 Songmi Wang
Songmi Wang Qun Hu
Qun Hu Yaxian Chen1
Yaxian Chen1 Ai Zhang
Ai Zhang Aiguo Liu
Aiguo Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 20 September 2022
Sec. Pediatric Hematology and Hematological Malignancies
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1013764
This article is part of the Research TopicCase Reports in Pediatric Hematology and Hematological Malignancies 2022View all 14 articles
Background: Acquired von Willebrand syndrome (AVWS) is a less common bleeding disorder, primarily manifested as mild to moderate mucocutaneous bleeding and laboratory tests are similar to hereditary von Willebrand disease (VWD). AVWS is secondary to other diseases, and systemic lupus erythematosus (SLE) is a relatively rare cause.
Case presentation: We report a case of AVWS as onset clinical presentation of SLE manifested as epistaxis and pulmonary hemorrhage. A 13-year-old male child presented to the hospital with a six-month history of recurrent epistaxis and a one-month history of anemia. Routine blood tests demonstrated severe normocytic anemia and normal platelet count. Von Willebrand test revealed a significantly lower level. High-resolution chest computed tomography (CT) showed patchy ground glass opacities consistent with hemorrhagic changes. After ruling out the family history, the patient was diagnosed with AVWS. Additional tests confirmed positive antinuclear and anti-Sm antibodies. The underlying SLE was diagnosed and treated with methylprednisolone with disease recovery.
Conclusion: We recommend screening for bleeding disorders in patients with recurrent epistaxis. AVWS should be considered when laboratory findings suggest hereditary von Willebrand disease without a personal or familial history of bleeding. In addition, the underlying disease should be explored.
Acquired von Willebrand syndrome (AVWS) is a rare hemorrhagic disease, similar to hereditary von Willebrand disease (VWD) in laboratory tests and clinical manifestations. AVWS primarily occurs in adults without a personal or family history of bleeding diathesis, characterized by mucocutaneous or gastrointestinal bleeding. Laboratory tests reveal prolonged bleeding time and low levels of plasma factor VIII (FVIII) and von Willebrand factor (VWF) measurements (1). Unlike VWD, AVWS is almost associated with an underlying disease. According to a survey of AVWS by the International Society of Thrombosis and Hemostasis (ISTH) in 2000, among the 186 cases, the associated diseases were lymphatic hyperplasia (48%), myeloproliferative diseases (15%), tumors (5%), immunology (2%), cardiovascular (21%) and other diseases (9%) related (2). Systemic lupus erythematosus (SLE) is a rare cause of AVWS. Herein, we report a case of AVWS as onset clinical presentation of SLE manifested as epistaxis and pulmonary hemorrhage.
A 13-year-old male child without significant past medical history or family history presented to the hematology department with a six-month history of recurrent epistaxis and a one-month history of anemia. Besides, he was prone to be bruised recently. The boy was found to be obese and pallor through physical examination. Old ecchymosis on the extremities and waist, blood scab in the bilateral nasal vestibule but without significant bleeding in the oropharynx could be noticed (Figure 1). Additionally, he had tachycardia (115 beats/min) and hepatosplenomegaly.
