1172/jci18714
87. Leslie EJ, O’Sullivan J, Cunningham ML, Singh A, Goudy SL, Ababneh F, et al. Expanding the genetic and phenotypic spectrum of popliteal pterygium disorders. Am J Med Genet A. (2015) 167:545–52. doi: 10.1002/ajmg.a.36896
88. Khandelwal KD, Ockeloen CW, Venselaar H, Boulanger C, Brichard B, Sokal E, et al. Identification of a de novo variant in CHUK in a patient with an EEC/AEC syndrome-like phenotype and hypogammaglobulinemia. Am J Med Genet A. (2017) 173:1813–20. doi: 10.1002/ajmg.a.38274
89. Cadieux-Dion M, Safina NP, Engleman K, Saunders C, Repnikova E, Raje N, et al. Novel heterozygous pathogenic variants in CHUK in a patient with AEC-like phenotype, immune deficiencies and 1q21.1 microdeletion syndrome: a case report. Bmc Med Genet. (2018) 19:41. doi: 10.1186/s12881-018-0556-2
90. Bainter W, Lougaris V, Wallace JG, Badran Y, Hoyos-Bachiloglu R, Peters Z, et al. Combined immunodeficiency with autoimmunity caused by a homozygous missense mutation in inhibitor of nuclear factor κB kinase alpha (IKKα). Sci Immunol. (2021) 6:eabf6723. doi: 10.1126/sciimmunol.abf6723
91. Pannicke U, Baumann B, Fuchs S, Henneke P, Rensing-Ehl A, Rizzi M, et al. Deficiency of innate and acquired immunity caused by an IKBKBMutation. N Engl J Med. (2013) 369:2504–14. doi: 10.1056/nejmoa1309199
92. Burns SO, Plagnol V, Gutierrez BM, Zahrani DA, Curtis J, Gaspar M, et al. Immunodeficiency and disseminated mycobacterial infection associated with homozygous nonsense mutation of IKKβ. J Allergy Clin Immun. (2014) 134:215–218.e3. doi: 10.1016/j.jaci.2013.12.1093
93. Cardinez C, Miraghazadeh B, Tanita K, da Silva E, Hoshino A, Okada S, et al. Gain-of-function IKBKB mutation causes human combined immune deficiency. J Exp Med. (2018) 215:2715–24. doi: 10.1084/jem.20180639
94. Abbott J. 2 ACE, 3 DJ, 3 PRR, 4 KO, 4 LM, gelfand EW. Heterozygous IKKb activation loop mutation results in a complex immunodeficiency syndrome. J Allergy Clin Immunol. (2021) 147:737–40. doi: 10.1016/j.jaci.2020.06.007
95. Tuijnenburg P, Allen HL, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in europeans. J Allergy Clin Immunol. (2018) 142:1285–96. doi: 10.1016/j.jaci.2018.01.039
96. Kaustio M, Haapaniemi E, Göös H, Hautala T, Park G, Syrjänen J, et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J Allergy Clin Immunol. (2017) 140:782–96. doi: 10.1016/j.jaci.2016.10.054
97. Fliegauf M, Kinnunen M, Posadas-Cantera S, Camacho-Ordonez N, Abolhassani H, Alsina L, et al. Detrimental NFKB1 missense variants affecting the rel-homology domain of p105/p50. Front Immunol. (2022) 13:965326. doi: 10.3389/fimmu.2022.965326
98. Fliegauf M, Bryant VL, Frede N, Slade C, Woon S-T, Lehnert K, et al. Haploinsufficiency of the NF-κB1 subunit p50 in common Variable immunodeficiency. Am J Hum Genet. (2015) 97:389–403. doi: 10.1016/j.ajhg.2015.07.008
99. Schröder C, Sogkas G, Fliegauf M, Dörk T, Liu D, Hanitsch LG, et al. Late-Onset antibody deficiency due to monoallelic alterations in NFKB1. Front Immunol. (2019) 10:2618. doi: 10.3389/fimmu.2019.02618
100. Lorenzini T, Fliegauf M, Klammer N, Frede N, Proietti M, Bulashevska A, et al. Characterization of the clinical and immunologic phenotype and management of 157 individuals with 56 distinct heterozygous NFKB1 mutations. J Allergy Clin Immunol. (2020) 146:901–11. doi: 10.1016/j.jaci.2019.11.051
101. Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun. (2014) 2:4172. doi: 10.1038/ncomms5172
102. Bernal GM, Wahlstrom JS, Crawley CD, Cahill KE, Pytel P, Liang H, et al. Loss of Nfkb1 leads to early onset aging. Aging Albany Ny. (2014) 6:931–42. doi: 10.18632/aging.100702
