
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 02 February 2023
Sec. Pediatric Endocrinology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1078853
This article is part of the Research TopicManagement of Bone Disorders in Children - Volume IIView all 9 articles
 Anthony Batte1,†
Anthony Batte1,† Philip Kasirye2Reagan Baluku2
Philip Kasirye2Reagan Baluku2 Sarah Kiguli2,†
Sarah Kiguli2,† Robert Kalyesubula3,†Chandy C. John4,5,†
Robert Kalyesubula3,†Chandy C. John4,5,† Andrew L. Schwaderer4
Andrew L. Schwaderer4 Erik A. Imel4
Erik A. Imel4 Andrea L. Conroy4,5*†
Andrea L. Conroy4,5*†
Background: Mineral bone disorders (MBD) are common in sickle cell anemia (SCA). Frequent vaso-occlusive crises (VOC) further impact MBD in children with SCA. We evaluated the prevalence of markers of SCA-related MBD (sMBD) in hospitalized children and assessed the relationship between sMBD and individual mineral abnormalities with kidney disease.
Methods: We prospectively recruited 185 children with SCA hospitalized with a VOC. Serum measures of mineral bone metabolism (calcium, phosphate, parathyroid hormone, 25-hydroxy vitamin D, FGF23, osteopontin) were measured at enrollment. The primary outcome was markers of sMBD defined as a composite of hypocalcemia, hyperphosphatemia, hyperparathyroidism, or deficiency in 25-OH vitamin D. Secondary outcomes included individual abnormalities in mineral metabolism. The Kidney Disease: Improving Global Outcomes (KDIGO) guidelines were used to define SCA-associated acute kidney injury (AKI). AKI was further assessed using urine NGAL as a marker of tubular injury. Acute kidney disease (AKD) was defined as a composite of AKI, an eGFR < 90 ml/min per 1.73 m2 using the Cystatin C GFR equation, or evidence of structural injury (positive biomarker test or albuminuria).
Results: The mean age of children was 8.9 years and 41.6% were female. The prevalence of sMBD was 47.6%, with hypocalcemia the most frequent abnormality (29.9%, 55/184) followed by hyperphosphatemia (20.7%, 38/184), hyperparathyroidism (8.7%, 16/185), and vitamin D deficiency (5.4%, 10/185). There was no association between sMBD and sKDIGO-defined AKI using serial changes in creatinine or when incorporating biomarkers to define AKI. However, the presence of AKD was associated with a 2.01-fold increased odds of sMBD (95% CI 1.05 to 3.83) and was driven by a decrease in eGFR (OR, 2.90 95% CI: 1.59 to 5.29). When evaluating individual mineral abnormalities, hypocalcemia was associated with AKD and low eGFR while hyperparathyroidism was associated with low eGFR, AKI and structural injury. Vitamin D deficiency was associated with structural kidney injury. Vitamin D deficiency, hyperparathryoidism, and increases in FGF23 and osteopontin predicted mortality (p < 0.05 for all).
Conclusion: MBD is common among children with SCA hospitalized with VOC. Biomarkers of kidney injury and bone health may help risk stratify children at risk of sMBD. Routine evaluation of sMBD in children with SCA may improve long-term bone health.
Sickle cell anemia (SCA) is among the commonest inherited hemoglobinopathies and results from a mutation in the β-globin gene (HBB) (1). The disease is autosomal recessive and characterized by Hemoglobin S polymerization, vaso-occlusion, and hemolysis which in turn leads to ischemia-reperfusion injury, endothelial dysfunction, oxidative stress, and immune activation (1, 2). SCA is a multi-system disease that causes repeated injury and progressive damage to various organs including the brain, kidneys and bones (2). Vaso-occlusive crises are one of the most common complications among children with SCA and are characterized by microvascular occlusions in the bone marrow leading to intense pain in one or more areas of the skeleton (3). Childhood and adolescence is a critical period of bone growth and bone mineral accrual with 90% of bone mass accumulated by 18 years of age (4, 5). Bone mass attained during childhood and adolescence is one of the most important determinants of skeletal health across the lifespan (6).
SCA is associated with growth disturbances and an increased risk of chronic bone problems including chronic pain, avascular necrosis, and vertebral collapse that lead to significant long-term morbidity (3). Mineral bone disorders (MBD) are common in the context of SCA with a number of mineral bone disease abnormalities reported, including hypocalcemia, hyperphosphatemia, hyperparathyroidism, and vitamin D deficiency (7–9). Additional biomarkers that modulate bone mineralization include osteopontin, a phosphorylated glycoprotein that can inhibit bone mineral deposition (10, 11), and fibroblast growth factor-23 (FGF23), a hormone that modulates phosphate handling of the kidneys (12). Acute kidney injury (AKI) develops in up to 36.2% of children with SCA hospitalized with a vaso-occlusive crisis (VOC) (13), and chronic kidney disease (CKD) is reported in up to 26.5% of children with SCA (14). Although MBD is well characterized in CKD (15), there is emerging evidence that MBD occurs during episodes of AKI (16) and may be associated with increased mortality in AKI (17). There are limited data on MBD in children with AKI and SCA thus highlighting the need for more studies.
In this prospective observational study, we evaluated the prevalence of markers of sickle-cell anemia related MBD (sMBD) in children with SCA hospitalized for a painful VOC and assessed the relationship between sMBD and the presence of AKI and other kidney abnormalities.
Between January and July in 2019, children with sickle cell anemia admitted with a VOC were screened and consecutively enrolled (18). Children were recruited from Mulago National Referral and Teaching Hospital, which is located in Central Uganda and has a high outpatient and inpatient volume. Most children admitted with a VOC are referred from the hospital's dedicated sickle cell clinic that attends to about 1,400 children per year. Routine care includes daily folic acid supplementation, three-monthly malaria prophylaxis with sulfadoxine-pyrimethamine, and penicillin V for children <5 years of age. Hydroxyurea is approved for use and is available in a limited capacity. Care for children with VOC involves hydration using oral or intravenous fluids and analgesic medications based on pain severity and typically includes paracetamol, non-steroidal anti-inflammatory drugs, and opioids (morphine, tramadol).
Eligibility criteria included an age between 2 and 18 years, documented SCA by hemoglobin electrophoresis (hemoglobin SS), hospitalization for a VOC with a pain score ≥2 using an age-specific pain scale. Pain was assessed using the face, legs, activity, cry, and consolability (FLACC) scale (1) in children aged 2–3 years, the Wong-Baker Faces pain scale in children 3–7 years and the numeric pain scale in children ≥ 8 years (2). Children were only eligible to be enrolled once during the recruitment period. The primary outcome was the presence of sMBD defined as a composite of one or more of the following mineral metabolism abnormalities; hypocalcemia, hyperphosphatemia, hyperparathyroidism, or vitamin D deficiency. The individual mineral metabolism abnormalities were secondary outcomes. A sample size of 185 children was generated assuming a prevalence of hypocalcemia of 14% (9). The sample size formula used was n = (z2 p(1 − p)/d2), where; z = standard normal variate corresponding to the 95% confidence interval and is 1.96, d = the required precision of the estimate (0.05), p = prevalence rate (19).
On enrollment all children had a complete history and physical exam to assess medication use, signs of infection, and the site and severity of pain. On admission children had venous blood drawn with serum collected in an additive free tube and allowed to clot at room temperature for 30–60 min before centrifugation at 1200 g for 20 min and storage at −80 °C for subsequent biomarker testing. A spot urine sample was collected using a urine bag or urine container for older children for urinalysis, processing, and storage. Urine samples were spun at room temperature for 5 min at 400 g to remove urine sediment. Urine samples were stored at −80 °C until ELISA testing.
Laboratory tests included complete blood count and serum studies to evaluate biochemical abnormalities in mineral metabolism [Ca2+, Mg2+, phosphorous, intact parathryroid hormone (iPTH)]. The complete blood counts were measured using the Sysmex automated haematology analyser, version XN 450. Serum laboratory tests were measured by the Johns Hopkins University Infectious Disease Institute Laboratory on serum samples using a Cobas machine (Roche Diagnostics, Indianapolis, USA). Intact PTH was measured in serum by the Ebenezer Clinical Laboratory Ltd. using an electrochemiluminescence immunassay using a Cobas e411 analyzer. Urine albumin and creatinine were tested by the Mulago Hospital Chemistry Laboratory using a Cobas analyzer (Roche Diagnostics, Indianapolis, USA).