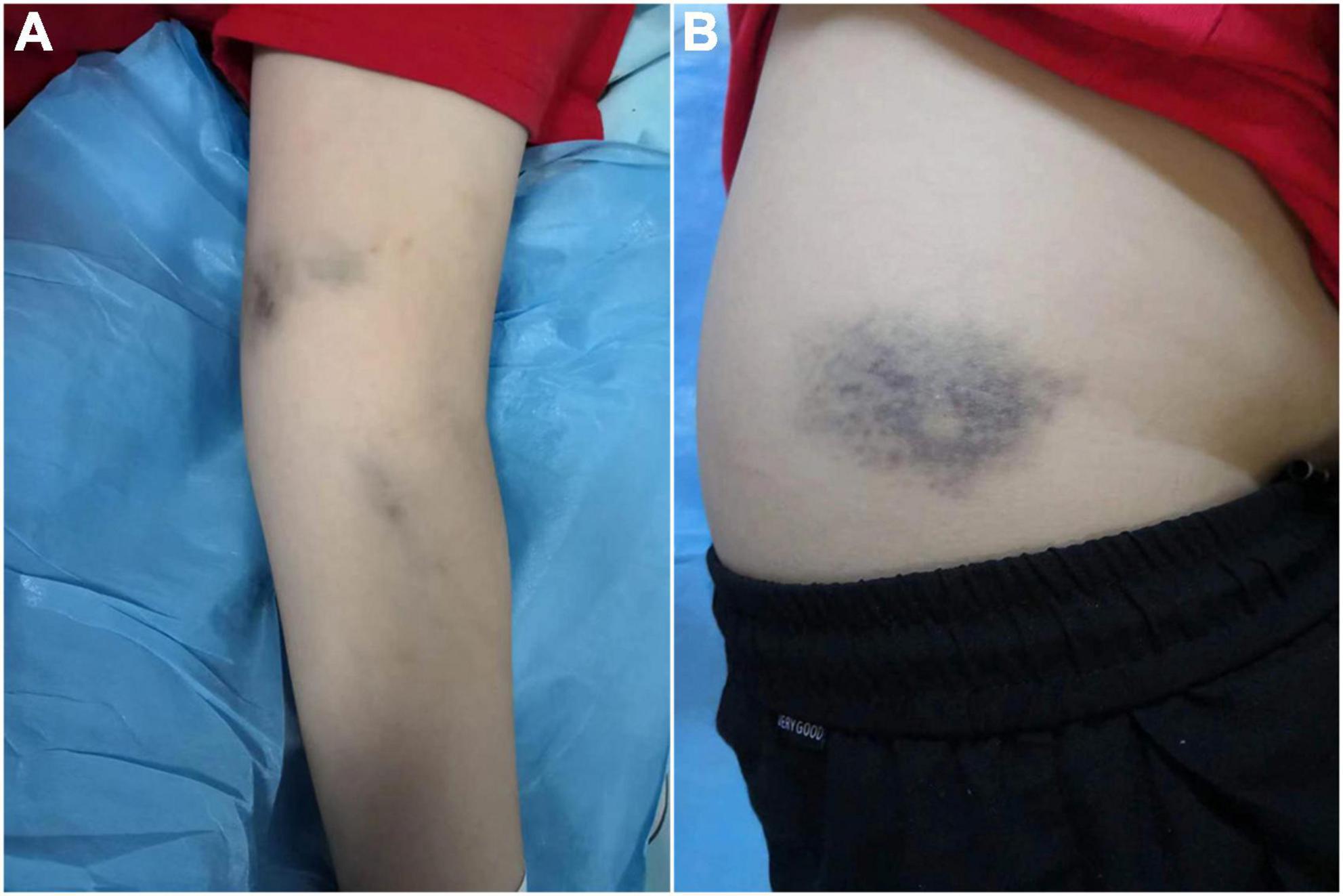
Figure 1. (A) Ecchymosis on the upper extremity. (B) Subcutaneous hemorrhage on the waist.
On initial laboratory tests, the patient had severe normocytic anemia with hemoglobin (Hb) of 58 g/L (MCV: 87.4fL, MCH: 27.0pg, MCHC: 309g/L, reticulocytes: 2.87%) and normal platelet count. Coagulation function detection showed normal activated partial thromboplastin time (APTT), prothrombin time, and fibrinogen but slightly elevated D-dimer and fibrin degradation products (FDP). As the child had a severe bleeding diathesis, further screening of platelet function tests was performed and turned out to be normal. Subsequent Von Willebrand test revealed a significantly lower level: von Willebrand factor antigen (VWF: Ag) was 19.9%, and von Willebrand factor ristocetin cofactor (VWF: RCo) was 22.7% (Table 1). His parents’ studies were negative for hereditary VWD. Therefore, AVWS was diagnosed.
