
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 23 December 2022
Sec. Pediatric Neurology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1073083
 Se Hee Kim1,†
Se Hee Kim1,† Soon Sung Kwon2,†
Soon Sung Kwon2,† Joon Soo Lee1
Joon Soo Lee1 Heung Dong Kim1
Heung Dong Kim1 Seung-Tae Lee2
Seung-Tae Lee2 Jong Rak Choi2
Jong Rak Choi2 Saeam Shin2*
Saeam Shin2* Hoon-Chul Kang1*
Hoon-Chul Kang1*
Background: Trio test has been widely used for diagnosis of various hereditary disorders. We aimed to investigate the contribution of trio test in genetically diagnosing neurodevelopmental disorders (NDD).
Methods: We retrospectively reviewed 2,059 NDD cases with genetic test results. The trio test was conducted in 563 cases. Clinical usefulness, optimal timing, and methods for the trio test were reviewed.
Results: Pathogenic or likely pathogenic variants were detected in 112 of 563 (19.9%) patients who underwent the trio test. With trio test results, the overall diagnostic yield increased by 5.4% (112/2,059). Of 165 de novo variants detected, 149 were pathogenic and we detected 85 novel pathogenic variants. Pathogenic, de novo variants were frequently detected in CDKL5, ATP1A3, and STXBP1.
Conclusion: The trio test is an efficient method for genetically diagnosing NDD. We identified specific situations where a certain trio test is more appropriate, thereby providing a guide for clinicians when confronted with variants of unknown significance of specific genes.
Neurodevelopmental disorders (NDD) are a group of disorders caused by abnormal brain development, which result in impaired brain functions. NDD include a heterogeneous group of diseases such as epilepsy, intellectual disability, autism, developmental delay, and other various neuropsychiatric diseases (1, 2). These disorders may cause serious life-long functional deficits. In the United States, a nationwide surveillance during 2014–2016 showed the following prevalence of NDD: 1.1%–1.3% for intellectual disability, 2.2%–2.7% for autism spectrum disorder, and 3.6%–4.6% for developmental delay (3). NDD may be caused by multiple factors affecting normal brain development, such as genetics, environment, toxins, infections, and injuries. Identifying the underlying genetic causes of NDD in patients is important for proper patient management, genetic counseling, and prediction of prognosis, as well as for understanding the pathophysiology of NDD.
To date, a large number of genes involved in the occurrence of NDD have been identified. However, there are still several patients with NDD for whom the causative variants have not yet been identified and only variants of unknown significance (VOUS) have been detected from several known or probable NDD-related genes. These variants cannot be determined as either pathogenic or benign owing to the lack of evidence. In practice, the easiest way of accessing the pathogenicity of detected variants is through a parental test with a proband test, also known as the trio test. When a certain variant is identified as a de novo variant, it provides powerful evidence that supports the pathogenicity of the variant.
In this study, we reviewed results of trio tests for suspected variants and identified NDD-causing pathogenic variants. The results of the trio tests of 563 patients with NDD are described, in which we detected 149 pathogenic de novo variants with several novel variants of various NDD-related genes. The results suggest the clinical usefulness of the trio test for the diagnosis of NDD with genetic causes. We also investigated whether there are differences in diagnostic yield according to the timing of trio test and trio test methods.
A total of 2,059 patients were referred for genetic diagnosis of various NDD at a tertiary hospital in Seoul, Korea, from 2016 to 2021. The clinical history and genetic test results of the patients were retrospectively reviewed for this study. Among them, individuals with trio test results were further investigated. A trio test is usually ordered by the primary care physician after a proband study if candidate genes or VOUS were found. However, sometimes a trio test is performed earlier for some patients if the family wanted to receive quick test result for reasons such as genetic counseling. Sometimes, the physician would not offer a trio test if no good gene candidate was identified in the first test report. In some cases, the parents refused to take the offered trio test because of the cost.
The types of trio tests included NGS-based trio test and Sanger sequencing (for each variant detected). The cases were discussed at a multidisciplinary meeting held with neurologists, geneticists, and bioinformaticians. Additional evaluations or the type of trio test were usually determined after the meeting.