103. Dissanayake D, Hall H, Berg-Brown N, Elford AR, Hamilton SR, Murakami K, et al. Nuclear factor-κB1 controls the functional maturation of dendritic cells and prevents the activation of autoreactive T cells. Nat Med. (2011) 17:1663–7. doi: 10.1038/nm.2556
104. Stanic AK, Bezbradica JS, Park J-J, Matsuki N, Mora AL, Kaer LV, et al. NF-κB Controls cell fate specification, survival, and molecular differentiation of immunoregulatory natural T lymphocytes. J Immunol. (2004) 172:2265–73. doi: 10.4049/jimmunol.172.4.2265
105. Badran YR, Dedeoglu F, Castillo JML, Bainter W, Ohsumi TK, Bousvaros A, et al. Human RELA haploinsufficiency results in autosomal-dominant chronic mucocutaneous ulceration. J Exp Med. (2017) 214:1937–47. doi: 10.1084/jem.20160724
106. Adeeb F, Dorris ER, Morgan NE, Lawless D, Maqsood A, Ng WL, et al. A novel RELA truncating mutation in a familial behçet’s disease–like mucocutaneous ulcerative condition. Arthritis Rheumatol. (2021) 73:490–7. doi: 10.1002/art.41531
107. Beaussant-Cohen S, Jaber F, Massaad MJ, Weeks S, Jones J, Alosaimi MF, et al. Combined immunodeficiency in a patient with c-rel deficiency. J Allergy Clin Immunol. (2019) 144:606–608.e4. doi: 10.1016/j.jaci.2019.05.003
108. Lee CE, Fulcher DA, Whittle B, Chand R, Fewings N, Field M, et al. Autosomal-dominant B-cell deficiency with alopecia due to a mutation in NFKB2 that results in nonprocessable p100. Blood. (2014) 124:2964–72. doi: 10.1182/blood-2014-06-578542
109. Chen K, Coonrod EM, Kumánovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline mutations in NFKB2 implicate the noncanonical NF-kappaB pathway in the pathogenesis of common variable immunodeficiency. Am J Hum Genet. (2013) 93:812–24. doi: 10.1016/j.ajhg.2013.09.009
110. Brue T, Quentien M-H, Khetchoumian K, Bensa M, Capo-Chichi J-M, Delemer B, et al. Mutations in NFKB2 and potential genetic heterogeneity in patients with DAVID syndrome, having variable endocrine and immune deficiencies. BMC Med Genet. (2014) 15:139. doi: 10.1186/s12881-014-0139-9
111. Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N, et al. Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front Immunol. (2019) 10:297. doi: 10.3389/fimmu.2019.00297
112. Wirasinha RC, Davies AR, Srivastava M, Sheridan JM, Sng XYX, Delmonte OM, et al. Nfkb2 variants reveal a p100-degradation threshold that defines autoimmune susceptibility. J Exp Med. (2021) 218:e20200476. doi: 10.1084/jem.20200476
113. Willmann KL, Klaver S, Dogu F, Santos-Valente E, Garncarz W, Bilic I, et al. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat Commun. (2014) 5:5360. doi: 10.1038/ncomms6360
114. Qiao Q, Yang C, Zheng C, Fontán L, David L, Yu X, et al. Structural architecture of the CARMA1/Bcl10/MALT1 signalosome: nucleation-induced filamentous assembly. Mol Cell. (2013) 51:766–79. doi: 10.1016/j.molcel.2013.08.032
115. Nakaya M, Xiao Y, Zhou X, Chang J-H, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. (2014) 40:692–705. doi: 10.1016/j.immuni.2014.04.007
116. Ruland J, Hartjes L. CARD–BCL-10–MALT1 signalling in protective and pathological immunity. Nat Rev Immunol. (2019) 19:118–34. doi: 10.1038/s41577-018-0087-2
117. Glocker E-O, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. NEJM. (2009) 361:1727–35. doi: 10.1056/NEJMoa0810719
118. Shields AM, Bauman BM, Hargreaves CE, Pollard AJ, Snow AL, Patel SY. A novel, heterozygous three base-pair deletion in CARD11 results in B cell expansion with NF-κB and T cell anergy disease. J Clin Immunol. (2020) 40:406–11. doi: 10.1007/s10875-019-00729-x