Additional markers related to bone mineral metabolism and kidney function were measured on stored serum and urine samples by enzyme linked immunoassays (ELISA). Serum cystatin C was measured using the R&D Systems Quantikine ELISA (R&D Systems, Minneapolis, MN) at 1:40 dilution with a reported range of 0.06 to 16 mg/L with pooled serum controls and commercial controls on every plate. The assay has been correlated against the Cystatin C reference standard provided by the Joint Research Centre Institute for Reference Material and Measurements (Catalog # ERM-DA471/ IFCC) with a slope of 1.07 and a R2 value of 0.998. Serum FGF23 was measured using a DuoSet by R&D Systems at a 1:5 dilution with a reported range of 200 to 50,000 pg/ml (R&D Systems, Minneapolis, MN). Serum and urine Osteopontin (OPN) were measured using a DuoSet by R&D Systems at a dilution of 1:100 and 1:1,000 respectively (R&D Systems, Minneapolis, MN). Urine NGAL was measured using an ELISA by BioPorto (Kit 036, BioPorto Diagnostics Inc., Hellerup, Denmark) at a 1:1,000 dilution with a reported range of 5–2000 ng/ml according to the manufacturer's protocol. Vitamin D was measured using a commercial assay (Eagle Biosciences Inc, NH, USA) that recognizes total 25-OH vitamin D (including vitamin D3 and D2) in serum with a reported range of 5–600 nmol/L. All sample testing was conducted by trained technicians blinded to patient details. Samples below the limit of detection were assigned the lower limit of the assay.
Acute kidney injury (AKI) was defined based on the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines as an increase in serum creatinine of ≥0.3 mg/dl within 48 h or a 50% increase in baseline creatinine within 7 days (3). Kidney function was assessed on enrollment, at 48 h and on day 7 or discharge (whichever happened earlier) using the i-STAT handheld blood analyzer (Abbott Point of Care Inc., Princeton, NJ) with assays traceable to the U.S. National Institute of Standards and Technology (NIST) standard reference material SRM909 with reportable range is 0.20–20.0 mg/dl. For the purposes of defining AKI a creatinine value below the reportable range (<0.20 mg/dl) was assigned a value of 0.19 mg/dl. The participants' lowest creatinine measure was taken as the baseline creatinine. In instances where only a single creatinine measure was available (n = 7), we used the Pottel-age based GFR equation assuming a normal GFR of 120 ml/min per 1.73 m2 (20, 21). As previously described, children with an increase in creatinine from 0.2 to 0.3 mg/dl were not considered to have AKI (13).
Defining AKI using creatinine in children with SCA has limitations as the children are likely to have hyperfiltration, increased tubular creatinine excretion (22) and creatinine levels may be affected by reduced muscle mass (23, 24). We thus used the 23rd Acute Disease Quality Initiative (ADQI-23) consensus guidelines which integrate the use of other functional biomarkers of kidney injury in addition to creatinine in defining and staging AKI (25). Based on the ADOQI-23 guidelines, AKI is staged based on presence of a positive or negative kidney injury biomarker on addition to the fold increase in creatinine as defined by the KDIGO guidelines. In our study, a uNGAL level ≥150 ng/ml was considered biomarker positive for tubular injury (BM+) (26). Admission eGFR was calculated using the CKiD cystatin C-based equation, eGFR = 70.69 * (cystatin C)−0.931 (27). Acute kidney disease was defined as one or more of the following: AKI or an eGFR < 90 ml/min per 1.73 m2 or signs of structural injury (macroalbuminuria or BM+) (25, 28).
Bone mineral abnormalities were defined using the following criteria: hypocalcemia (<2.2 mmol/L following correction for serum albumin) (29), hyperphosphatemia (phosphorous > upper limit of normal for age and sex) (30), hyperparathyroid hormone (iPTH > 70 pg/ml) (31), vitamin D deficiency (25-OH vitamin D < 30 nmol/L) (32). A child meeting any of the above criteria was considered to have sickle-cell anemia related MBD (sMBD). In addition, children were classified as having elevated FGF23 and elevated osteopointin if the concentrations exceeded the 95th percentile measured using a population of children with SCA in steady state with the cut-offs as follows: FGF23 > 535 pg/ml and osteopontin >375 ng/ml.
Data were entered into REDCap electronic data capture tools hosted at Indiana University. Data were analyzed using STATA v17.0 (StataCorp) and GraphPad Prism v9. Data are presented descriptively using median and interquartile range (IQR) for continuous variables and the number and frequency for discrete variables. To examine the relationship between continuous variables and dichotomized measures of sMBD, a Wilcoxon rank-sum test was used for continuous variables. For categorical variables, Pearson's Chi-square or Fisher's exact test were used, as appropriate. Spearman's rank correlation was used to evaluate relationships between measures of bone metabolism and kidney function. Logistic regression was used to evaluate the relationship between AKI and AKD with sMBD or individual mineral metabolism abnormalities and models were adjusted for participant age and sex. Bonferroni correction was used to adjust for multiple testing.
We assessed sMBD in 185 children with sickle cell anemia hospitalized for a painful VOC as outlined in Figure 1. The median age of children was 8.9 years [interquartile range (IQR), 5.9 to 11.8] and 41.6% of participants were female (Table 1). The median duration of pain prior to presentation was 3 days (IQR, 2 to 4) with a median pain score of 6 (IQR, 4 to 8). Overall, the prevalence of sMBD was 47.6% (Figure 1). Hypocalcemia was the most frequent mineral bone abnormality observed at 29.9% (55/184) followed by hyperphosphatemia at 20.7% (38/184), hyperparathyroidism at 8.7% (16/185) and vitamin D deficiency at 5.4% (10/185). Vitamin D insufficiency (30–50 nmol/L) was present in 24.3% (45/185) of the children.
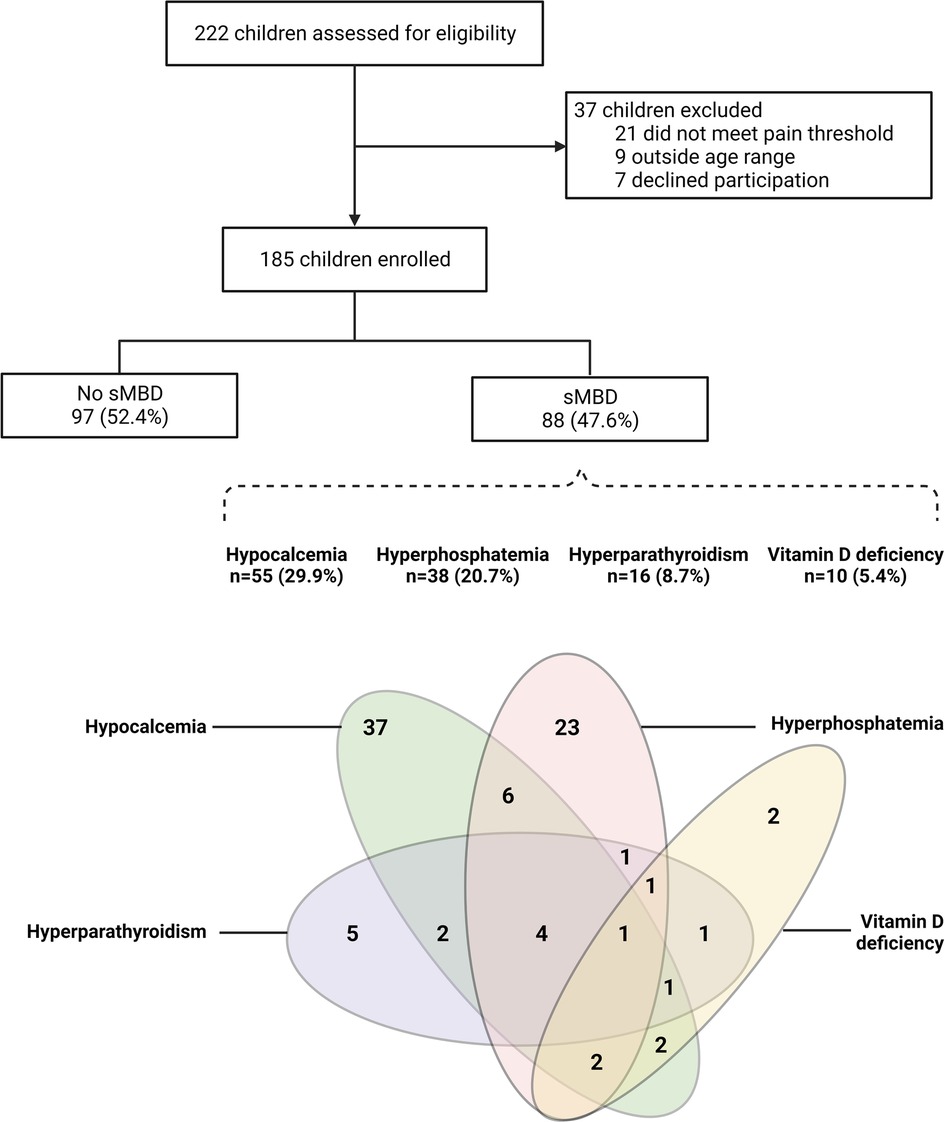
Figure 1. Flowchart of the study population. Flow chart depicting study participants based on sickle cell anemia related mineral bone disorder (sMBD). Below the flowchart is a Venn diagram showing the inter-relationships between parameters used to define sMBD with hypocalcemia (corrected calcium <2.2 mmol/L) present in 55 children (29.9%), hyperphosphatemia (phosphate levels > upper limit of normal for age and sex) present in 38 children (20.7%), hyperparathyroidism (parathyroid hormone >70 pg/ml) present in 16 study participants (8.7%) and Vitamin D deficiency (<30 nmol/L) present in 10 (5.4%) of study participants. Created with BioRender.com.