Clinical variables, such as age, sex, and diagnosis, were collected from the electronic medical record review.
This study was conducted following the standards of the Declaration of Helsinki and was approved by the Institutional Review Board (IRB) of the Yonsei College of Medicine (4-2022-0327). The IRB also approved a waiver of informed consent because all patients information were de-identified and this study was conducted as a retrospective study.
Genomic DNA was extracted from the peripheral blood of patients and their parents. For an NGS-based trio test, libraries were prepared, and targets were captured using various custom panels for candidate genes (gene panels and genes included in each panel are listed in Supplementary Table S1). The prepared and pooled libraries were sequenced using NextSeq 550Dx System (Illumina, San Diego, CA, United States). The sequencing data were analyzed using the custom bioinformatics pipeline as previously described (4, 5). For the test, the parents underwent the same NGS panel testing that the patient received.
For a Sanger sequencing trio test, gene regions that included the target variants were amplified by target-specific primers and further sequenced using a 3,730 DNA Analyzer with BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA).
Variants were classified based on the recommendation of the American College of Medical Genetics and Genomics (6). Among the variants identified from the bioinformatics analyses, the benign and likely benign variants were sorted based on the location of the variant and population frequency reported from the 1,000 Genomes project, Genome Aggregation Database, Exome Sequencing Project, and Korean Reference Genome Database. Variants that were not classified as benign or likely benign were further reviewed by geneticists to determine the pathogenicity of the variant. Literature and database reviews and in silico analyses were performed by geneticists. Genotype–phenotype correlation of the variants was additionally performed by pediatric neurologists and geneticists. The interpretations of the variants were reviewed and reclassified based on the result of the trio test.
The time for the trio test was presented as median, minimum, maximum, and interquartile ranges (IQR). Descriptive statistics were calculated using Microsoft Excel (Microsoft, Redmond, WA, USA).
During 2016–2021, a total of 2,059 patients with NDD were requested for an NGS test to determine the causal pathogenic variants (Figure 1). In this study, 1,212 patients underwent clinical exome, 755 patients underwent epilepsy-targeted gene panel, and 92 patients underwent malformation of cortical development-targeted gene panel testing. Of the 2,059 patients, 1,184 were male (57.5%), and the median age was 54.8 months (0–794.8).
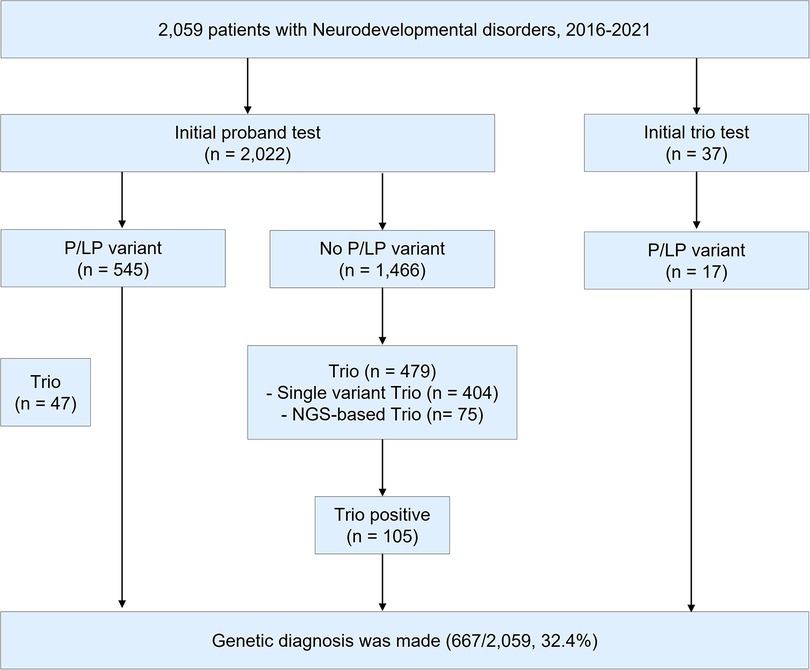
Figure 1. Study design. During 2016–2021, a total of 2,059 patients were tested for the diagnosis of neurodevelopmental disorders (NDD). P/LP variant; pathogenic/likely pathogenic variant. NGS; next-generation sequencing.
