 Yonglin Yu
Yonglin Yu Hongwei Yin1,†
Hongwei Yin1,† Ke Wu
Ke Wu- 1Department of Rehabilitation, The Children’s Hospital Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, China
- 2Chigene (Beijing) Translational Medical Research Center Co Ltd, Beijing, China
- 3Prenatal Diagnosis Center, Yiwu Maternity and Child Health Care Hospital, Yiwu, China
Background: Pathogenic variants in the FIG4 gene have been described to be associated with a diverse spectrum of syndromes, such as autosomal recessive bilateral temporooccipital polymicrogyria (OMIM 612691), autosomal dominant amyotrophic lateral sclerosis-11 (ALS11; OMIM 612577), autosomal recessive Charcot-Marie-Tooth disease, type 4J (CMT4J; OMIM 611228), and autosomal recessive Yunis-Varon syndrome (YVS; OMIM 216340). Heterozygous FIG4 variants are responsible for ALS11 characterized by progressive muscular weakness, atrophy, and bulbar palsy. CMT4J is a disorder of peripheral nervous system defects mainly presenting with a highly variable onset of proximal and/or distal muscle weakness. YVS is a disorder of severe neurological involvement with central nervous system (CNS) dysfunction and extensive skeletal anomalies.
Case Presentation: We reported two Chinese siblings born with a weakness in all limbs. They experienced rapidly progressive weakness in distal limbs. At the age of 6 years, the elder brother presented with severe scoliosis and cervical kyphosis. They both had global developmental delay and a CNS involvement with cognitive deficits and swallowing problems. Genetic screening in the patients' family for inherited diseases was recommended. Novel compound heterozygous variants in the FIG4 gene (c.2148delTinsAA and c.317A > G) were found by whole-exome sequencing in the patients. These variants were confirmed by Sanger sequencing in family members.
Conclusions: Herein, we reported two Chinese male patients with CMT4J who presented with abnormal CNS features. CMT4J with CNS involvement has been very rarely reported. We hoped this study could expand the phenotypic and genetic spectrum of FIG4-related diseases. And we helped physicians to understand the genotype–phenotype correlation.
Introduction
Patients with Charcot-Marie-Tooth disease, type 4J (CMT4J) predominantly presented with progressive weakness of distal and/or proximal muscle, significant motor dysfunction, and variably progressive sensory loss. Nerve biopsies indicated demyelination and axonal loss (1). CMT4J accounted for 0.3% of 17,880 individuals with neuropathy (2). The age of onset varied from early childhood to the sixth decade. The congenital or infantile onset of CMT4J has been rarely reported. Yunis-Varon syndrome (YVS) was a rare and severe heterogeneous autosomal recessive disorder characterized by severe skeletal abnormalities, brain malformations, retinopathy, and facial dysmorphisms. YVS also presented with central nervous system (CNS) anomalies like global developmental delay, and feeding and swallowing difficulties. Less than 30 cases of YVS have been reported (3). YVS usually led to lethality in infancy or early childhood. While most CMT4J patients did not exhibit CNS deficits, recent studies reported that rare CMT4J cases involved abnormal CNS. Zimmermann et al. (4) described five patients with a phenotypical continuum between CMT4J and YVS. FIG4 gene encoded the protein polyphosphoinositide phosphatase, which catalyzed the dephosphorylation of phosphatidylinositol 3,5-bisphosphate [PtdIns(3,5)P2] to form phosphatidylinositol 3-phosphate (PtdIns3P) (5). Fig4−/− null mice studies showed defective repair of CNS myelin and damage of myelinated axons in the mature peripheral nervous system (PNS) (6). Homozygous or compound heterozygous deleterious FIG4 variants causing a phenotypical of combined PNS and CNS defects have been very rarely reported. Herein, we report two Chinese siblings with compound heterozygous deleterious FIG4 variants presented with abnormal PNS and CNS features.
Case presentation
The pedigree had two male patients (Figure 1), who were the children of phenotypically normal Chinese parents. The parents did not have consanguineous relation and their family history was not notable. No neurologic disease was reported in the parent's family. The two brothers were naturally conceived with uneventful normal pregnancy. The elder brother (II1) was not treated and did not receive rehabilitation therapy in a local hospital since he was born with a weakness in all limbs, so we could not get his detailed clinical data. At the age of 6 years, the proband (II2) was born at 40 weeks gestation.