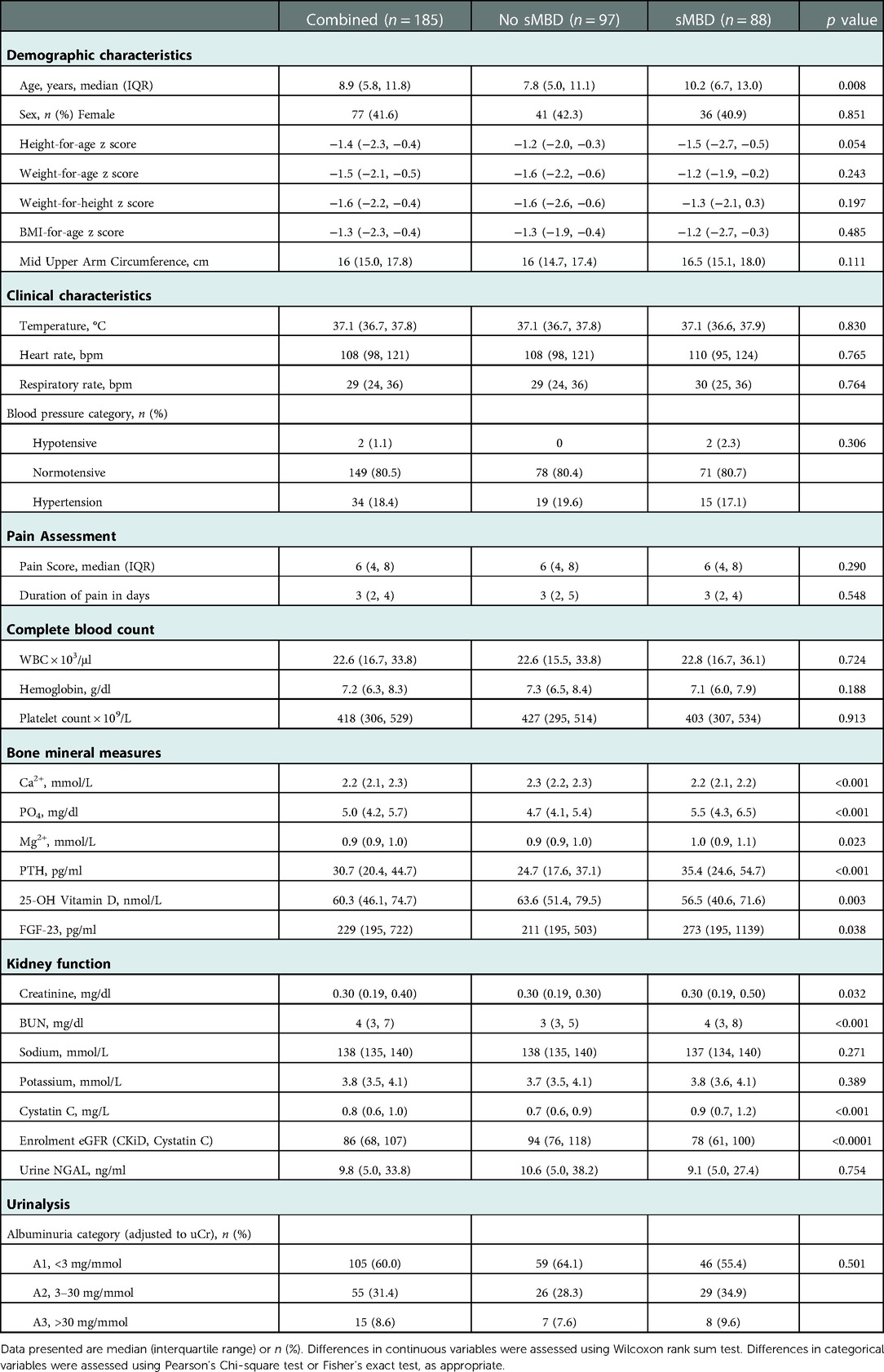
Table 1. Demographic, clinical and laboratory characteristics on admission associated with bone mineral disease.
Demographic, clinical, and laboratory characteristics are presented in Table 1. Children with sMBD were significantly older than children without sMBD (p = 0.008) but were similar in nutritional status and clinical characteristics on admission. There were no differences in the history of pain duration or the severity of pain on admission based on sMBD (p > 0.05). In addition to differences in the measures used to define sMBD (Ca2+, PO4−, PTH, 25-OH vitamin D), children with sMBD had higher levels of Mg2+ and FGF23 compared to children without sMBD (p < 0.05 for both). There were differences in kidney function (BUN, Cystatin C) in children with sMBD compared to children without sMBD.
We conducted additional analyses evaluating differences in the same measures in children with and without hypocalcemia and hyperphosphatemia as the most frequent findings in children with sMBD (Table 2) and vitamin D deficiency and hyperparathyroidism as the least frequent findings (Table 2). Participants with hypocalcemia, hyperphosphatemia, and hyperparathyroidism all had increased levels of Cystatin C and BUN compared to children with normal values for the same marker of mineral metabolism. Further, children with both vitamin D deficiency or with hyperparathyroidism had higher levels of urine NGAL as a measure of proximal tubular injury, higher potassium, osteopontin, and FGF23 compared to children without vitamin D deficiency or hyperparathyroidsm. When evaluating mineral abnormalities by age, children with hyperparathyroidism and vitamin D deficiency were older (Table 2) and this was further supported by an increase in the frequency of mineral abnormalities across age categories (Figure 2). The earliest abnormality to present was hypocalcemia, present in 22% of children <5 years of age followed by hyperphosphatemia that emerged in children between 5 and 10 years of age, while both hyperparathyroidism and vitamin D deficiency were relatively infrequent before 10 years of age (Figure 2).
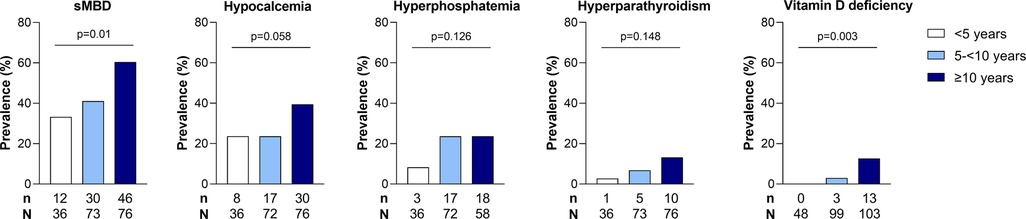
Figure 2. Relationship between age and sMBD in children with SCA. Bar graph depicting the frequency of sMBD prevalence and individual mineral abnormalities across age categories (<5 years, 5- <10 years, ≥10 years). Differences in the frequency of sMBD and mineral abnormalities assessed using Pearson's chi-square test or Fisher's exact test, as appropriate. The p values are presented on the bar graph.