Of the 2,059 patients, trio test was performed for 563 patients to improve interpretation. Of the 563 patients, 332 were male (59.0%), and the median age was 44.4 months (0.3–321.6). A total of 410 out of the 563 patients (72.8%) were patients with NDD with epilepsy. Clinical exome was the most frequently used for diagnosis (342/563, 60.7%), followed by an epilepsy panel (206/563, 36.6%) and malformation of cortical development panel (15/563, 2.7%). A single gene trio test was performed using Sanger sequencing in 442 cases (78.5%), while an NGS-based trio was performed in 121 cases (21.5%). Detailed demographics of the patients are presented in Supplementary Table S2.
The overall diagnostic yield of NGS for NDD was 32.4% (667/2,059). Among these, 112 cases were confirmed to have pathogenic or likely pathogenic variants by trio test, which improved the diagnostic yield by 5.4% (112/2,059). The median time for the trio test performed from the initial test report to the final report was 2.1 months (0–48.9, IQR: 1.3–3.5). The diagnostic yield, number of cases with trio tests, number of cases solved by trio tests, and de novo variants of each targeted gene panel are presented in Table 1. Among those 563 patients with trio test, trio test was conducted at the initial molecular diagnosis in 37 patients (Supplementary Table S3).
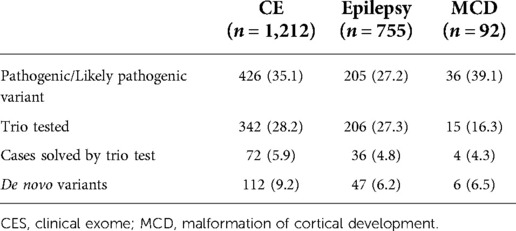
Table 1. Statistics of targeted gene panels.
