 Zhengrong Wang1,2
Zhengrong Wang1,2 Yuqing Xu
Yuqing Xu Yixi Sun
Yixi Sun Minyue Dong
Minyue Dong
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 10 January 2023
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1062900
Glanzmann thrombasthenia (GT) is a rare inherited disease characterized by mucocutaneous bleeding due to the abnormalities in quantity or quality of platelet membrane GP IIb (CD41) or GP IIIa (CD61). GP IIb and GP IIIa are encoded by the ITGA2B and ITGB3 genes, respectively. Herein, we described a 7-year-old Chinese boy of the consanguineous couple who was diagnosed with GT based on the typical clinical manifestations, absence of blood clot retraction and the reduced expression of CD41 and CD61 in platelets. A homozygous silent variant c.1431C > T (p. G477=) of the ITGB3 gene was identified by the Whole-exome sequencing and confirmed by Sanger sequencing. The variant was predicted to affect the splicing. RT-PCR and sequencing revealed that the variant caused a deletion of 95 base pairs and frameshift, and subsequently created a premature stop codon in exon 10 of ITGB3 (p. G477Afs*30). It was indicated that the variant c.1431C > T (p. G477=) of ITGB3 was the cause for Glanzmann thrombasthenia. Our findings expanded the mutation spectrum and provided the information for the genetic counseling, prenatal diagnosis and preimplantation genetic testing (PGT).
Glanzmann thrombasthenia (GT) is an autosomal recessive disorder characterized by mucocutaneous bleeding symptoms due to platelet defects. Ecchymosis, haematomas, petechiae, nose and gum bleeding and menorrhagia are the most common clinical manifestations (1, 2). According to previous publications, the prevalence of GT is estimated to be approximately 1 in million, with increased rates in high-consanguinity areas (3, 4). The main etiology of the disease is due to the qualitative or quantitative reduction of platelet membrane GP IIb/IIIa (integrin αIIbβ3). αIIbβ3 is a common dimeric complex, which promotes platelet adhesion, platelet aggregation, and is involved in hemostasis (5). GP IIb (CD41) and GP IIIa (CD61) are encoded by the ITGA2B and ITGB3 genes, respectively (6). Mutations in the ITGB3 or ITGA2B genes damage the synthesis of GPIIb/IIIa, inhibit platelet aggregation, and subsequently lead to the development of hereditary platelet incompetence (7, 8).
In the current investigation, we described a 7-year-old Chinese boy with GT caused by a homozygous silent mutation of the ITGB3 gene. The mutation c.1431C > T (p. G477=) was identified by Whole-exome sequencing (WES) and validated by Sanger sequencing. Moreover, RT-PCR and sequencing demonstrated that the mutation created a premature stop codon and the truncation of the ITGB3.
The consanguineous couple came to the Department of Reproductive Genetics, Women's Hospital, School of Medicine, Zhejiang University for genetic counseling because they had two children with generalized mucocutaneous bleeding. The first child of the healthy couple was a female who suffered from generalized mucocutaneous bleeding and died when she was 5 years old. The couple provided no detailed medical documents of the first child. The second child was a healthy female. The third child was a male (proband) at 7 years of age at the time of genetic counseling. He presented with generalized scattered skin, petechiae, and spots on his face after his birth. A blood clot retraction test revealed that the clots did not shrink over 24 h. Furthermore, the expression levels of CD41 and CD61 were significantly reduced as compared with control while the expression of CD42 was comparable with control (Supplementary Figure S1).
The use of medical records of this family was approved by the Institutional Review Board of the Women’s Hospital, School of Medicine, Zhejiang University and the participants provided their written informed consents.
Genomic DNA was extracted by a QIAamp DNA blood mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions and then was fragmented by Covaris LE220 (Massachusetts, USA) to generate a paired-end library (200–250 bp). All amplified libraries were performed on the BGISEQ-500 platform (BGI, Shenzhen, China), the single-strand DNA was mixed with MGIEasy™ DNA Library Prep Kit V1 (BGI, Shenzhen, China) and then sequenced using 100SR chemistry with BGISEQ-500RS high-throughput sequencing Kit (BGI, Shenzhen, China).