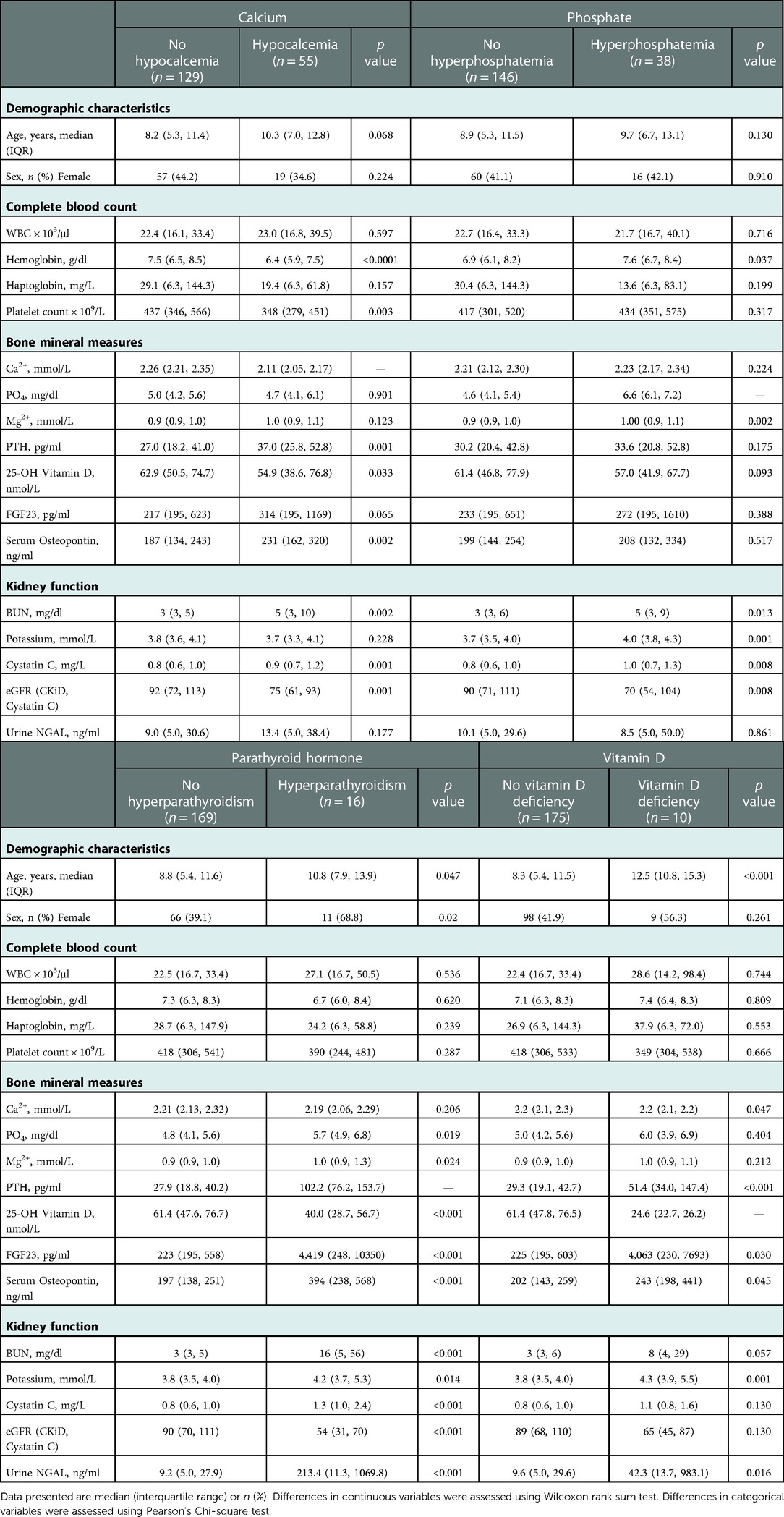
Table 2. Demographic, clinical and laboratory characteristics on admission associated with mineral bone disorders.
We further assessed the relationship between kidney function and sMBD based on the presence of AKI and AKD defined during hospitalization. Overall, 36.2% of children had AKI defined based on serial changes in serum creatinine, with 16 children BM + (8.6%) based on a positive uNGAL test, and 69.7% had AKD (Figure 3). There was no difference in sMBD based on the presence of sub-clinical AKI or AKI status (BM− vs. BM+). However, there was an increase in sMBD in children with AKD that encompassed reduced GFR using the Cystatin C based eGFR equation or structural abnormalities including BM + or macroalbuminuria. A low eGFR defined using Cystatin C was also independently associated with sMBD (Figure 3). We further assessed the relationship between individual mineral abnormalities and AKI and AKD. Both BM + AKI and BM− AKI were associated with hyperparathyroidism while BM + AKI was associated with vitamin D deficiency. There was no association between AKI and hypocalcemia or hyperphosphatemia. Children with AKD had increased odds for having hypocalcemia while a low Cystatin C defined eGFR was associated with both hypocalcemia and hyperparathyroidism. Presence of a marker of structural kidney damage was associated with a 9-fold increased odds of having vitamin D deficiency (95% CI 2.37 to 34.17) (Figure 4).
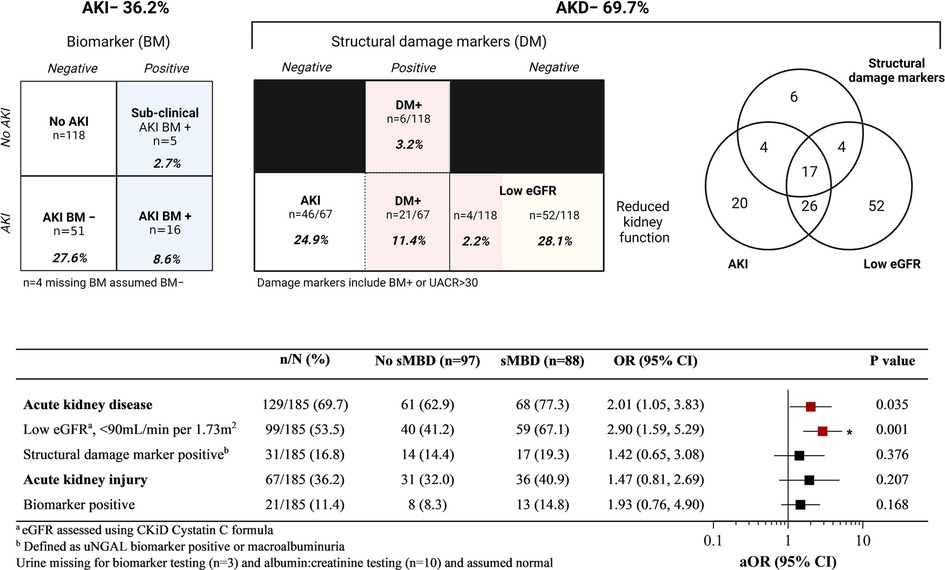
Figure 3. Relationship between acute kidney injury (AKI) and disease (AKD) and mineral bone disease in children with sickle cell anemia hospitalized for a vaso-occlusive crisis. Top. AKI defined using acute changes in creatinine based on sKDIGO definition. AKD was defined based on presence of either a low GFR, AKI or presence of structural damage markers (DM). Structural damage markers included a positive biomarker test (BM+) (urine NGAL ≥ 150 ng/ml), or urine albumin to creatinine ratio (UACR) > 30 mg/mmol. The relationships between measures of kidney injury and disease are presented in the Venn diagram to the right. Bottom. A Forest plot depicting the frequency between different measures of kidney disease and the presence of sMBD. The adjusted odds ratio (aOR) is generated using logistic regression adjusting for participant age and sex. Relationships that were significant (p < 0.05) are presented in red and those significant following adjustment for multiple testing are indicated with an asterisk. The adjusted p value to account for multiple testing was p < 0.01 based on 5 comparisons. Created with BioRender.com.