The trio test detected 165 de novo variants from 159 patients, of which 149 variants proved to be pathogenic or likely pathogenic. Missense variants were the most frequent type (99/149, 66.4%), followed by nonsense (11/149, 7.4%), splicing site (10/149, 6.7%), gene or chromosomal deletion (10/149, 6.7%), duplication (5/149, 3.4%), and indel (4/149, 2.7%) variants. Among the detected de novo variants, 85 variants were novel, which were listed in Table 2. The novel variants were frequently identified from the following genes: STXBP1 (7 cases), SCN2A (7 cases), CDKL5 (4 cases), GABRA1 (3 cases), DNM1 (3 cases), and SCN1A (3 cases).
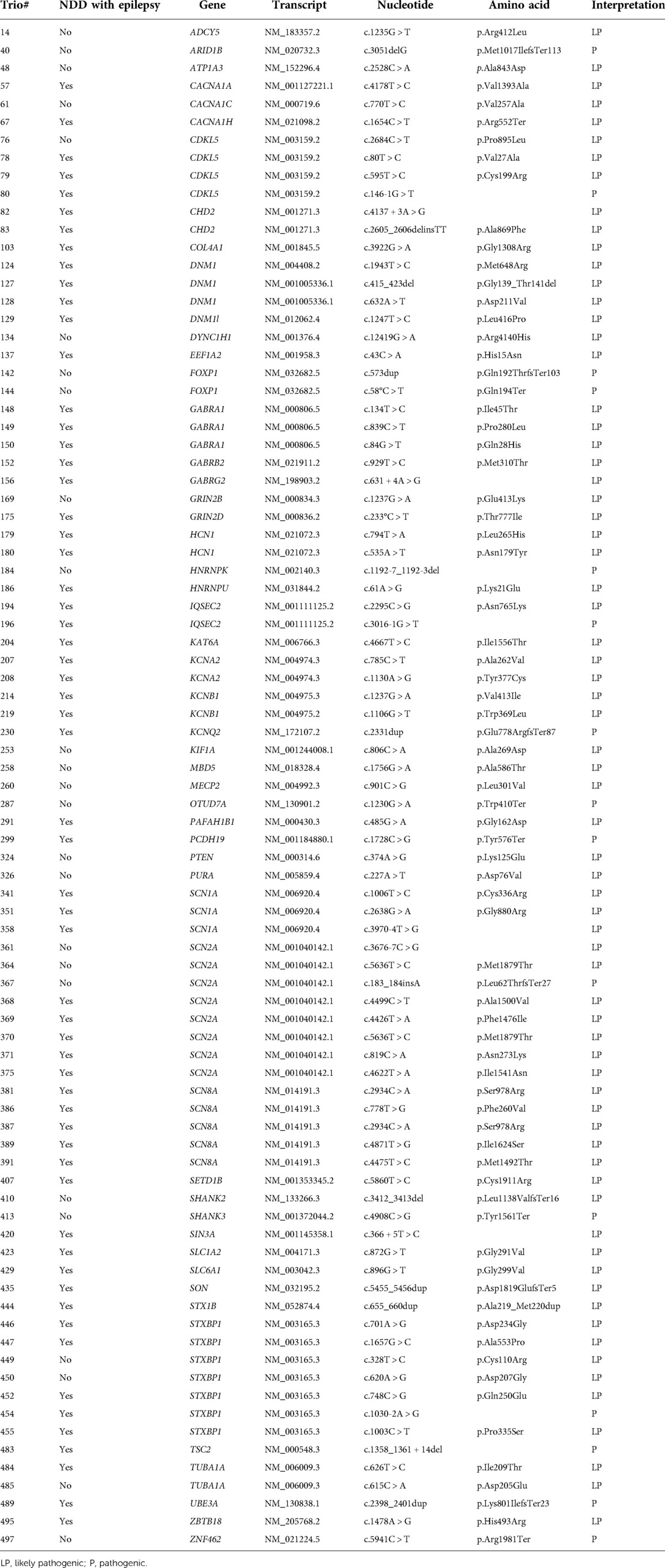
Table 2. Novel de novo pathogenic/likely pathogenic variants detected in patients with NDD.
A total of 16 de novo variants remained as VOUS after the trio test (Supplementary Table S4). Most of these variants were not reported in the general population and related patients, and the phenotypic correlations were weak. The variants could not be classified as pathogenic or likely pathogenic although they were confirmed to occur de novo.
The target variants and genes requested for trio testing are listed in Table 3. The most frequently requested gene was SCN1A, followed by SCN2A, ADGRV1, SCN8A, TSC2, and CHD2. Notably, all the variants of three genes—CDKL5 (6/6, 100%), ATP1A3 (3/3, 100%), and STXBP1 (9/11, 81.8%)—were confirmed as de novo and causative. By contrast, the variants of the following genes were rarely confirmed to be de novo: ADGRV1 (1/14), SCN9A (0/11) and SETBP1 (0/10). For 37 genes, no de novo variants were detected.