Variants were assessed according to the protocol issued by the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). DECIPHER (http://decipher.sanger.ac.uk), OMIM (http://omim.org/), PubMed (http://www.ncbi.nlm.nih.gov/pubmed), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and HGMD (http://www.hgmd.cf.ac.uk/ac/index.php) databases were used to investigate the clinical relevance of the mutations (9).
Sanger sequencing was carried out to confirm the variant of ITGB3 gene. The primers were designed using Oligo Primer Designer (Forward: 5′-GATACTATTCCCGTGCTTG-3′; Reverse: 5′-CACATTGACCACAGAGGC-3′). The DNA was amplified using the following procedure: 95°C for 10 min; 35 cycles at 95°C for 30 s, 60°C for 30 s, 72°C for 30 s; 72°C for 10 min. Sequencing was performed by an ABI 3130 DNA analyzer (10).
Total RNA was extracted from the peripheral blood cells of proband and a healthy control using TRIzol (Takara, Japan) and reverse-transcribed using RT Kit (Takara, Japan) following the manufacturer's instructions. RT-PCR was performed using GoldStar Best Master Mix (CWBIO, Beijing) with the primers designed using Oligo Primer Designer as following: Forward:5′-AAGATTGGAGACACGGTGAG-3′ and Reverse: 5′-GCAGTAACGGTTGCAGGTAT-3′. The procedure of the PCR was as follows: 94°C for 10 min followed by 35 cycles at 94°C for 30 s, 60°C for 30 s, 72°C for 30 s, and a final extension step at 72°C for 10 min. Sequencing was performed by an ABI 3130 DNA analyzer.
A homozygous silent mutation on exon 10 of ITGB3: c.1431C > T (p. G477=) was identified in proband by WES and confirmed by Sanger sequencing. The parents and the second elder sister were heterozygous carriers, in accordance with the autosomal recessive inheritance pattern (Figure 1). The mutation has never been reported in any database (gnomAD, ClinVar or HGMD) or literature.
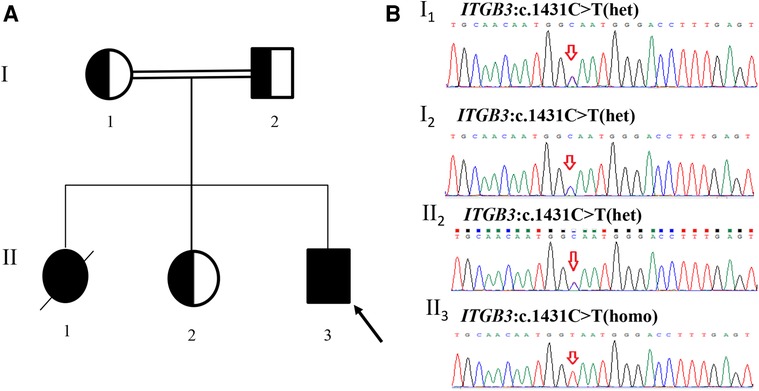
Figure 1. Pedigree of the family and sanger sequencing validation. (A) II3(proband) had a homozygous mutation (c.1431C > T) in exon 10 of ITGB3 gene. I1(mother), I2(father) and II2(the second elder sister) were all heterozygous carriers. II1 died at 5 years old and did not carry out any genetic test. (B) Sanger sequencing results of the family members (red arrows indicate the mutation site).
NetGene2 Server (http://www.cbs.dtu.dk/services/NetGene2/) was used to predict the effects of the variation c.1431C > T (p. G477=) on splicing. A new splicing site was found with confidence score of 0.82 (Figures 2A,B). Alternative Splice Site Predictor (ASSP) (http://wangcomputing.com/assp/index.html) was also used to predict the mutation, which was in accordance with the NetGene2 Server (Figures 2C,D).