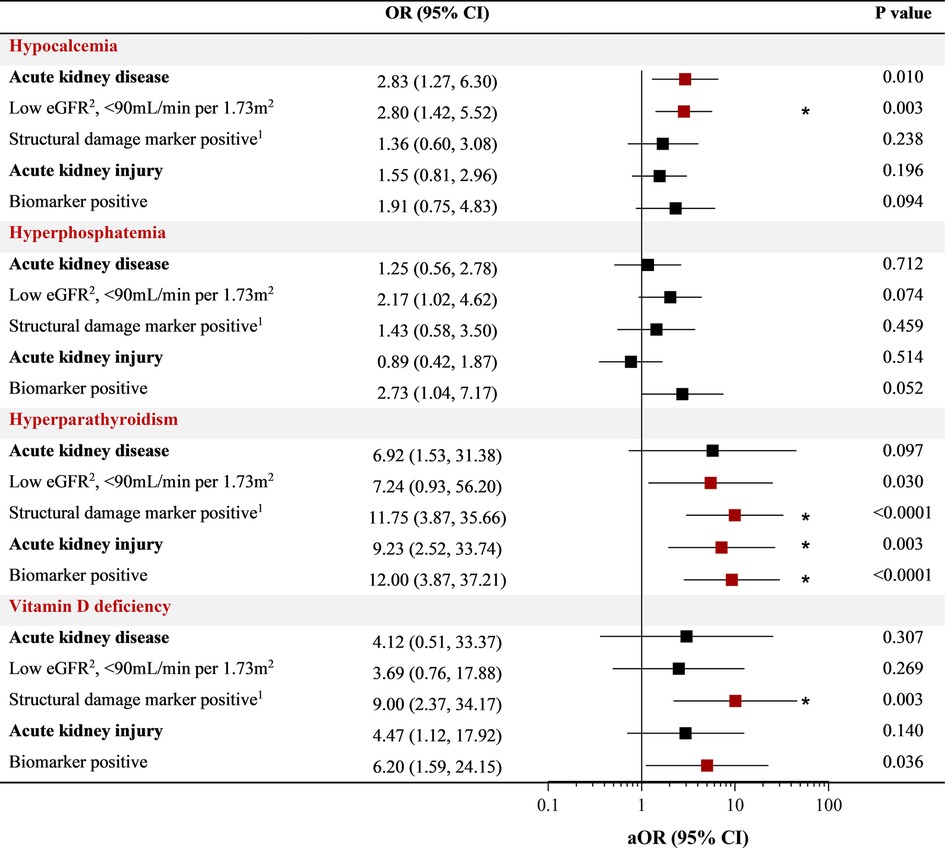
Figure 4. Relationship between kidney function and individual markers of mineral metabolism. Forest plot depicting the odds ratio (OR) and 95% CI between individual abnormalities in bone mineral metabolism (hypocalcemia, hyperphosphatemia, hyperparathyroidism, vitamin D deficiency) and measures of kidney disease using logistic regression. The adjusted OR (aOR) adjusts for participant age and sex with relationships with a p < 0.05 in red and an asterisk indicating relationships significant after adjusting for multiple testing at an adjusted alpha of 0.01 within each mineral abnormality category (*).
In this study, 6/185 (3.2%) of participants died in-hospital with a similar frequency of mortality among children with sMBD 3/88 (3.4%) compared to children without sMBD 3/97 (3.1%). While infrequent, the presence of hyperparathyroidism and vitamin D deficiency were associated with increased mortality (Figure 5). There were significant increases in the median level of serum bone biomarkers FGF23 (p < 0.0001) and osteopontin (p < 0.01) in children who died in-hospital compared to survivors (p < 0.001) (Figure 5). Both elevated FGF23 and osteopontin were associated with higher mortality (p < 0.001) (Figure 5).
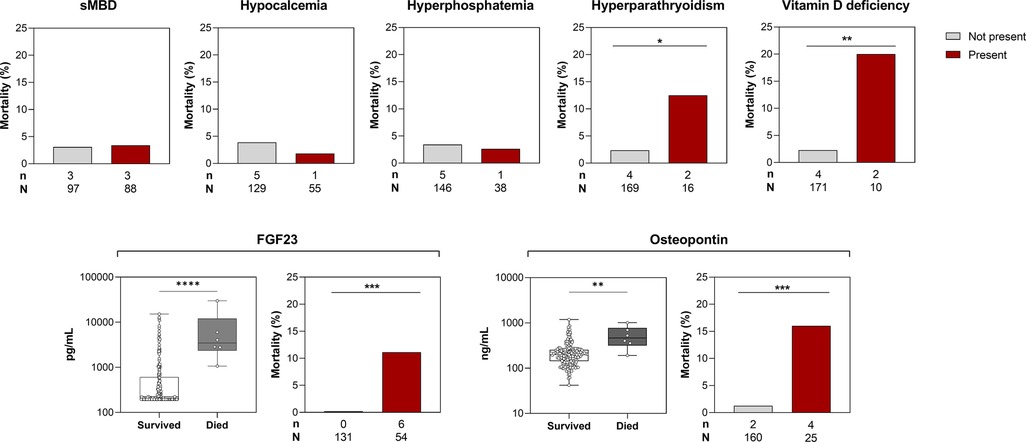
Figure 5. Relationship between sMBD and mortality in children with SCA. Top. Bar graphs depicting the frequency of children who died in-hospital based on the presence of sMBD or individual mineral abnormalities. Bottom. Box plots depicting serum levels of FGF23 and osteopontin at enrollment in children based on in-hospital mortality with differences between groups assessed using the Wilcoxon rank sum test. The corresponding bar graphs depict the frequency of in-hospital death among children with elevated levels of FGF23 and osteopontin using the 95th percentiles from Ugandan children with SCA in steady state to define the reference range. Differences in dichotomous variables and mortality assessed using Fisher's exact test. *p < 0.05, ** p < 0.01, ***p < 0.001, and ****p < 0.0001.
Finally, we conducted correlation analysis to evaluate relationships between individual measures of mineral bone metabolism and kidney function (Figure 6). Within markers of mineral metabolism, parathyroid hormone was positively correlated with phosphate levels and negatively correlated with both calcium and vitamin D levels. FGF23 was negatively correlated with calcium and vitamin D and positively correlated with parathyroid hormone levels, BUN and cystatin C. Parathryroid hormone levels were strongly correlated with markers of reduced kidney filtration (creatinine, BUN, Cystatin C) as well as structural injury to the kidney (urine NGAL) (Figures 4, 6). Osteopontin was negatively correlated with calcium and positively correlated with markers of reduced kidney filtration (creatinine, BUN, Cystatin C) and FGF23.