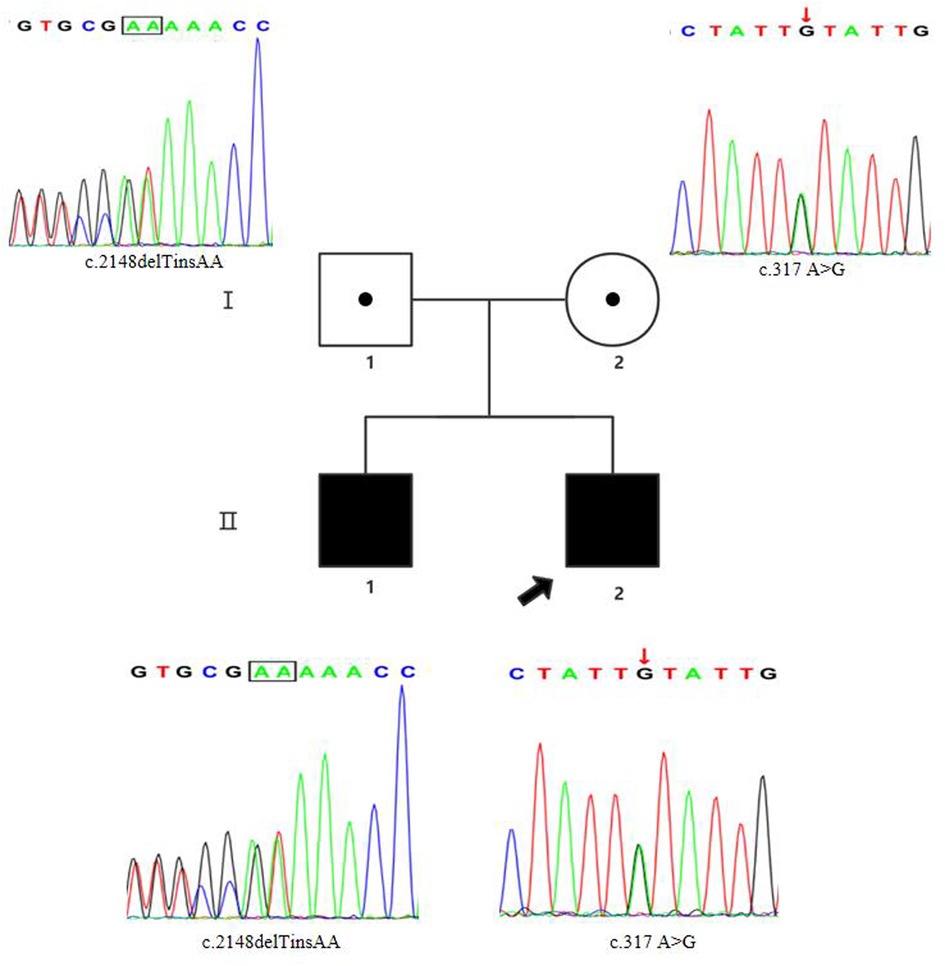
Figure 1. Pedigree chart and Sanger sequencing results of the family members. The proband was the younger brother (marked with the black arrow). The two siblings carried the compound heterozygous FIG4 variants. The results of Sanger sequencing indicated that the father was a heterozygous carrier of the c.2148delTinsAA (p.P718Tfs*3) variant (marked with the black box), and the mother was a heterozygous carrier of the c.317A > G (p.Y106C) variant (marked with the red arrow).
Since the proband was born, he was hospitalized for weakness of all extremities and received treatment for 7 days in the local neonatology department. At the age of 3 months, he was treated in a local rehabilitation institution for “developmental retardation”, and there was no significant improvement. At the age of 10 months, the boy was referred to the rehabilitation department of our hospital. He had an occipital frontal circumference (OFC) of 44.5 cm. No seizures occurred. Scattered red macules could be seen on the skin of the whole body. There were no distinctive facial features except for a high-arched palate and dental dysplasia. He had problems with swallowing and dental growth. Now, he could take small amounts of solids and liquids. No congenital malformations of the scrotum and cryptorchidism were found in both patients. His eyes could track objects, and he responded to sound or simple verbal commands. He could not lift his head, roll over, stand on four limbs, or sit alone without support. He was not able to make a fist or grasp objects. His hands showed thumb adduction deformity. For now, he still suffered from constipation. As their mother recalled, the elder brother had similar symptoms and disease processes. Now he could not walk alone (Figure 2). A nerve biopsy was offered but refused by the parent.