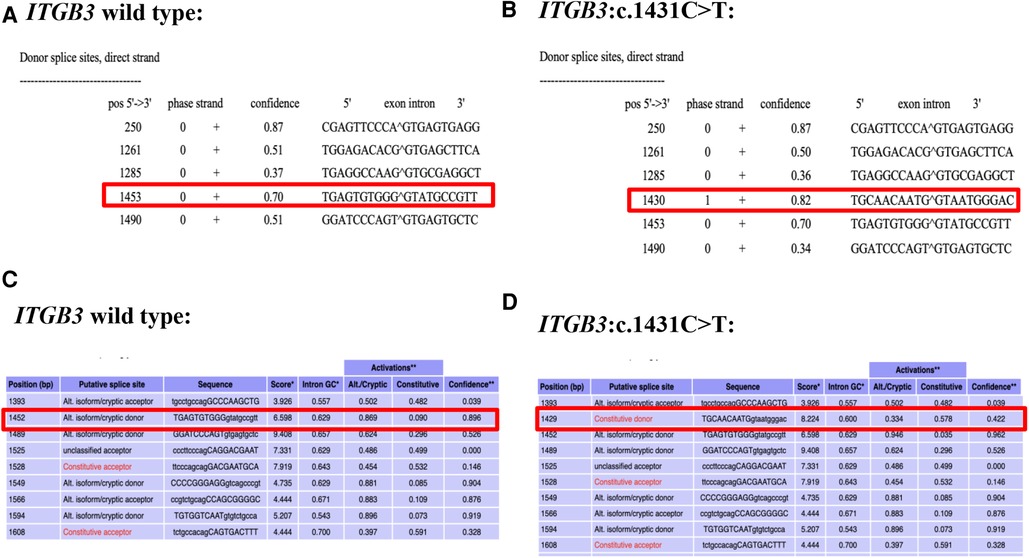
Figure 2. Predict results of the c.1431C > T variant site. (A) The predict result of wild type ITGB3 by using NetGene2 Server (the red square represented). (B) The predict result of ITGB3: c.1431C > T by using NetGene2 Server (the red square represented). (C) The predict result of wild type ITGB3 by using ASSP (the red square represented). (D) The predict result of ITGB3: c.1431C > T by using ASSP (the red square represented).
Based on the highly consistent genotype–phenotype correlation and splicing prediction, RT-PCR was performed to verify the pathogenicity. With the designed primers, the exon 10 of ITGB3 was amplified. It was showed that the mutation c.1431C > T resulted in a new donor splice site at the position of 1,430–1,431 and a new acceptor splice site at the position of 1,523–1,524 base pairs, which caused a deletion of 95 base pairs (c.1430_1524del, Figure 3A,B). Therefore, the amino acid encoded at the position 477 was changed from Glycine to Alanine and then frameshift was found. ITGB3 was truncated by a creation of a premature stop codon after 30 amino acids translation (p. G477Afs*30) (Figure 3C,D). According to ACMG recommendations (11), the mutation ITGB3: c.1431C > T (p. G477=) was classified as likely pathogenic.