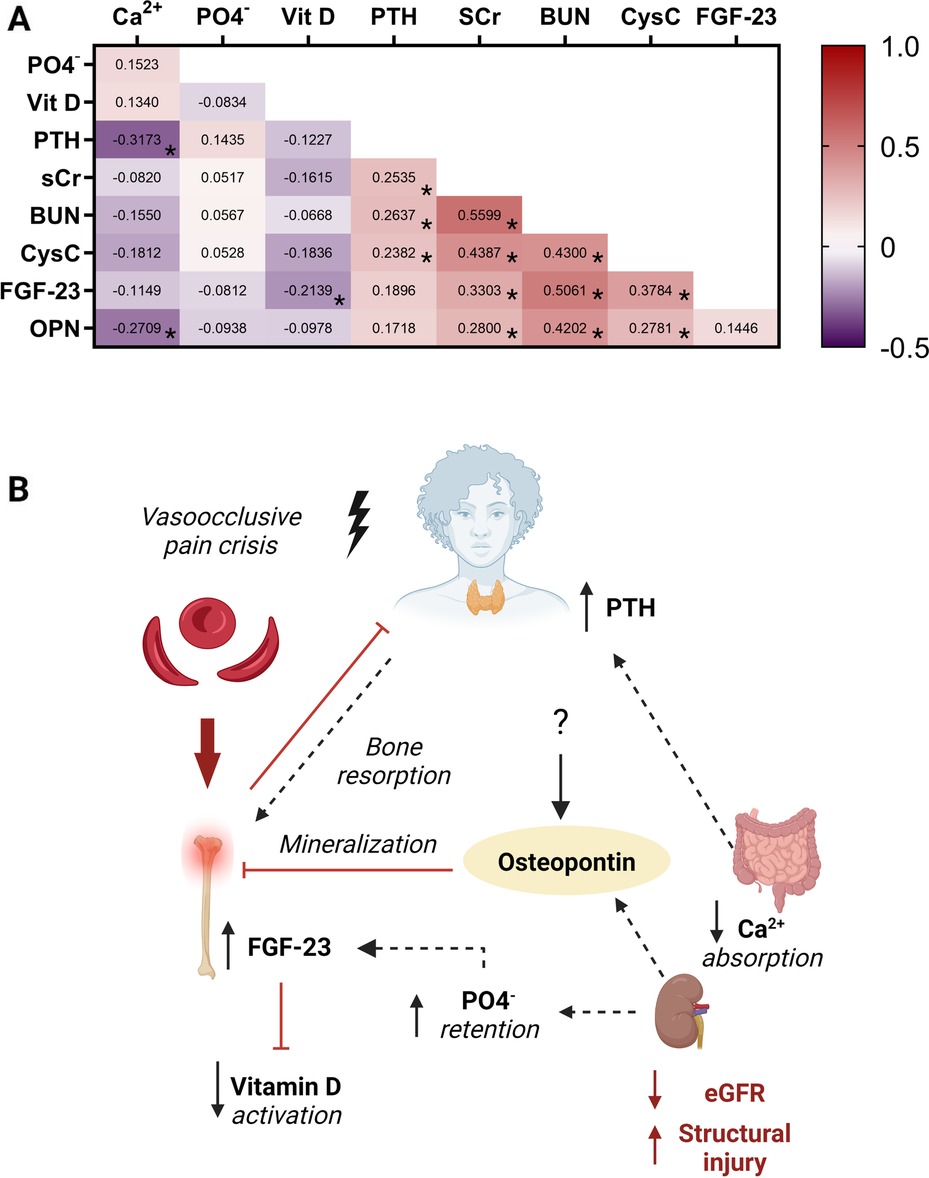
Figure 6. The bone-kidney axis during a vaso-occlusive event. (A) Heatmap depicting the Spearman correlation coefficients between serum measures of mineral metabolism and kidney function. Positive relationships are depicted in red while negative relationships are shown in purple. Relationships significant following adjustment for multiple testing are indicated with an asterisk based on an adjusted alpha of 0.005. (B) Conceptual diagram depicting the relationship between measures of bone mineral metabolism and kidney function during a vaso-occlusive crisis demonstrating the relationship depicted in the heatmap. Low serum calcium levels and increased phosphate levels lead to hyperparathyroidism with resultant bone resorption. The high phosphate levels due to reduced kidney clearance led to high FGF23 which is known to inhibit vitamin D activation. Osteopontin released by various cells and upregulated in AKI, inhibits bone mineralization. Abbreviations: calcium (Ca2+), phosphate (PO4−), Vitamin D (Vit D), parathyroid hormone (PTH), serum creatinine (SCr), blood urea nitrogen (BUN), Cystatin C (CysC), fibroblast growth factor 23 (FGF23), Osteopontin (OPN). Created with BioRender.com.
In this prospective observational study, we demonstrated a high prevalence of sMBD in children hospitalized with a VOC. Hypocalcemia was the most common mineral bone abnormality followed by hyperphosphatemia, hyperparathyroidism and vitamin D deficiency, respectively. With the exception of hyperphosphatemia, all mineral bone abnormalities were associated with kidney disease. sMBD and hypocalcemia were associated with AKD and low eGFR while hyperparathyroidism and vitamin D deficiency were associated with structural injury to the kidney. Although individual features of sMBD varied by age, features associated with mortality during hospitalization (hyperparathryoidism and vitamin D deficiency) were most common in older children. In this population, two additional markers of sMBD, FGF23 and osteopontin, were associated with kidney disease and also strongly associated with mortality.
Mineral bone abnormalities are well known complications of CKD (15, 33); however, there is emerging evidence that AKI is also associated with dysregulation of bone minerals that can impact bone health (16). Data on mineral bone abnormalities in children with AKI and SCA are limited and complicated by challenges in assessing kidney disease in children with SCA due to glomerular hyperfiltration and muscle wasting which affect serum creatinine levels (22–24). In the present study, AKI was assessed using both serum creatinine and urine NGAL as a biomarker of structural kidney injury (25). There were differences in the frequency of sMBD based on AKI status. However, when AKD was evaluated incorporating Cystatin C, a more stable biomarker of glomerular filtration in the context of SCA, and markers of structural kidney injury were incorporated (i.e., macroalbuminuria, positive NGAL), AKD was identified as a risk factor for sMBD. As children with SCA have altered tubular handling of creatinine, alternative biomarkers of kidney function are useful in defining the presence of kidney disease and identifying acute structural injury (34). When defining AKI based on changes in serum creatinine alone (13, 28), children with kidney disease may be misclassified. Thus, the use of a broader AKD definition was able to identify children with AKI and pre-existing kidney disease. As CKD is defined based on a follow-up period of not less than 3 months, a criterion which was not fulfilled in our study, we were unable to differentiate between AKD and CKD.
The most common mineral abnormality in our study was hypocalcemia which occurred in over a quarter of the population and was frequent across the age span. In the context of AKI, low calcium is thought to be secondary to reduced activity of 1 alpha hydroxylase enzyme and vitamin D deficiency (16). Vitamin D deficiency was comparatively uncommon in our cohort, occurring in 5.4% of children although a large proportion of children (24%) had vitamin D insufficiency. A recent multi-country study of 4,509 African children <8 years of age identified vitamin D deficiency in 0.6% of children with seasonal variations in vitamin D levels and increasing vitamin D deficiency in older children (35). Hypocalcemia may also occur as a result of increased intracellular uptake of calcium in sickled cells (36) and as a result of CKD in children with SCA (14, 31). Hypocalcemia and hyperphosphatemia are drivers for elevated parathyroid hormone levels, which were also observed in the population. Parathyroid hormone levels were negatively correlated with serum calcium and positively correlated with serum phosphate levels. The hyperphosphatemia was likely due to reduced renal clearance and was correlated with higher Cystatin C levels as a measure of reduced glomerular filtration in these children (37). In children with sickle cell anemia, hyperphosphatemia may also be secondary to increased tubular reabsorption of phosphorus and resistance to FGF23 (38, 39).
Hyperparathyroidism was associated with multiple measures of kidney function, including structural abnormalities in kidney function and AKI. This suggests that AKI may exacerbate hyperparathyroidism present in SCA (40, 41). Hyperparathyroidism occurred in 8.7% of children with SCA, was more frequent in children ≥10 years of age (13.2%) and was associated with a marked increase in both Cystatin C and urine NGAL levels (Table 2). While death was relatively infrequent in the study population, there was a substantial increase in mortality among children with hyperphosphatemia and vitamin D deficiency. Vitamin D deficiency was not seen in children <5 years of age but increased to 12.6% in children ≥10 years of age and was more frequent in the context of structural kidney injury (such as children who were biomarker positive for NGAL).
We demonstrated that FGF23 and osteopontin were elevated in sMBD and strongly associated with mortality. FGF23 is a hormone excreted by osteoblasts and osteoclasts and regulates serum phosphate through the FGF receptor /Klotho complex in the kidneys (12). FGF23 has been found to predict mortality in patients with AKI following cardiac surgery (42), critically ill patients (43, 44), and patients with CKD (45). There is limited evidence that FGF23 may predict SCA-related mortality in adults (46), and our study provides additional evidence that increased FGF23 is associated with mortality in children with SCA. FGF23 reduces circulating plasma phosphate by decreasing renal reabsorption of phosphate and by blocking vitamin D activation. There was an 18-fold increase in median FGF23 levels in children with vitamin D deficiency and a negative correlation between FGF23 and 25-hydroxyvitamin D. However, we did not have measures of active 1,25 dihydroxy vitamin D to assess whether elevated FGF23 was inversely correlated with activated vitamin D. Osteopontin is a glycoprotein expressed mostly by the bones and in varying amounts from other cells such as macrophages, smooth muscle, endothelial and epithelial cells and is involved in inhibiting bone mineralization (10, 11). Studies among patients with sepsis (47) and critical illness (48, 49) reported elevated osteopontin among patients with a fatal outcome although there is limited data on the role of osteopontin in patients with SCA. Osteopontin is upregulated in vasculature in the setting of ischemia, and may also be released from renal tubular cells during acute kidney injury which can influence acute lung injury in mice (50–52). With findings from our study that highlight the role of both FGF23 and osteopontin in predicting mortality in SCA, there is need for further research to understand the interplay between bone biomarkers and kidney function in predicting mortality in children during a VOC.