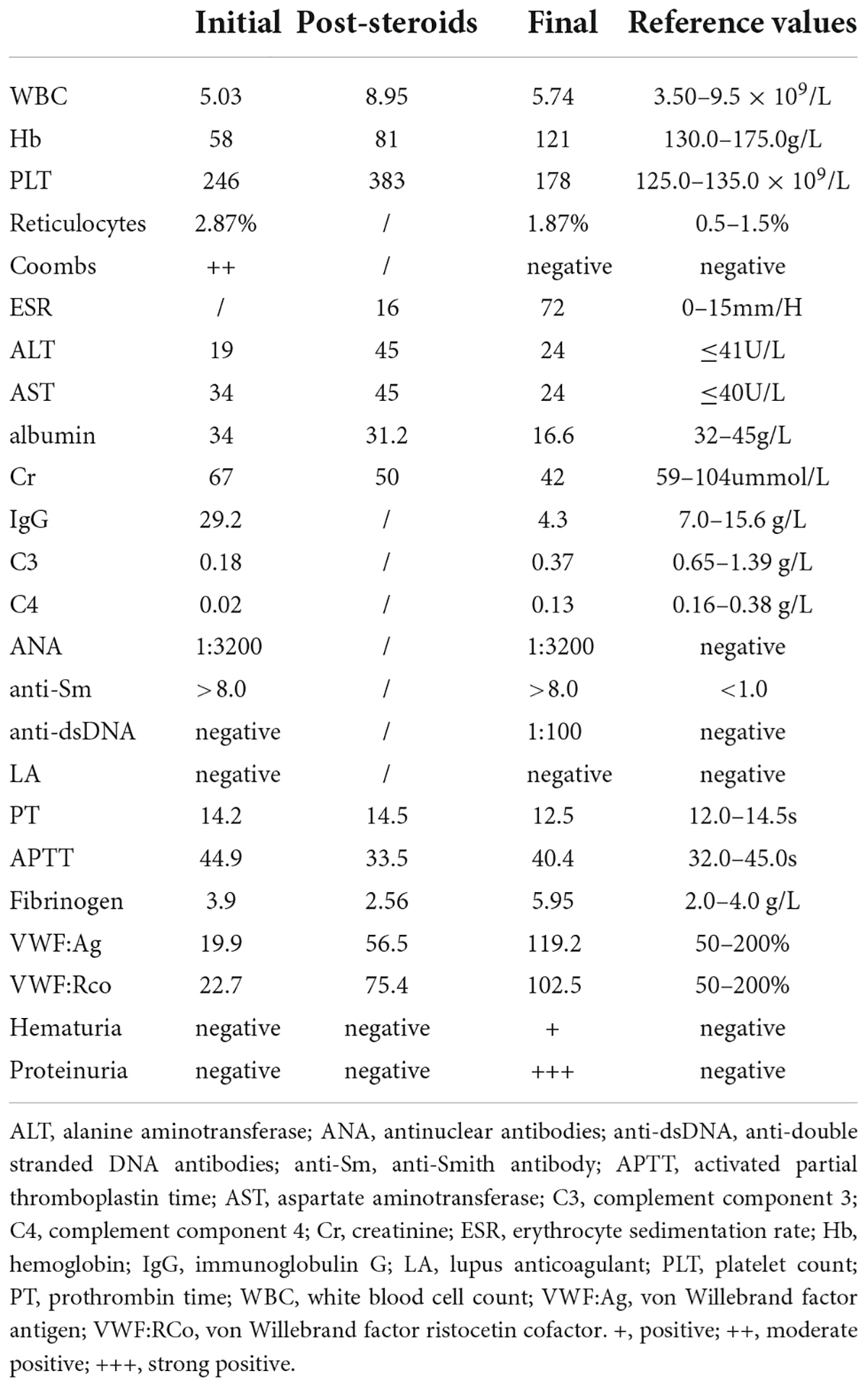
Table 1. Laboratory data of the patient during his hospitalization.
The underlying cause required further investigation. Considering that the patient had normocytic anemia with elevated reticulocytes, the Coombs test was carried out with a positive result. Additional tests confirmed positive antinuclear antibody (1:3200) and anti-Sm antibody. Lupus anticoagulants were negative. The immunologic testing revealed a higher immunoglobulin G level and significantly lower complement levels (C3: 0.18g/L, C4: 0.02g/L). According to Systemic Lupus International Collaborating Clinics (SLICC) classification criteria for SLE (3), the underlying SLE was diagnosed. To further clarify the disease activity index, the following examinations were performed. Although the child had no symptoms such as fever, cough, dyspnea, chest pain, or hemoptysis, lung CT showed diffuse ground-glass nodules consistent with hemorrhagic changes. With the exclusion of infectious lesions, pulmonary manifestations in Systemic Lupus Erythematosus were considered (Figures 2A,B). Urinalysis and color Doppler ultrasound of the urinary system, and magnetic resonance imaging (MRI) of the head, showed no abnormalities. Echocardiography revealed left atrial enlargement and a small amount of pericardial effusion. Mild fatty liver and splenomegaly were found in color ultrasound of the liver and spleen. According to the above, this patient’s systemic lupus erythematosus disease activity index (SLEDAI) (4) was 4 points.