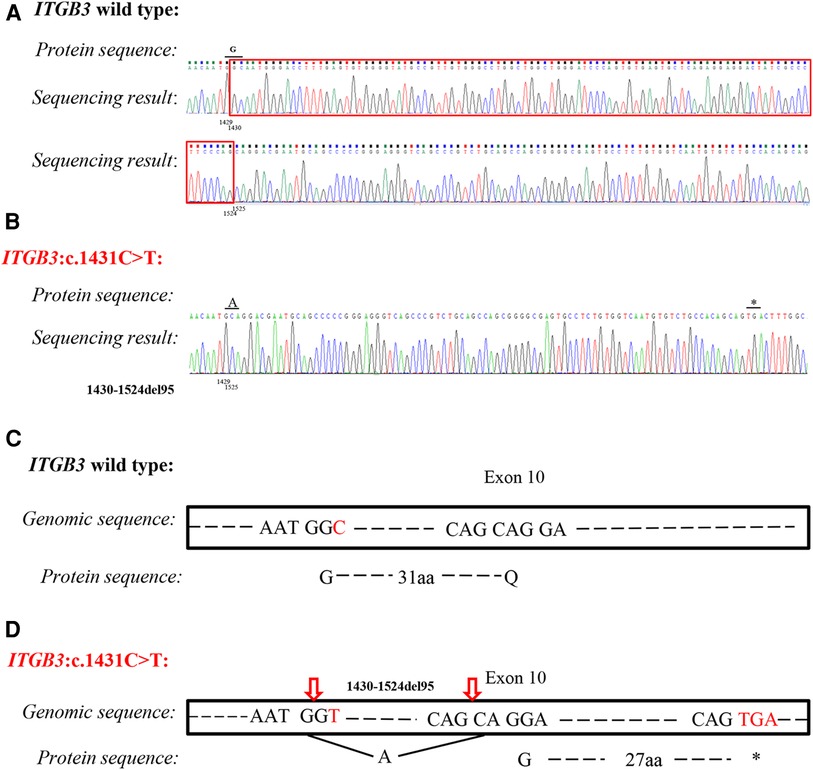
Figure 3. Analysis of silent variant c.1431C > T (p. G477=) of ITGB3 gene. Sequencing results of ITGB3 cDNA from a healthy control (A) and proband (B). The mutation caused 95 base pairs deletion (c.1430_1524del) and frameshift and then ITGB3 was truncated by a creation of a premature stop codon. (C) Splicing schematic representation of exon 10 organization in wild type of ITGB3 gene. (D) Splicing schematic representation of exon 10 organization in mutant type (ITGB3: c.1431C > T). The amino acid encoded at position 477 was changed from Glycine to Alanine, and caused frameshift. ITGB3 was truncated after 30 amino acids translation (p. G477Afs*30).
In the current investigation, we described a Chinese boy with GT due to the homozygous silent mutation of the ITGB3 gene. A heterozygous mutation was detected in his healthy parents and sister. The silent mutation c.1431C > T (p. G477=) was proved to affect splicing, which resulted in a premature stop codon and truncation of the ITGB3. In addition, the CD41 and CD61 of the proband were nearly absent, which significantly damaged the function of αIIbβ3. Therefore, the platelet adhesion, platelet aggregation and clot retraction were affected and consequently caused GT. The mutation has never been reported in any database or literature, indicating our findings expand the spectrum of the diagnosis for the GT and provide insight and information for the genetic counseling.
GT is a rare inherited abnormal platelet function disorder (12, 13). Patients with GT may experience mild to severe bleeding symptoms, including easy bruising, epistaxis, mucosal bleeding, and increased bleeding after trauma or surgery (14, 15). Most of the children may die at their early age due to severe excessive bleeding (3). Heavy menstrual bleeding (HMB) is common in adolescent and adult females (16). In this study, the 7-year-old boy diagnosed with GT presented generalized scattered skin, petechiae and spots on his face after his birth. A blood clot retraction test revealed that the clots did not shrink over 24 h. Furthermore, the expression levels of CD41 and CD61 were significantly lower than those in normal while the expression level of CD42 was the same as healthy people.