Our study had several strengths including the comprehensive evaluation of sMBD parameters (calcium, phosphate, PTH, vitamin D, FGF23) in a relatively large cohort of children with SCA alongside a comprehensive assessment of kidney function (23, 24, 53). While the study was underpowered to assess biomarkers of mortality as there were only six deaths in the study population, the data were consistent with bone biomarkers being elevated in children who died while experiencing an acute pain crisis requiring hospitalization. A limitation of the study was that bone biopsies and bone density assessments were not conducted due to resource constraints (15). Additional studies are needed to evaluate serum biochemical abnormalities in conjunction with biopsy findings and to delineate the dynamics of biochemical markers of sMBD in children over clinical recovery, as well as regarding long term bone changes in relation to child growth.
In summary, mineral bone abnormalities were common in children with SCA hospitalized with vaso-occlusion and were associated with the presence of kidney disease and increased with age. As children with SCA experience frequent VOC that can accelerate the development of chronic bone disease over childhood (53), routine evaluation of bone mineral disorders is warranted. Osteopontin is a biomarker of bone disease or of acute ischemic injury and is associated with sMBD and mortality and may represent a novel biomarker to assess bone health or predict mortality in SCA. Finally, this study presents critical insight into sMBD in children with SCA in Africa, where the majority of people living with SCA reside but for which there are limited data. Recommendations regarding the appropriate management of mineral bone disorders remain problematic because they differ based on location, resource availability, the context in which it occurs and is further complicated in pediatric populations with unique needs across development. Based on the early age of sMBD onset in this cohort, we recommend policies promoting earlier assessment of kidney and bone health in this vulnerable population and the development of context-specific guidelines to support evidence-based care.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
This study involved human participants and was reviewed and approved by the Institutional Review Board from Makerere University School of Biomedical Sciences Research and Ethics Committee (SBSREC) (first approval date 13th May 2018, IRB number; SBS-S46) and the Uganda National Council for Science and Technology (approval date 07th September 2018, approval number; HS 2443). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin and assent was obtained for children 8 years of age and older.
AB, SK, JL, RK: participated in the conceptualization and design of the study. ALC and AB: designed the laboratory assessments and AB: wrote the first draft of the manuscript. Data analysis was conducted by ALC and AB. All authors participated in data interpretation, editing and writing of the manuscript. All authors contributed to the article and approved the submitted version.
Research reported in this publication was supported by the Fogarty International Center (FIC) of the National Institutes of Health and the National Heart, Lung, And Blood Institute (NHLBI) under grant #D43TW009345 awarded to the Northern Pacific Global Health Fellows Program. This work was also supported by Grant Number D43TW010132 supported by Office of the Director, National Institutes of Health (OD), National Institute of Dental & Craniofacial Research (NIDCR), National Institute of Neurological Disorders and Stroke (NINDS), National Heart, Lung, and Blood Institute (NHLBI), Fogarty International Center (FIC), National Institute on Minority Health and Health Disparities (NIMHD). The study was also supported by Grant number 1R25TW011213 [Fogarty International Center of the National Institutes of Health, U.S. Department of State's Office of the U.S. Global AIDS Coordinator and Health Diplomacy (S/GAC), and President's Emergency Plan for AIDS Relief, PEPFAR]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We would like to thank the study participants and their families for their participation and the study team for their efforts as well as the CHILD laboratory team (Caroline Kazinga, Ivan Mufumba, Kristin Murphy, Geoffrey Situma, Ronald Wasswa) for conducting the biomarker testing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. (2010) 376(9757):2018–31. doi: 10.1016/S0140-6736(10)61029-X
2. Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. (2017) 376(16):1561–73. doi: 10.1056/NEJMra1510865
3. Almeida A, Roberts I. Bone involvement in sickle cell disease. Br J Haematol. (2005) 129(4):482–90. doi: 10.1111/j.1365-2141.2005.05476.x
4. Baxter-Jones AD, Faulkner RA, Forwood MR, Mirwald RL, Bailey DA. Bone mineral accrual from 8 to 30 years of age: an estimation of peak bone mass. J Bone Miner Res. (2011) 26(8):1729–39. doi: 10.1002/jbmr.412
5. Bailey DA, McKay HA, Mirwald RL, Crocker PR, Faulkner RA. A six-year longitudinal study of the relationship of physical activity to bone mineral accrual in growing children: the university of Saskatchewan bone mineral accrual study. J Bone Miner Res. (1999) 14(10):1672–9. doi: 10.1359/jbmr.1999.14.10.1672
6. Nih Consensus Development Panel on Osteoporosis Prevention D, Therapy. Osteoporosis prevention, diagnosis, and therapy. Jama. (2001) 285(6):785–95. doi: 10.1001/jama.285.6.785
7. Rovner AJ, Stallings VA, Kawchak DA, Schall JI, Ohene-Frempong K, Zemel BS. High risk of vitamin D deficiency in children with sickle cell disease. J Am Diet Assoc. (2008) 108(9):1512–6. doi: 10.1016/j.jada.2008.06.433
8. Oladipo OO, Temiye EO, Ezeaka VC, Obomanu P. Serum magnesium, phosphate and calcium in Nigerian children with sickle cell disease. West Afr J Med. (2005) 24(2):120–3. doi: 10.4314/wajm.v24i2.28180
9. Mohammed S, Addae S, Suleiman S, et al. Serum calcium, parathyroid hormone, and vitamin D status in children and young adults with sickle cell disease. Ann Clin Biochem. (1993) 30(Pt 1):45–51. doi: 10.1177/000456329303000108
10. Beck GR Jr, Zerler B, Moran E. Phosphate is a specific signal for induction of osteopontin gene expression. Proc Natl Acad Sci USA. (2000) 97(15):8352–7. doi: 10.1073/pnas.140021997
11. Mazzali M, Kipari T, Ophascharoensuk V, Wesson J, Johnson R, Hughes J. Osteopontin—a molecule for all seasons. QJM. (2002) 95(1):3–13. doi: 10.1093/qjmed/95.1.3
13. Batte A, Menon S, Ssenkusu JM, et al. Acute kidney injury in hospitalized children with sickle cell anemia. BMC Nephrol. (2022) 23(1):110. doi: 10.1186/s12882-022-02731-9
14. Yee MM, Jabbar SF, Osunkwo I, et al. Chronic kidney disease and albuminuria in children with sickle cell disease. Clin J Am Soc Nephrol. (2011) 6(11):2628–33. doi: 10.2215/CJN.01600211
15. Wheeler DC, Winkelmayer WC. KDIGO 2017 Clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD) foreword. Kidney Int Suppl (2011). (2017) 7(1):1–59. doi: 10.1016/j.kisu.2017.04.001
16. Hegde A, Denburg MR, Glenn DA. Acute kidney injury and pediatric bone health. Front Pediatr. (2020) 8:635628. doi: 10.3389/fped.2020.635628
17. Yang T, Wang W, Tang X, et al. Association between mineral and bone disorder in patients with acute kidney injury following cardiac surgery and adverse outcomes. BMC Nephrol. (2019) 20(1):1–9. doi: 10.1186/s12882-018-1181-1
18. Akrawinthawong K, Shaw MK, Kachner J, et al. Urine catalytic iron and neutrophil gelatinase-associated lipocalin as companion early markers of acute kidney injury after cardiac surgery: a prospective pilot study. Cardiorenal Med. (2013) 3(1):7–16. doi: 10.1159/000346815
20. Pottel H, Hoste L, Martens F. A simple height-independent equation for estimating glomerular filtration rate in children. Pediatr Nephrol. (2012) 27(6):973–9. doi: 10.1007/s00467-011-2081-9
21. Batte A, Starr MC, Schwaderer AL, et al. Methods to estimate baseline creatinine and define acute kidney injury in lean Ugandan children with severe malaria: a prospective cohort study. BMC Nephrol. (2020) 21(1):417. doi: 10.1186/s12882-020-02076-1
22. Nath KA, Hebbel RP. Sickle cell disease: renal manifestations and mechanisms. Nat Rev Nephrol. (2015) 11(3):161–71. doi: 10.1038/nrneph.2015.8
23. Martensson J, Martling CR, Bell M. Novel biomarkers of acute kidney injury and failure: clinical applicability. Br J Anaesth. (2012) 109(6):843–50. doi: 10.1093/bja/aes357
24. Greenberg JH, Parikh CR. Biomarkers for diagnosis and prognosis of AKI in children: one size does not fit all. Clin J Am Soc Nephrol. (2017) 12(9):1551–7. doi: 10.2215/CJN.12851216
25. Ostermann M, Zarbock A, Goldstein S, et al. Recommendations on acute kidney injury biomarkers from the acute disease quality initiative consensus conference: a consensus statement. JAMA Netw Open. (2020) 3(10):e2019209. doi: 10.1001/jamanetworkopen.2020.19209
26. Stanski N, Menon S, Goldstein SL, Basu RK. Integration of urinary neutrophil gelatinase-associated lipocalin with serum creatinine delineates acute kidney injury phenotypes in critically ill children. J Crit Care. (2019) 53:1–7. doi: 10.1016/j.jcrc.2019.05.017
27. Pierce CB, Muñoz A, Ng DK, Warady BA, Furth SL, Schwartz GJ. Age- and sex-dependent clinical equations to estimate glomerular filtration rates in children and young adults with chronic kidney disease. Kidney Int. (2021) 99(4):948–56. doi: 10.1016/j.kint.2020.10.047
28. KDIGO. KDIGO clinical practice guideline for acute kidney injury. Kidney Int. (2012) 2(1):124–38. doi: 10.1038/kisup.2012.1