119. Dadi H, Jones TA, Merico D, Sharfe N, Ovadia A, Schejter Y, et al. Combined immunodeficiency and atopy caused by a dominant negative mutation in caspase activation and recruitment domain family member 11 (CARD11). J Allergy Clin Immun. (2018) 141:1818–1830.e2. doi: 10.1016/j.jaci.2017.06.047
120. Stepensky P, Keller B, Buchta M, Kienzler A-K, Elpeleg O, Somech R, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol. (2013) 131:477–85.e1. doi: 10.1016/j.jaci.2012.11.050
121. Greil J, Rausch T, Giese T, Bandapalli OR, Daniel V, Bekeredjian-Ding I, et al. Whole-exome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immunol. (2013) 131:1376–83.e3. doi: 10.1016/j.jaci.2013.02.012
122. Peled A, Sarig O, Sun G, Samuelov L, Ma CA, Zhang Y, et al. Loss-of-function mutations in caspase recruitment domain-containing protein 14 (CARD14) are associated with a severe variant of atopic dermatitis. J Allergy Clin Immun. (2019) 143:173–181.e10. doi: 10.1016/j.jaci.2018.09.002
123. Torres JM, Martinez-Barricarte R, García-Gómez S, Mazariegos MS, Itan Y, Boisson B, et al. Inherited BCL10 deficiency impairs hematopoietic and nonhematopoietic immunity. J Clin Invest. (2014) 124:5239–48. doi: 10.1172/JCI77493
124. Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, et al. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immunol. (2013) 132:151–8. doi: 10.1016/j.jaci.2013.04.047
125. Wiegmann H, Reunert J, Metze D, Marquardt T, Engel T, Kunde V, et al. Refining the dermatological spectrum in primary immunodeficiency: mucosa-associated lymphoid tissue lymphoma translocation protein 1 deficiency mimicking netherton/omenn syndromes. Brit J Dermatol. (2020) 182:202–7. doi: 10.1111/bjd.18091
126. Sefer AP, Abolhassani H, Ober F, Kayaoglu B, Eltan SB, Kara A, et al. Expanding the clinical and immunological phenotypes and natural history of MALT1 deficiency. J Clin Immunol. (2022) 42:634–52. doi: 10.1007/s10875-021-01191-4
127. Rym AVD, Taur P, Martinez-Barricarte R, Lorenzo L, Puel A, Gonzalez-Navarro P, et al. Human BCL10 deficiency due to homozygosity for a rare allele. J Clin Immunol. (2020) 40:388–98. doi: 10.1007/s10875-020-00760-3
128. García-Gómez S, Chaparro R, Safa A, Rym AVD, Martinez-Barricarte R, Lorenzo L, et al. Double-strand break repair through homologous recombination in autosomal recessive BCL10 deficiency. J Allergy Clin Immun. (2019) 143:1931–1934.e1. doi: 10.1016/j.jaci.2018.12.1000
129. Turvey SE, Durandy A, Fischer A, Fung S-Y, Geha RS, Gewies A, et al. The CARD11-BCL10-MALT1 (CBM) signalosome complex: stepping into the limelight of human primary immunodeficiency. J Allergy Clin Immun. (2014) 134:276–84. doi: 10.1016/j.jaci.2014.06.015
130. McKinnon ML, Rozmus J, Fung S-Y, Hirschfeld AF, Bel KLD, Thomas L, et al. Combined immunodeficiency associated with homozygous MALT1 mutations. J Allergy Clin Immun. (2014) 133:1458–1462.e7. doi: 10.1016/j.jaci.2013.10.045
131. Demeyer A, Nuffel EV, Baudelet G, Driege Y, Kreike M, Muyllaert D, et al. MALT1-Deficient Mice develop atopic-like dermatitis upon aging. Front Immunol. (2019) 10:2330. doi: 10.3389/fimmu.2019.02330
132. Dorjbal B, Stinson JR, Ma CA, Weinreich MA, Miraghazadeh B, Hartberger JM, et al. Hypomorphic caspase activation and recruitment domain 11 (CARD11) mutations associated with diverse immunologic phenotypes with or without atopic disease. J Allergy Clin Immunol. (2019) 143:1482–95. doi: 10.1016/j.jaci.2018.08.013
133. Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline hypomorphic CARD11 mutations in severe atopic disease. Nat Genet. (2017) 49:1192–201. doi: 10.1038/ng.3898