GT is caused by the mutations of ITGA2B or ITGB3 genes encoding the αIIbβ3 integrin (6, 11). αIIbβ3 integrin is made up of CD41 and CD61. Low expression of CD41 and CD61 on platelets severely influence platelets functions, including platelet adhesion, platelet aggregation and clot retraction (17, 18). Autosomal recessive inheritance is the general rule. Clinically, people with GT are classified into three groups out of the different expression and function of αIIbβ3. Most patients are originally identified as type I, with less than 5% αIIbβ3. Some people belonging to type II GT express low but residual αIIbβ3 (5%–20%) (19, 20). Furthermore, in type III GT patients, also named variant-type patients, αIIbβ3 expression reaches 100% of normal level while the αIIbβ3 fails to function. The most common subtype is GT type I which accounts for 78% of patients, while GT type II and type III constitute 14% and 8% of cases, respectively (11). As a result of loss of αIIbβ3 function, platelets are unable to bind fibrinogen (Fg) and other adhesive proteins after vessel injury, which may lead to loss of thrombus formation and clot retraction in some cases (21, 22). In the present study, a homozygous silent mutation c.1431C > T (p. G477=) of ITGB3 gene was identified and confirmed in proband by WES and Sanger sequencing. His parents and sister carried the heterozygous ITGB3 c.1431C > T (p. G477=) mutation and all of them did not present bleeding symptoms. Based on the genotype–phenotype correlation and the results of the effect on splicing by two online prediction tools (NetGene2 and ASSP), RT-PCR was carried out to identify the pathology of the mutation. It showed that the mutation caused 95 pairs base deletion and a premature termination codon of ITGB3, which might lead to absence of ITGB3 protein out of nonsense-mediated decay (NMD) or truncated ITGB3 protein. To our best acknowledgement, the silent mutation c.1431C > T (p. G477=) has never been reported before. Taken together, the homozygous silent mutation c.1431C > T (p. G477=) of ITGB3 in proband may explain the cause for GT and the proband belongs to the type I GT.
Nowadays, platelet transfusions, anti-fibrinolytic agents infusion (aminocaproic acid or tranexamic), recombinant activated factor VII (rFVIIa) infusion and hematopoietic stem cell transplantation (HSCT) have been the mainstay of therapies for GT patients (23, 24). However, the treatment of GT patients remains unsatisfactory. Patients’ quality of life is significantly impaired by multiple, spontaneous mucocutaneous bleeding episodes, and the high risk of hemorrhaging with surgery or any trauma (3). Because the therapies mentioned above focus on bleeding relief episodes rather than providing a cure except the costly HSCT (3). More importantly, the diagnosis of GT is often overlooked, as it shares common clinical and laboratory features with other platelet disorders. Therefore, it is of great value to carry out genetic diagnosis as early as possible (25). In addition, prenatal diagnosis or PGT is significant in families with GT history.
The gene ITGB3 was located on chromosome 17q21.32 with 15 exons. According to HGMD (released February 2022), all 223 mutations have been reported in ITGB3. Among them, are 144 missense or nonsense mutations (118 pathogenic mutations, 21 uncertain significance mutation and 5 polymorphic mutations), 18 splicing mutations (17 pathogenic mutations and 1 uncertain significance mutation), 3 regulatory substitutions (3 polymorphic mutations), 37 small deletions (36 pathogenic mutations and 1 uncertain significance mutation), 8 small insertions/duplications mutations (pathogenic mutations), 4 small insertions mutation (pathogenic mutations), 4 gross deletions (pathogenic mutations), 4 gross insertions (pathogenic mutations), 1 complex rearrangement (pathogenic mutation). More than 70% of the mutations are associated with GT.
In conclusion, we report a novel homozygous silent variant c.1431C > T (p. G477=) in exon 10 of the ITGB3 gene in a GT family by the combined applications of WES, Sanger sequencing and bioinformatics analysis. Furthermore, RT-PCR is necessary to perform if the genotype–phenotype correlation is consistent while only a homozygous silent mutation in autosomal recessive disease is detected. The RT-PCR and sequencing verify that the mutation causes a premature termination codon of the ITGB3, upgrading pathogenicity evidence of the silent mutation. These findings are helpful for prenatal diagnosis and preimplantation testing for GT.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by Institutional Review Board of the Women’s Hospital, School of Medicine, Zhejiang University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
MD conceived of the study and revised the manuscript. ZW participated in study design, drafted the manuscript. YX extracted the genomic DNA, designed primers and drafted the manuscript. YS carried out the RT-PCR and revised the manuscript. SW analyzed the clinical data. All authors contributed to the article and approved the submitted version.