29. Pagana KD, Pagana TJ, Pagana TN. Mosby's diagnostic and laboratory tests. Fifteenth edn. Elsevier (2021).
30. Colantonio DA, Kyriakopoulou L, Chan MK, et al. Closing the gaps in pediatric laboratory reference intervals: a CALIPER database of 40 biochemical markers in a healthy and multiethnic population of children. Clin Chem. (2012) 58(5):854–68. doi: 10.1373/clinchem.2011.177741
31. KDIGO. KDIGO 2012 Clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int (Supplement). (2013) 3(1):1–150. doi: 10.1038/kisup.2012.73
32. Munns CF, Shaw N, Kiely M, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. (2016) 101(2):394–415. doi: 10.1210/jc.2015-2175
33. Wesseling-Perry K, Salusky IB. Chronic kidney disease: mineral and bone disorder in children. Elsevier. (2013):169–79.
34. Audard V, Moutereau S, Vandemelebrouck G, et al. First evidence of subclinical renal tubular injury during sickle-cell crisis. Orphanet J Rare Dis. (2014) 9(1):67. doi: 10.1186/1750-1172-9-67
35. Mogire RM, Morovat A, Muriuki JM, et al. Prevalence and predictors of vitamin D deficiency in young African children. BMC Med. (2021) 19(1):115. doi: 10.1186/s12916-021-01985-8
36. Etzion Z, Tiffert T, Bookchin RM, Lew VL. Effects of deoxygenation on active and passive Ca2 + transport and on the cytoplasmic Ca2+levels of sickle cell anemia red cells. J Clin Invest. (1993) 92(5):2489–98. doi: 10.1172/JCI116857
37. Goyal R, Jialal I. Hyperphosphatemia. In: editors. Statpearls. Treasure island (FL): StatPearls Publishing (2022).
38. Raj VM, Freundlich M, Hamideh D, et al. Abnormalities in renal tubular phosphate handling in children with sickle cell disease. Pediatr Blood Cancer. (2014) 61(12):2267–70. doi: 10.1002/pbc.25188
39. Sharpe CC, Thein SL. Sickle cell nephropathy—a practical approach. Br J Haematol. (2011) 155(3):287–97. doi: 10.1111/j.1365-2141.2011.08853.x
40. Krishnamoorthy P, Alyaarubi S, Abish S, Gale M, Albuquerque P, Jabado N. Primary hyperparathyroidism mimicking vaso-occlusive crises in sickle cell disease. Pediatrics. (2006) 118(2):e537–9. doi: 10.1542/peds.2006-0337
41. Denoix E, Bomahou C, Clavier L, et al. Primary hyperparathyroidism in sickle cell disease: an unknown complication of the disease in adulthood. J Clin Med. (2020) 9(2):308. doi: 10.3390/jcm9020308
42. Leaf DE, Christov M, Juppner H, et al. Fibroblast growth factor 23 levels are elevated and associated with severe acute kidney injury and death following cardiac surgery. Kidney Int. (2016) 89(4):939–48. doi: 10.1016/j.kint.2015.12.035
43. Rygasiewicz K, Hryszko T, Siemiatkowski A, Brzosko S, Rydzewska-Rosolowska A, Naumnik B. C-terminal and intact FGF23 in critical illness and their associations with acute kidney injury and in-hospital mortality. Cytokine. (2018) 103:15–9. doi: 10.1016/j.cyto.2017.12.024
44. Leaf DE, Jacob KA, Srivastava A, et al. Fibroblast growth factor 23 levels associate with AKI and death in critical illness. J Am Soc Nephrol. (2017) 28(6):1877–85. doi: 10.1681/ASN.2016080836
45. Isakova T, Xie H, Yang W, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. Jama. (2011) 305(23):2432–9. doi: 10.1001/jama.2011.826
46. Liu C-H, Laja O, Mendelsohn L, et al. Plasma FGF23 is a biomarker for left ventricular hypertrophy and mortality in adults with sickle cell anemia chronic kidney disease. Blood. (2015) 126(23):2171. doi: 10.1182/blood.V126.23.2171.2171
47. Carbone F, Bonaventura A, Vecchie A, et al. Early osteopontin levels predict mortality in patients with septic shock. Eur J Intern Med. (2020) 78:113–20. doi: 10.1016/j.ejim.2020.04.035
48. Roderburg C, Benz F, Cardenas DV, et al. Persistently elevated osteopontin serum levels predict mortality in critically ill patients. Crit Care. (2015) 19(1):271. doi: 10.1186/s13054-015-0988-4
49. Lorenzen JM, Hafer C, Faulhaber-Walter R, et al. Osteopontin predicts survival in critically ill patients with acute kidney injury. Nephrol Dial Transplant. (2011) 26(2):531–7. doi: 10.1093/ndt/gfq498
50. Cen C, Aziz M, Yang WL, Nicastro JM, Coppa GF, Wang P. Osteopontin blockade attenuates renal injury after ischemia reperfusion by inhibiting NK cell infiltration. Shock. (2017) 47(1):52–60. doi: 10.1097/SHK.0000000000000721
51. Lok ZSY, Lyle AN. Osteopontin in vascular disease: friend or foe? Arterioscler, Thromb, Vasc Biol. (2019) 39(4):613–22. doi: 10.1161/ATVBAHA.118.311577
52. Khamissi FZ, Ning L, Kefaloyianni E, et al. Identification of kidney injury–released circulating osteopontin as causal agent of respiratory failure. Sci Adv. (2022) 8(8):eabm5900. doi: 10.1126/sciadv.abm5900
Keywords: mineral bone disease, acute kidney injury, sickle cell anemia (SCA), mortality, pediatrics, acute kidney disease (AKD)
Citation: Batte A, Kasirye P, Baluku R, Kiguli S, Kalyesubula R, John CC, Schwaderer AL, Imel EA and Conroy AL (2023) Mineral bone disorders and kidney disease in hospitalized children with sickle cell anemia. Front. Pediatr. 10:1078853. doi: 10.3389/fped.2022.1078853
Received: 24 October 2022; Accepted: 19 December 2022;
Published: 2 February 2023.
Edited by:
Madhusmita Misra, Harvard Medical School, United StatesReviewed by:
Mark Hanudel, University of California, United States© 2023 Batte, Kasiyre, Baluku, Kiguli, Kalyesubula, John, Schwaderer, Imel and Conroy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea L. Conroy Y29ucm95YUBpdS5lZHU=
†ORCID Anthony Batte orcid.org/0000-0001-7463-5277 Sarah Kiguli orcid.org/0000-0002-8764-4161 Robert Kalyesubula orcid.org/0000-0003-3211-163X Chandy C. John orcid.org/0000-0002-1634-1411 Andrea L. Conroy orcid.org/0000-0002-5328-6511
Specialty Section: This article was submitted to Pediatric Endocrinology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.