134. Izadi N, Bauman BM, Dabbah G, Thauland TJ, Butte MJ, Snow AL, et al. CADINS In an adult with chronic sinusitis and atopic disease. J Clin Immunol. (2021) 41:256–8. doi: 10.1007/s10875-020-00893-5
135. Fuchs S, Rensing-Ehl A, Pannicke U, Lorenz MR, Fisch P, Jeelall Y, et al. Omenn syndrome associated with a functional reversion due to a somatic second-site mutation in CARD11 deficiency. Blood. (2015) 126:1658–69. doi: 10.1182/blood-2015-03-631374
136. Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, et al. Identifying the MAGUK protein carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. (2003) 18:751–62. doi: 10.1016/s1074-7613(03)00141-9
137. Altin JA, Tian L, Liston A, Bertram EM, Goodnow CC, Cook MC. Decreased T-cell receptor signaling through CARD11 differentially compromises forkhead box protein 3-positive regulatory versus TH2 effector cells to cause allergy. J Allergy Clin Immunol. (2011) 127:1277–85.e5. doi: 10.1016/j.jaci.2010.12.1081
138. Hutcherson SM, Bedsaul JR, Pomerantz JL. Pathway-Specific defects in T, B, and NK cells and age-dependent development of high IgE in mice heterozygous for a CADINS-associated dominant negative CARD11 allele. J Immunol. (2021) 207:1150–64. doi: 10.4049/jimmunol.2001233
139. Hara H, Wada T, Bakal C, Kozieradzki I, Suzuki S, Suzuki N, et al. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity. (2003) 18:763–75. doi: 10.1016/s1074-7613(03)00148-1
140. Tian L, Altin JA, Makaroff LE, Franckaert D, Cook MC, Goodnow CC, et al. Foxp3 + regulatory T cells exert asymmetric control over murine helper responses by inducing Th2 cell apoptosis. Blood. (2011) 118:1845–53. doi: 10.1182/blood-2011-04-346056
141. Wang M, Zhang S, Zheng G, Huang J, Songyang Z, Zhao X, et al. Gain-of-Function mutation of Card14 leads to spontaneous psoriasis-like skin inflammation through enhanced keratinocyte response to IL-17A. Immunity. (2018) 49:66–79.e5. doi: 10.1016/j.immuni.2018.05.012
142. Jordan CT, Cao L, Roberson EDO, Pierson KC, Yang C-F, Joyce CE, et al. PSORS2 Is due to mutations in CARD14. Am J Hum Genet. (2012) 90:784–95. doi: 10.1016/j.ajhg.2012.03.012
143. Bähr C, Rohwer A, Stempka L, Rincke G, Marks F, Gschwendt M. DIK, a novel protein kinase that interacts with protein kinase cδ CLONING, CHARACTERIZATION, AND GENE ANALYSIS*. J Biol Chem. (2000) 275:36350–7. doi: 10.1074/jbc.m004771200
144. Xu J, Wei Q, He Z. Insight into the function of RIPK4 in keratinocyte differentiation and carcinogenesis. Frontiers Oncol. (2020) 10:1562. doi: 10.3389/fonc.2020.01562
145. Rountree RB, Willis CR, Dinh H, Blumberg H, Bailey K, Dean C, et al. RIP4 Regulates epidermal differentiation and cutaneous inflammation. J Invest Dermatol. (2010) 130:102–12. doi: 10.1038/jid.2009.223
146. Bartsocas CS, Papas CV. Popliteal pterygium syndrome. Evidence for a severe autosomal recessive form. J Med Genet. (1972) 9:222. doi: 10.1136/jmg.9.2.222
147. Mitchell K, O’Sullivan J, Missero C, Blair E, Richardson R, Anderson B, et al. Exome sequence identifies RIPK4 as the bartsocas- papas syndrome locus. Am J Hum Genetics. (2012) 90:69–75. doi: 10.1016/j.ajhg.2011.11.013
148. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. (2016) 48:67–73. doi: 10.1038/ng.3459
149. Gans MD, Wang H, Moura NS, Aksentijevich I, Rubinstein A. A20 haploinsufficiency presenting with a combined immunodeficiency. J Clin Immunol. (2020) 40:1041–4. doi: 10.1007/s10875-020-00823-5
150. Duncan CJA, Dinnigan E, Theobald R, Grainger A, Skelton AJ, Hussain R, et al. Early-onset autoimmune disease due to a heterozygous loss-of-function mutation in TNFAIP3 (A20). Ann Rheum Dis. (2018) 77:783–6. doi: 10.1136/annrheumdis-2016-210944
151. Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. (2000) 25:160–5. doi: 10.1038/76006
152. Biggs PJ, Wooster R, Ford D, Chapman P, Mangion J, Quirk Y, et al. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12–q13: evidence for its role as a tumour suppressor gene. Nat Genet. (1995) 11:441–3. doi: 10.1038/ng1295-441
153. Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fässler R. Cyld inhibits tumor cell proliferation by blocking bcl-3-dependent NF-kappaB signaling. Cell. (2006) 125:665–77. doi: 10.1016/j.cell.2006.03.041
154. Häcker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. (2006) 2006:re13. doi: 10.1126/stke.3572006re13
155. Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD Is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature. (2003) 424:793–6. doi: 10.1038/nature01803
156. Hutti JE, Shen RR, Abbott DW, Zhou AY, Sprott KM, Asara JM, et al. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKɛ promotes cell transformation. Mol Cell. (2009) 34:461–72. doi: 10.1016/j.molcel.2009.04.031
157. Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature. (2003) 424:801–5. doi: 10.1038/nature01802
158. Mathis B, Lai Y, Qu C, Janicki J, Cui T. CYLD-Mediated Signaling and diseases. Curr Drug Targets. (2015) 16:284–94. doi: 10.2174/1389450115666141024152421
159. Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. (2008) 135:1311–23. doi: 10.1016/j.cell.2008.10.044
160. Schworer SA, Smirnova II, Kurbatova I, Bagina U, Churova M, Fowler T, et al. Toll-like receptor-mediated down-regulation of the deubiquitinase cylindromatosis (CYLD) protects macrophages from necroptosis in wild-derived mice*. J Biol Chem. (2014) 289:14422–33. doi: 10.1074/jbc.m114.547547
161. Massoumi R, Paus R. Cylindromatosis and the CYLD gene: new lessons on the molecular principles of epithelial growth control. Bioessays. (2007) 29:1203–14. doi: 10.1002/bies.20677
162. Zhang J, Stirling B, Temmerman ST, Ma CA, Fuss IJ, Derry JMJ, et al. Impaired regulation of NF-κB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest. (2006) 116:3042–9. doi: 10.1172/jci28746
163. Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, et al. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell. (2016) 166:1215–1230.e20. doi: 10.1016/j.cell.2016.07.019
164. Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc National Acad Sci. (2016) 113:10127–32. doi: 10.1073/pnas.1612594113
165. Damgaard RB, Elliott PR, Swatek KN, Maher ER, Stepensky P, Elpeleg O, et al. OTULIN Deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol Med. (2019) 11:972. doi: 10.15252/emmm.201809324
166. Spaan AN, Neehus A-L, Laplantine E, Staels F, Ogishi M, Seeleuthner Y, et al. Human OTULIN haploinsufficiency impairs cell-intrinsic immunity to staphylococcal α-toxin. Science. (2022) 376:eabm6380. doi: 10.1126/science.abm6380
Keywords: NF-κB, immunodeficiency, genetics, skin, ectoderm abnormalities
Citation: Shen Y, Boulton APR, Yellon RL and Cook MC (2023) Skin manifestations of inborn errors of NF-κB. Front. Pediatr. 10:1098426. doi: 10.3389/fped.2022.1098426
Received: 14 November 2022; Accepted: 23 December 2022;
Published: 17 January 2023.
Edited by:
Oded Shamriz, Hadassah Medical Center, IsraelReviewed by:
Antonio Condino-Neto, University of São Paulo, Brazil© 2023 Shen, Boulton, Yellon and Cook. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew C. Cook bWMyMzg2QGNhbS5hYy51aw==
Specialty Section: This article was submitted to Pediatric Immunology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.