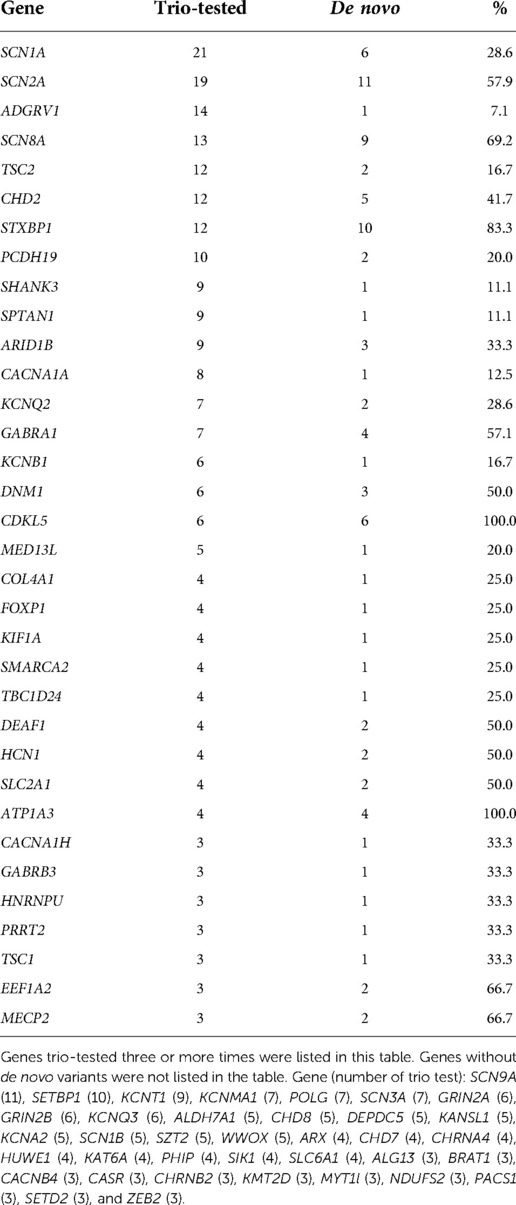
Table 3. Trio-tested genes and the number of de novo variants detected.
Recent progress in NGS tests has improved the cost-effectiveness of the NGS-based trio tests, thereby encouraging its use. NGS-based trio tests can determine novel candidate genes and variants and find causal pathogenic variants, which can be overlooked during variant interpretation. Exome-trio has recently been widely used, and studies have reported its clinical usefulness for the diagnosis of NDD (7–9). In these studies, various number of patients with NDD were trio-tested (54–244 patients). Diagnostic yields ranged from 25.2% to 57.4%. In this study, the trio test detected pathogenic causal variants in 22.3% (112/503) of the cases, which might not have been interpreted as pathogenic without the trio test. This resulted in an increase in the overall diagnostic yield of NDD from 27.0% to 32.4%, which encouraged the use of trio tests for the genetic diagnosis of NDD. A trio test is a powerful tool that offers a high diagnostic yield, while proband-only clinical exome sequencing provides a cost-effective approach to NDD (5).
De novo variants rarely occur in an individual, and the occurrence of de novo variants in the coding region of a certain gene is even rarer (10, 11). For this reason, a de novo variant in a patient with a family history consistent with de novo inheritance is highly suspected as the causal pathogenic variant for NDD. Guidelines for the interpretation of sequence variants regarded de novo occurrence of a variant as strong evidence for pathogenicity (6). The results also supported the high correlation between de novo occurrence of the variants and their pathogenicity, with 86.1% of the trio-tested pathogenic variants (149/173) occurring de novo. This high rate of de novo variants was consistent with previous studies, which reported the rate of de novo variants 80% and 61.5%, respectively (8, 9).
To have a high detection yield with a single gene trio, variants to be trio-tested need to be selected carefully. First, the genotype-phenotype correlation between the gene and patient should exist. Second, variants that are not detected in a normal healthy population were selected. Third, the previous test results were reviewed and checked if the target gene has been frequently reported with de novo variants. These genes included CDKL5, ATP1A3, and STXBP1. These genes were already known for their high frequency of de novo variants (12–17). On the contrary, there are situations where the trio test is less likely to be helpful when genes with low rates of de novo variants are detected. There are genes in which pathogenic variants were rarely found. In this study, de novo variants were rarely detected in ADGRV1, SCN9A, SETBP1, and KCNT1. Non-pathogenic, inherited VOUS are frequently reported in these genes, probably owing to their relatively large gene size. Calculated scoring systems for the prediction of pathogenicity, such as the Z-score and MutScore, can aid in determining the need for trio testing (18, 19).
In this study, most of the trio tests were performed after the initial NGS test results were determined negative. Only 37 cases underwent an initial trio. For the cases with initial trio, the diagnostic yield was high, reaching approximately 50% (17/37), and the diagnosis could be made immediately. By contrast, a retrospective review of these cases also demonstrated that some of those variants (10/17, 58.8%) could have been classified as pathogenic without the trio test. In addition, there was no difference in the diagnostic rate of cases solved by the trio test between the initial trio and delayed trio tests (18.9% vs. 20.0%). The optimal timing of the trio test has not yet been established. The clinical setting, phenotypes, socio-economic status of parents, and experience of the clinicians would have to be considered when pediatric neurologists and geneticists decide when to perform the trio test.