This work was supported by the Key Research and Development Program of Zhejiang Province (grant numbers 2019C03025), Zhejiang Provincial Natural Science Foundation of China (grant numbers LY22H110004) and the National Natural Science Foundation of China (grant numbers 81901382).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.1062900/full#supplementary-material.
1. Bacci M, Ferretti A, Marchetti M, Alberelli MA, Falanga A, Lodigiani C, et al. Autoimmune disorders of platelet function: systematic review of cases of acquired glanzmann thrombasthenia and acquired delta storage pool disease. Blood Transfus. (2022) 20(5):420–32. doi: 10.2450/2021.0119-21
2. Sandrock-Lang K, Oldenburg J, Wiegering V, Halimeh S, Santoso S, Kurnik K, et al. Characterisation of patients with glanzmann thrombasthenia and identification of 17 novel mutations. Thromb Haemost. (2015) 113(4):782–91. doi: 10.1160/TH14-05-0479
3. Solh T, Botsford A, Solh M. Glanzmann's thrombasthenia: pathogenesis, diagnosis, and current and emerging treatment options. J Blood Med. (2015) 6:219–27. doi: 10.2147/JBM.S71319
4. Poon MC, Di Minno G, d'Oiron R, Zotz R. New insights into the treatment of Glanzmann thrombasthenia. Transfus Med Rev. (2016) 30(2):92–9. doi: 10.1016/j.tmrv.2016.01.001
5. Pillois X, Peters P, Segers K, Nurden AT. In silico analysis of structural modifications in and around the integrin αIIb genu caused by ITGA2B variants in human platelets with emphasis on Glanzmann thrombasthenia. Mol Genet Genomic Med. (2018) 6(2):249–60. doi: 10.1002/mgg3.365
6. Nurden AT, Fiore M, Nurden P, Pillois X. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. (2011) 118(23):5996–6005. doi: 10.1182/blood-2011-07-365635
7. Botero JP, Lee K, Branchford BR, Bray PF, Freson K, Lambert MP, et al. Glanzmann thrombasthenia: genetic basis and clinical correlates. Haematologica. (2020) 105(4):888–94. doi: 10.3324/haematol.2018.214239
8. Zafarghandi Motlagh F, Fallah MS, Bagherian H, Shirzadeh T, Ghasri S, Dabbagh S, et al. Molecular genetic diagnosis of Glanzmann syndrome in Iranian population; reporting novel and recurrent mutations. Orphanet J Rare Dis. (2019) 14(1):87. doi: 10.1186/s13023-019-1042-4
9. Sun Y, Li Y, Chen M, Luo Y, Qian Y, Yang Y, et al. A novel silent mutation in the L1CAM gene causing fetal hydrocephalus detected by whole-exome sequencing. Front Genet. (2019) 10:817. doi: 10.3389/fgene.2019.00817
10. Fan L, Jin P, Qian Y, Shen G, Shen X, Dong M. Case report: prenatal diagnosis of postaxial polydactyly with bi-allelic variants in smoothened (SMO). Front Genet. (2022) 13:887082. doi: 10.3389/fgene.2022.887082
11. George JN, Caen JP, Nurden AT. Glanzmann's thrombasthenia: the spectrum of clinical disease. Blood. (1990) 75(7):1383–95. doi: 10.1182/blood.V75.7.1383.1383
12. Kasinathan G, Sathar J. Blood management strategies in congenital Glanzmann thrombasthenia at a hematology referral center. Blood Res. (2021) 56(4):315–21. doi: 10.5045/br.2021.2021165
13. Alharbi A, Hashmi JA, Alharby E, Albalawi AM, Ramzan K, Basit S. A novel frameshift mutation in the ITGB3 gene leading to glanzmann's thrombasthenia in a Saudi Arabian family. Hematol Oncol Stem Cell Ther. (2022) 15(1):21–6. doi: 10.1016/j.hemonc.2021.01.003