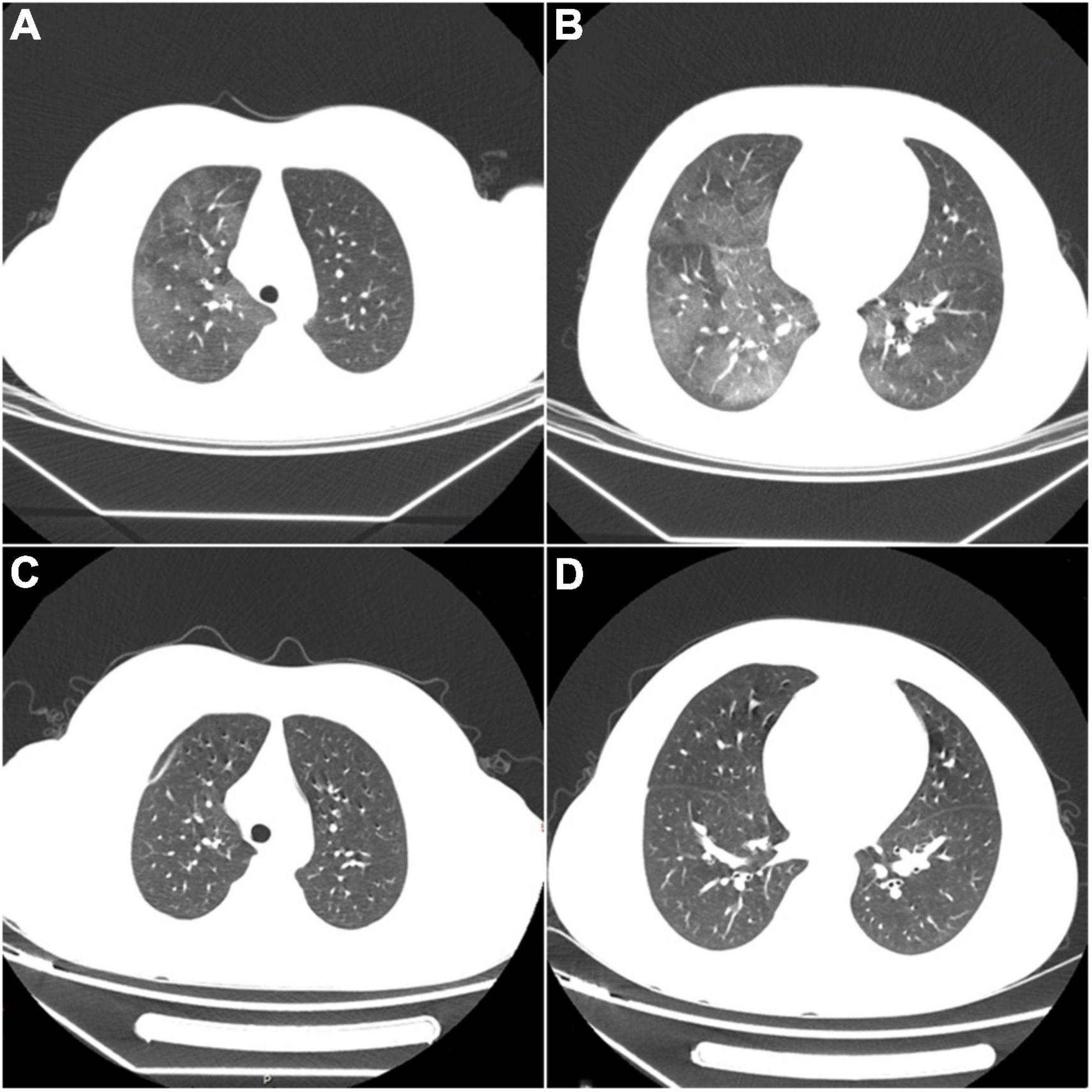
Figure 2. (A,B) Diffuse ground-glass opacities consistent with hemorrhagic changes in both lungs. (C,D) After treatment, repeated CT shows complete disappearance of the abnormal lesions.