Trio test identified cases with parental mosaicism. Parental mosaicism of SLC2A1 has been reported (20, 21), and the level of mosaicism appears to be related to symptoms, which is consistent with the results of this study. Parental mosaicisms of SPTAN1 and RORA have not been reported yet to the best of our knowledge. Parental pathogenic mosaic variants should be addressed considering that they increase the risk of disease recurrence in future offsprings (22, 23). A recent study reported the contribution of low-level parental mosaic disease-causing variants to NDD (24). In cases of parental testing conducted by Sanger sequencing or low-depth NGS tests, low-level parental mosaicisms may be missed during diagnosis. A sensitive NGS-based trio test along with proper noting of family history can identify such cases.
In this study, we reported 16 de novo VOUS. In these cases, variants could not be classified as pathogenic owing to the lack of evidence. For example, a frameshift variant of RBM12 was detected in a patient with epilepsy and mildly delayed development. The correlation between pediatric NDD and RBM12 has not been reported yet, which led to this variant being classified as VOUS. However, one report demonstrated that a truncating variant of RBM12 is associated with psychosis (25), which remains to be a possible relation between the RBM12 variant and the phenotype of the patient. A missense de novo variant of CUX1 was detected in a patient with epilepsy, autism, and intellectual disability. Although the de novo variant of CUX1 had been reported in patients with delayed development (26), all the reported cases were of truncating variants. Thus, the variant was reported as VOUS. There were de novo variants that lacked phenotypic consistency; a splicing site variant of ATP8A1 was detected in a patient with autism spectrum disorder and delayed development who did not have any symptoms and signs related to cholestasis. A splicing site variant of COPA, a gene-related to autoimmune diseases (27), was identified as a de novo occurrence in a patient with delayed development and epilepsy without other abnormalities. Pathogenic variants of DNM2 were known to cause myopathy and Charcot–Marie–Tooth disease (28, 29). Additionally, a frameshift variant occurred de novo in a patient with epilepsy who did not demonstrate the phenotype related to DNM2-related diseases. Detected de novo variants of other genes (CTC1, TDRD7, RANBP17, RABL6, DLX6, LDHA, VPS13A, LAMA3, GAL3ST2, and GATA3) were classified as VOUS, considering these genes lack disorder-related reports. Although these kinds of variants should be interpreted with caution, they may be novel variants of NDD, which may be identified as causal variants in future re-analyses.
Advances in the genetic diagnostic methods for NDD have now led to the identification of numerous variants, which requires development of methods to interpret their clinical meaning. This study demonstrated the results of the trio test with several novel pathogenic variants causing NDD. The data demonstrated that the trio test is a powerful tool to determine causal pathogenic variants among the variants from patients with NDD. The study also describes specific situations where the trio test is more or less useful, which can provide clinicians with a guide when confronted with VOUS of specific genes.
The data presented in the study are deposited in the ClinVar repository, accession number SCV002761259 to SCV002761341.
This study was conducted following the standards of the Declaration of Helsinki and was approved by the Institutional Review Board (IRB) of the Yonsei College of Medicine (4- 2022-0327). The IRB also approved a waiver of informed consent because all patients information were de-identified and this study was conducted as a retrospective study.
Conceptualization: SHK, SSK, SS, H-CK. Data curation: SH-K, SS-K, JL, HK, S-TL, JC. Funding acquisition: H-CK. Investigation: SH-K, SS-K. Methodology: SH-K, SS-K, SS, H-CK. Project administration: ST-L, JC, SS, H-CK. Supervision: SS, H-CK. Writing—original draft: SH-K, SS-K. Writing—review and editing: SH-K, SS-K, JS-L, HK, ST-L, JR-C, S-S, HC-K. All authors contributed to the article and approved the submitted version.