14. Sharma R, Jamwal M, Senee HK, Uppal V, Hira JK, Bose P, et al. Next-generation sequencing based approach to identify underlying genetic defects of Glanzmann thrombasthenia. Indian J Hematol Blood Transfus. (2021) 37(3):414–21. doi: 10.1007/s12288-020-01368-8
15. Xie Z, Jiang J, Cao L, Jiang M, Yang F, Ma Z, et al. Nonsense-mediated mRNA decay efficiency influences bleeding severity in ITGA2B c.2659C > T (p.Q887X) knock-in mice. Clin Genet. (2021) 100(2):213–8. doi: 10.1111/cge.13975
16. Kadowaki S, Makino S, Mohri Y, Awaguni H, Shinozuka J, Imashuku S. Difficulty in controlling heavy menstrual bleeding at menarche in a patient with Glanzmann's thrombasthenia. Blood Coagul Fibrinolysis. (2021) 32(2):155–8. doi: 10.1097/MBC.0000000000000977
17. Shattil SJ, Kashiwagi H, Pampori N. Integrin signaling: the platelet paradigm. Blood. (1998) 91(8):2645–57. doi: 10.1182/blood.V91.8.2645.2645_2645_2657
18. Li X, Xu J, Li Z, Song Y, Fei Y, Yang G, et al. A novel homozygous frameshift mutation in ITGB3 causes Glanzmann's thrombasthenia. Acta Haematol. (2022) 145(1):78–83. doi: 10.1159/000517050
19. Phillips DR, Agin PP. Platelet membrane defects in Glanzmann's thrombasthenia. Evidence for decreased amounts of two major glycoproteins. J Clin Invest. (1977) 60(3):535–45. doi: 10.1172/JCI108805
20. Xie J, Pabón D, Jayo A, Butta N, González-Manchón C. Type I Glanzmann thrombasthenia caused by an apparently silent beta3 mutation that results in aberrant splicing and reduced beta3 mRNA. Thromb Haemost. (2005) 93(5):897–903. doi: 10.1160/TH04-09-0633
21. Buitrago L, Lefkowitz S, Bentur O, Padovan J, Coller B. Platelet binding to polymerizing fibrin is avidity driven and requires activated αIIbβ3 but not fibrin cross-linking. Blood Adv. (2021) 5(20):3986–4002. doi: 10.1182/bloodadvances.2021005142
22. Mansour W, Einav Y, Hauschner H, Koren A, Seligsohn U, Rosenberg N. An αIIb mutation in patients with Glanzmann thrombasthenia located in the N-terminus of blade 1 of the β-propeller (Asn2Asp) disrupts a calcium binding site in blade 6. J Thromb Haemost. (2011) 9(1):192–200. doi: 10.1111/j.1538-7836.2010.04087.x
23. Poon MC. The use of recombinant activated factor VII in patients with Glanzmann's thrombasthenia. Thromb Haemost. (2021) 121(3):332–40. doi: 10.1055/s-0040-1718373
24. Friend BD, Roach GD, Kempert PH, Moore TB. Successful use of hematopoietic stem cell transplantation for 2 pediatric cases of Glanzmann thrombasthenia and review of the literature. J Pediatr Hematol Oncol. (2020) 42(6):e521–e6. doi: 10.1097/MPH.0000000000001646
Keywords: Glanzmann thrombasthenia, integrin αIIbβ3, ITGB3, silent mutation, whole exome sequencing
Citation: Wang Z, Xu Y, Sun Y, Wang S and Dong M (2023) Novel homozygous silent mutation of ITGB3 gene caused Glanzmann thrombasthenia. Front. Pediatr. 10:1062900. doi: 10.3389/fped.2022.1062900
Received: 6 October 2022; Accepted: 12 December 2022;
Published: 10 January 2023.
Edited by:
Loredana Bury, University of Perugia, ItalyReviewed by:
Ana Marín Quílez, University of Murcia, Spain© 2023 Wang, Xu, Sun, Wang and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minyue Dong ZG9uZ215QHpqdS5lZHUuY24=
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.