We did not choose desmopressin, VWF-containing concentrates, or intravenous immunoglobulin for hemorrhage control, considering that the child had no acute bleeding or adequate funding. The patient received pulse therapy with methylprednisolone (500 mg/d) for five days, and VWF activity (VWF: Ag 56.5%, VWF: RCo: 75.4%) increased to the normal level. Then the patient had no further active bleeding. In a follow-up to the nine months since he was diagnosed, no further epistaxis or ecchymosis occurred during this period, which was confirmed by his subsequent normal coagulation function and VWF levels. His chest CT also recovered (Figures 2C,D). Unfortunately, the serologic tests suggested higher levels of antinuclear antibodies (1:3200) and strongly positive anti-double-stranded DNA antibodies (1:100). What was worse, his kidneys were affected, shown as nephrotic syndrome included heavy proteinuria (urine protein: + + +, 24-h urinary protein quantification: 5.09g), hypoalbuminemia (16.6g/L), peripheral edema, and hyperlipidemia (total cholesterol 11.97mmol/L, triglycerides 2.30 mmol/L). SLEDAI’s score was as high as 22 points. Renal biopsy showed lupus nephritis (WHO type V).
Acquired von Willebrand syndrome (AVWS) is a less common bleeding disorder, often unrecognized or mistaken for von Willebrand disease. In 1968 Dr. Simone et al. (5) first reported an adolescent with SLE and AVWS. Subsequently, AVWS has regained attention due to its relevance in cardiovascular diseases, including congenital heart defects, aortic stenosis, and using left ventricular assist devices (6). Additionally, AVWS is associated with many other underlying disorders, such as solid tumors, hematological cancers, and autoimmune diseases. More than 20 patients with AVWS and SLE have been reported (7–10).
Most cases with AVWS present mild to moderate mucosal bleeding without a personal or familial history of bleeding. Laboratory tests are similar to hereditary VWD, characterized by prolonged bleeding time and decreased von Willebrand factor levels (e.g., decreased VWF: Ag, decreased VWF: RCo, and/or abnormal high-molecular-weight multimers). FVIII levels may be decreased, and APTT may be prolonged. In this case, the detection of VWF was not performed until six months after the onset of symptoms, despite skin ecchymosis and epistaxis at presentation to the hospital.
Nosebleeds are common in children, with 75% of children experiencing at least 1 episode of epistaxis (11). The nose is well vascularized. The Kiesselbach plexus, located in the anterior nasal cavity, is the main source of epistaxis in children (12). The nasal mucosa provides little anatomical support and protection to the underlying blood vessels, resulting in nasal vascular congestion or any factor that dries or irritates the nasal mucosa can increase the likelihood of epistaxis. Common causes of epistaxis include dry nasal mucosa, sinusitis, trauma, foreign bodies, etc. However, repeated epistaxis should consider the possibility of systemic diseases such as bleeding disorders and tumors (13, 14). Unfortunately, the boy’s parents and the attending physician were likely falsely reassured by his young age and a large amount of exercise.
The pathophysiology of AVWS is not fully understood. Most AVWS patients have normal or even increased VWF synthesis and release, but the removal of VWF from plasma is significantly increased, except for patients with hypothyroidism. Current studies suggest the pathogenic mechanisms underlying AVWS may be related to the following mechanisms: autoantibodies to VWF or FVIII inhibit VWF function and increase the clearance of the FVIII-VWF complex (15); adsorption of VWF by tumor cells, or activated platelets increase the clearance of the FVIII-VWF complex (16); cell-mediated or drug-induced increase proteolysis of VWF multimers (1, 6). Currently, the first mechanism involving circulating antibodies to VWF is considered the primary mechanism of AVWS in SLE patients.
For AVWS, the effective treatment of hemorrhage occurs when the underlying disease and the autoantibodies are controlled (17, 18). Current treatments are mainly for primary diseases and pathogenic mechanisms, including desmopressin, VWF-containing concentrates, intravenous immunoglobulin, recombinant factor VIIa, antifibrinolytics, and plasmapheresis (19–24). For AVWS and SLE, steroids and cyclophosphamide are effective in controlling hemorrhage (25–28). Recent studies have reported that rituximab is used to treat AVWS in adolescent SLE (29–31), and bleeding in both patients has been effectively controlled. In this case, the activity of VWF and FVIII returned to normal levels after methylprednisolone pulse therapy. The primary disease control of the child in a later stage is not ideal, but there is no bleeding diathesis. It suggests that the remission of the primary disease is not always consistent with AVWS (17).
Therefore, it is necessary to perform the relevant examinations for unexplained bleeding. Patients with clinical manifestations and laboratory findings similar to hereditary VWD should be considered AVWS after excluding family history. It is essential to explore the underlying disorders. The key to controlling bleeding is the diagnosis and treatment of the primary disease.