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2022R1A2C1012522), a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HI21C1659) and the Team Science Award of Yonsei University College of Medicine (6-2021-0007).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.1073083/full#supplementary-material.
1. Cardoso AR, Lopes-Marques M, Silva RM, Serrano C, Amorim A, Prata MJ, et al. Essential genetic findings in neurodevelopmental disorders. Hum Genomics. (2019) 13(1):31. doi: 10.1186/s40246-019-0216-4
2. Parenti I, Rabaneda LG, Schoen H, Novarino G. Neurodevelopmental disorders: from genetics to functional pathways. Trends Neurosci. (2020) 43(8):608–21. doi: 10.1016/j.tins.2020.05.004
3. Zablotsky B, Black LI, Blumberg SJ. Estimated prevalence of children with diagnosed developmental disabilities in the United States, 2014–2016. NCHS Data Brief, no 291. Hyattsville, MD: National Center for Health Statistics. 2017.
4. Rim JH, Kim SH, Hwang IS, Kwon SS, Kim J, Kim HW, et al. Efficient strategy for the molecular diagnosis of intractable early-onset epilepsy using targeted gene sequencing. BMC Med Genomics. (2018) 11(1):6. doi: 10.1186/s12920-018-0320-7
5. Kim SH, Kim B, Lee JS, Kim HD, Choi JR, Lee ST, et al. Proband-only clinical exome sequencing for neurodevelopmental disabilities. Pediatr Neurol. (2019) 99:47–54. doi: 10.1016/j.pediatrneurol.2019.02.017
6. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
7. Gao C, Wang X, Mei S, Li D, Duan J, Zhang P, et al. Diagnostic yields of trio-WES accompanied by CNVseq for rare neurodevelopmental disorders. Front Genet. (2019) 10:485. doi: 10.3389/fgene.2019.00485
8. Brunet T, Jech R, Brugger M, Kovacs R, Alhaddad B, Leszinski G, et al. De novo variants in neurodevelopmental disorders-experiences from a tertiary care center. Clin Genet. (2021) 100(1):14–28. doi: 10.1111/cge.13946
9. Brea-Fernandez AJ, Alvarez-Barona M, Amigo J, Tubio-Fungueirino M, Caamano P, Fernandez-Prieto M, et al. Trio-based exome sequencing reveals a high rate of the de novo variants in intellectual disability. Eur J Hum Genet. (2022) 30(8):938–45. doi: 10.1038/s41431-022-01087-w
10. Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. (2016) 17(1):241. doi: 10.1186/s13059-016-1110-1
11. Goldmann JM, Wong WS, Pinelli M, Farrah T, Bodian D, Stittrich AB, et al. Parent-of-origin-specific signatures of de novo mutations. Nat Genet. (2016) 48(8):935–9. doi: 10.1038/ng.3597
12. Hamdan FF, Piton A, Gauthier J, Lortie A, Dubeau F, Dobrzeniecka S, et al. De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann Neurol. (2009) 65(6):748–53. doi: 10.1002/ana.21625
13. Heinzen EL, Swoboda KJ, Hitomi Y, Gurrieri F, Nicole S, de Vries B, et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet. (2012) 44(9):1030–4. doi: 10.1038/ng.2358
14. Rosewich H, Thiele H, Ohlenbusch A, Maschke U, Altmuller J, Frommolt P, et al. Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene-identification study. Lancet Neurol. (2012) 11(9):764–73. doi: 10.1016/S1474-4422(12)70182-5
15. Zhao Y, Zhang X, Bao X, Zhang Q, Zhang J, Cao G, et al. Clinical features and gene mutational spectrum of CDKL5-related diseases in a cohort of Chinese patients. BMC Med Genet. (2014) 15:24. doi: 10.1186/1471-2350-15-24
16. Di Meglio C, Lesca G, Villeneuve N, Lacoste C, Abidi A, Cacciagli P, et al. Epileptic patients with de novo STXBP1 mutations: key clinical features based on 24 cases. Epilepsia. (2015) 56(12):1931–40. doi: 10.1111/epi.13214
17. Yamamoto T, Shimojima K, Kimura N, Mogami Y, Usui D, Takayama R, et al. Recurrent occurrences of CDKL5 mutations in patients with epileptic encephalopathy. Hum Genome Var. (2015) 2:15042. doi: 10.1038/hgv.2015.42
18. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536(7616):285–91. doi: 10.1038/nature19057
19. Quinodoz M, Peter VG, Cisarova K, Royer-Bertrand B, Stenson PD, Cooper DN, et al. Analysis of missense variants in the human genome reveals widespread gene-specific clustering and improves prediction of pathogenicity. Am J Hum Genet. (2022) 109(3):457–70. doi: 10.1016/j.ajhg.2022.01.006
20. Wang D, Ho YY, Pascual J, Hinton VJ, Yang H, Anolik M, et al. GLUT1 deficiency syndrome: R333W genotype and paternal mosaicism. Ann Neurol. (2001) 50:S124.