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
SW contributed to the acquisition, analysis of data, and writing the manuscript. NT, YC, AZ, XH, and QH performed the diagnostics and treatment of the patient. AL, QH, and AZ conceived the idea and reviewed the manuscript. All authors have substantively revised the work and approved the final submitted version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
APTT, activated partial thromboplastin time; AVWS, acquired von Willebrand syndrome; CT, computed tomography; FDP, fibrin degradation products; FVIII, factor VIII; MRI, magnetic resonance imaging; SLE, systemic lupus erythematosus; SLEDAI, systemic lupus erythematosus disease activity index; SLICC, systemic lupus international collaborating clinics; VWF, von Willebrand factor; VWD, von Willebrand disease; VWF: Ag, von Willebrand factor antigen; VWF: RCo, von Willebrand factor ristocetin cofactor.
1. Franchini M, Lippi G. Acquired von Willebrand syndrome: an update. Am J Hematol. (2007) 82:368–75. doi: 10.1002/ajh.20830
2. Federici AB, Rand JH, Bucciarelli P, Budde U, van Genderen PJ, Mohri H, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost. (2000) 84:345–9.
3. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. (2012) 64:2677–86. doi: 10.1002/art.34473
4. Gladman DD, Ibañez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. (2002) 29:288–91.
5. Simone JV, Cornet JA, Abildgaard CF. Acquired von Willebrand’s syndrome in systemic lupus erythematosus. Blood. (1968) 31:806–12.
6. Franchini M, Mannucci PM. Acquired von Willebrand syndrome: focused for hematologists. Haematologica. (2020) 105:2032–7. doi: 10.3324/haematol.2020.255117
7. Dicke C, Holstein K, Schneppenheim S, Dittmer R, Schneppenheim R, Bokemeyer C, et al. Acquired hemophilia A and von Willebrand syndrome in a patient with late-onset systemic lupus erythematosus. Exp Hematol Oncol. (2014) 3:21. doi: 10.1186/2162-3619-3-21
8. Stufano F, Baronciani L, Biguzzi E, Cozzi G, Colpani P, Chisini M, et al. Severe acquired von Willebrand syndrome secondary to systemic lupus erythematosus. Haemophilia. (2019) 25:e30–2. doi: 10.1111/hae.13626
9. Lee A, Sinclair G, Valentine K, James P, Poon MC. Acquired von Willebrand syndrome: von Willebrand factor propeptide to von Willebrand factor antigen ratio predicts remission status. Blood. (2014) 124:e1–3. doi: 10.1182/blood-2014-02-557132
10. Cao XY, Li MT, Zhang X, Zhao Y, Zeng XF, Zhang FC, et al. Characteristics of acquired inhibitors to factor VIII and von Willebrand factor secondary to systemic lupus erythematosus: experiences from a Chinese tertiary medical center. J Clin Rheumatol. (2021) 27:201–5. doi: 10.1097/RHU.0000000000001284
11. Béquignon E, Teissier N, Gauthier A, Brugel L, De Kermadec H, Coste A, et al. Emergency department care of childhood epistaxis. Emerg Med J. (2017) 34:543–8. doi: 10.1136/emermed-2015-205528
12. Bernius M, Perlin D. Pediatric ear, nose, and throat emergencies. Pediatr Clin North Am. (2006) 53:195–214. doi: 10.1016/j.pcl.2005.10.002
14. Tunkel DE, Anne S, Payne SC, Ishman SL, Rosenfeld RM, Abramson PJ, et al. Clinical practice guideline: nosebleed (epistaxis). Otolaryngol Head Neck Surg. (2020) 162:S1–38. doi: 10.1177/0194599819890327
15. Mannucci P, Lombardi R, Bader R, Horellou M, Finazzi G, Besana C, et al. Studies of the pathophysiology of acquired von Willebrand’s disease in seven patients with lymphoproliferative disorders or benign monoclonal gammopathies. Blood. (1984) 64:614–21.