21. Takahashi S, Matsufuji M, Yonee C, Tsuru H, Sano N, Oguni H. Somatic mosaicism for a SLC2A1 mutation: implications for genetic counseling for GLUT1 deficiency syndrome. Clin Genet. (2017) 91(6):932–3. doi: 10.1111/cge.12902
22. Wright CF, Prigmore E, Rajan D, Handsaker J, McRae J, Kaplanis J, et al. Clinically-relevant postzygotic mosaicism in parents and children with developmental disorders in trio exome sequencing data. Nat Commun. (2019) 10(1):2985. doi: 10.1038/s41467-019-11059-2
23. Domogala DD, Gambin T, Zemet R, Wu CW, Schulze KV, Yang Y, et al. Detection of low-level parental somatic mosaicism for clinically relevant SNVs and indels identified in a large exome sequencing dataset. Hum Genomics. (2021) 15(1):72. doi: 10.1186/s40246-021-00369-6
24. Shu L, Zhang Q, Tian Q, Yang S, Peng X, Mao X, et al. Parental mosaicism in de novo neurodevelopmental diseases. Am J Med Genet A. (2021) 185(7):2119–25. doi: 10.1002/ajmg.a.62174
25. Steinberg S, Gudmundsdottir S, Sveinbjornsson G, Suvisaari J, Paunio T, Torniainen-Holm M, et al. Truncating mutations in RBM12 are associated with psychosis. Nat Genet. (2017) 49(8):1251–4. doi: 10.1038/ng.3894
26. Platzer K, Cogne B, Hague J, Marcelis CL, Mitter D, Oberndorff K, et al. Haploinsufficiency of CUX1 causes nonsyndromic global developmental delay with possible catch-up development. Ann Neurol. (2018) 84(2):200–7. doi: 10.1002/ana.25278
27. Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, et al. COPA mutations impair ER-golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. (2015) 47(6):654–60. doi: 10.1038/ng.3279
28. Bitoun M, Bevilacqua JA, Prudhon B, Maugenre S, Taratuto AL, Monges S, et al. Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann Neurol. (2007) 62(6):666–70. doi: 10.1002/ana.21235
Keywords: neurodevelopmental disorder, genetic diagnosis, next-generation sequencing, trio test, de novo variant
Citation: Kim SH, Kwon SS, Lee JS, Kim HD, Lee S, Choi JR, Shin S and Kang H (2022) Analysis of trio test in neurodevelopmental disorders. Front. Pediatr. 10:1073083. doi: 10.3389/fped.2022.1073083
Received: 18 October 2022; Accepted: 21 November 2022;
Published: 23 December 2022.
Edited by:
Pasquale Striano, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Aldamaria Puliti, University of Genoa, Italy© 2022 Kim, Kwon, Lee, Kim, Lee, Choi, Shin and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saeam Shin c2FlYW0wMzA0QHl1aHMuYWM= Hoon-Chul Kang aGlwbzAyMDdAeXVocy5hYw==
†These authors have contributed equally to this work
Specialty Section: This article was submitted to Pediatric Neurology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.