16. Richard C, Cuadrado MA, Prieto M, Batlle J, Lopez Fernandez MF, Rodriguez Salazar ML, et al. Acquired von Willebrand disease in multiple myeloma secondary to absorption of von Willebrand factor by plasma cells. Am J Hematol. (1990) 35:114–7. doi: 10.1002/ajh.2830350210
17. Tiede A, Rand JH, Budde U, Ganser A, Federici AB. How I treat the acquired von Willebrand syndrome. Blood. (2011) 117:6777–85. doi: 10.1182/blood-2010-11-297580
18. Federici AB. Therapeutic approaches to acquired von Willebrand syndrome. Expert Opin Investig Drugs. (2000) 9:347–54. doi: 10.1517/13543784.9.2.347
19. Federici AB, Stabile F, Castaman G, Canciani MT, Mannucci PM. Treatment of acquired von Willebrand syndrome in patients with monoclonal gammopathy of uncertain significance: comparison of three different therapeutic approaches. Blood. (1998) 92:2707–11.
20. Franchini M, Veneri D, Lippi G. The use of recombinant activated factor VII in congenital and acquired von Willebrand disease. Blood Coagul Fibrinolysis. (2006) 17:615–9. doi: 10.1097/MBC.0b013e3280100d1e
21. Hvas A-M, Sørensen HT, Norengaard L, Christiansen K, Ingerslev J, Sørensen B. Tranexamic acid combined with recombinant factor VIII increases clot resistance to accelerated fibrinolysis in severe hemophilia A. J Thromb Haemost. (2007) 5:2408–14. doi: 10.1111/j.1538-7836.2007.02755.x
22. Basso IN, Keeling D. Myocardial infarction following recombinant activated factor VII in a patient with type 2A von Willebrand disease. Blood Coagul Fibrinolysis. (2004) 15:503–4. doi: 10.1097/00001721-200408000-00010
23. Franchini M. The use of desmopressin as a hemostatic agent: a concise review. Am J Hematol. (2007) 82:731–5. doi: 10.1002/ajh.20940
24. Biguzzi E, Siboni SM, Peyvandi F. Acquired von Willebrand syndrome and response to desmopressin. Haemophilia. (2018) 24:e25–8. doi: 10.1111/hae.13382
25. Franchini M, Lippi G, Favaloro EJ. Advances in hematology. Etiology and diagnosis of acquired von Willebrand syndrome. Clin Adv Hematol Oncol. (2010) 8:20–4.
26. Michiels JJ, Budde U, van der Planken M, van Vliet HH, Schroyens W, Berneman Z. Acquired von Willebrand syndromes: clinical features, aetiology, pathophysiology, classification and management. Best Pract Res Clin Haematol. (2001) 14:401–36. doi: 10.1053/beha.2001.0141
27. Michiels JJ, Schroyens W, van der Planken M, Berneman Z. Acquired von Willebrand syndrome in systemic lupus erythematodes. Clin Appl Thromb Hemost. (2001) 7:106–12. doi: 10.1177/107602960100700205
28. Kasatkar P, Ghosh K, Shetty S. Acquired von Willebrand syndrome: a rare disorder of heterogeneous etiology. J Postgrad Med. (2013) 59:98–101. doi: 10.4103/0022-3859.113816
29. Jimenez AR, Vallejo ES, Cruz MZ, Cruz AC, Miramontes JV, Jara BS. Rituximab effectiveness in a patient with juvenile systemic lupus erythematosus complicated with acquired von Willebrand syndrome. Lupus. (2013) 22:1514–7. doi: 10.1177/0961203313502862
30. Taveras Alam S, Alexis K, Sridharan A, Strakhan M, Elrafei T, Gralla RJ, et al. Acquired von Willebrand’s syndrome in systemic lupus erythematosus. Case Rep Hematol. (2014) 2014:208597. doi: 10.1155/2014/208597
Keywords: epistaxis, pulmonary hemorrhage, pediatrics, acquired von Willebrand syndrome, systemic lupus erythematosus
Citation: Wang S, Hu Q, Chen Y, Hu X, Tang N, Zhang A and Liu A (2022) Case report: A case of acquired von Willebrand syndrome as onset clinical presentation of systemic lupus erythematosus manifested as epistaxis and pulmonary hemorrhage. Front. Pediatr. 10:1013764. doi: 10.3389/fped.2022.1013764
Received: 07 August 2022; Accepted: 02 September 2022;
Published: 20 September 2022.
Edited by:
Paulo Sérgio da Silva Santos, University of São Paulo, BrazilReviewed by:
Bernadete Liphaus, University of São Paulo, BrazilCopyright © 2022 Wang, Hu, Chen, Hu, Tang, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aiguo Liu, ZHJsaXVhaWd1b0AxNjMuY29t; Ai Zhang, YWl6aGFuZzExMDlAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.