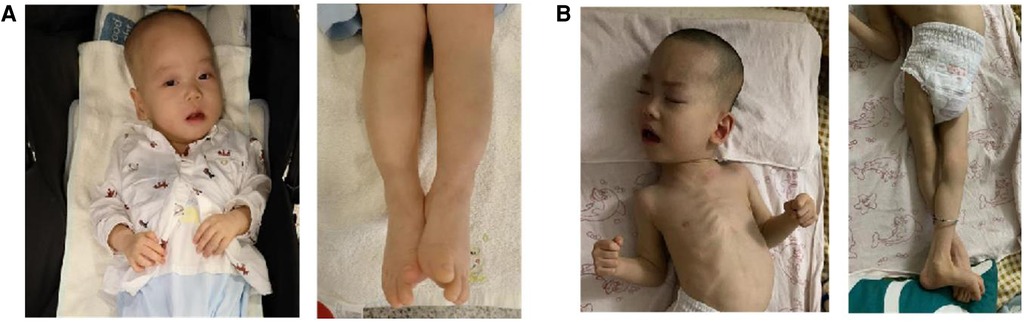
Figure 2. Upper and lower limbs of the two siblings. (A) The proband (1 year old); (B) the elder brother (7 years old).
The modified Ashworth scale (MAS) (7) is clinically used for measuring the increase of muscle tone. The MAS of both upper and lower limbs was 1. A neurological examination revealed decreased muscle strength (Medical Research Council Scale for Muscle Strength was Grade 4/5 in all extremities). Bilateral knee tendon reflexes were positive. The Babinski sign, the Moro reflex, and the asymmetrical tonic neck reflex (ATNR) were all present. The ankle clonus reflex was absent.
Blood laboratory tests revealed a low hemoglobin count of 80 g/L (120–165 g/L), low mean corpuscular volume (MCV) of 48.2 fL (82–100 fL), low mean corpuscular hemoglobin (MCH) of 13.6 pg (26–32 pg), low mean corpuscular hemoglobin concentration (MCHC) of 282 g/L (316–354 g/L), a low IgG level of 1.60 g/L (7–16 g/L), and an elevated IgE level of 1020.0 IU/mL (20–200 IU/mL). Allergy testing showed that he was allergic to the dander of dogs. Thyroid function testing revealed that an elevated triiodothyronine (T3) level of 2.39 nmol/L (0.98–2.33 nmol/L) and an increased thyroxine level of 180.03 nmol/L (62.68–150.80 nmol/L).
Echocardiography indicated the mildly regurgitant tricuspid valve. A nerve conduction study of all four limbs revealed the slightly decreased motor nerve conduction velocity (MCV) in the left tibial and bilateral median nerves and the prolonged F-wave latencies of the left tibial nerve. Brain magnetic resonance imaging (MRI) revealed a slightly thinner corpus callosum, and symmetrical long T1 and T2 signal changes in the ventral medulla oblongata and bilateral thalamus (Figure 3). Brain MRI of the elder brother was not available.
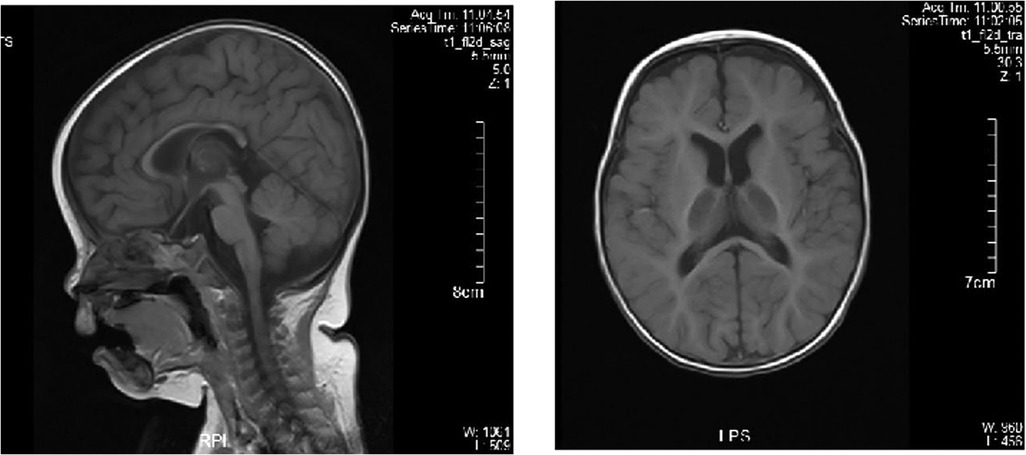
Figure 3. Brain MRI of the proband. Brain MRI showed the normal bilateral lateral ventricle and slightly thinner corpus callosum.
The spine x-rays of the two siblings showed that the spine of the proband was normal (Figure 4A) and the elder brother had severe scoliosis and cervical kyphosis (Figure 4B). The hand x-rays of the two siblings did not show abnormalities.
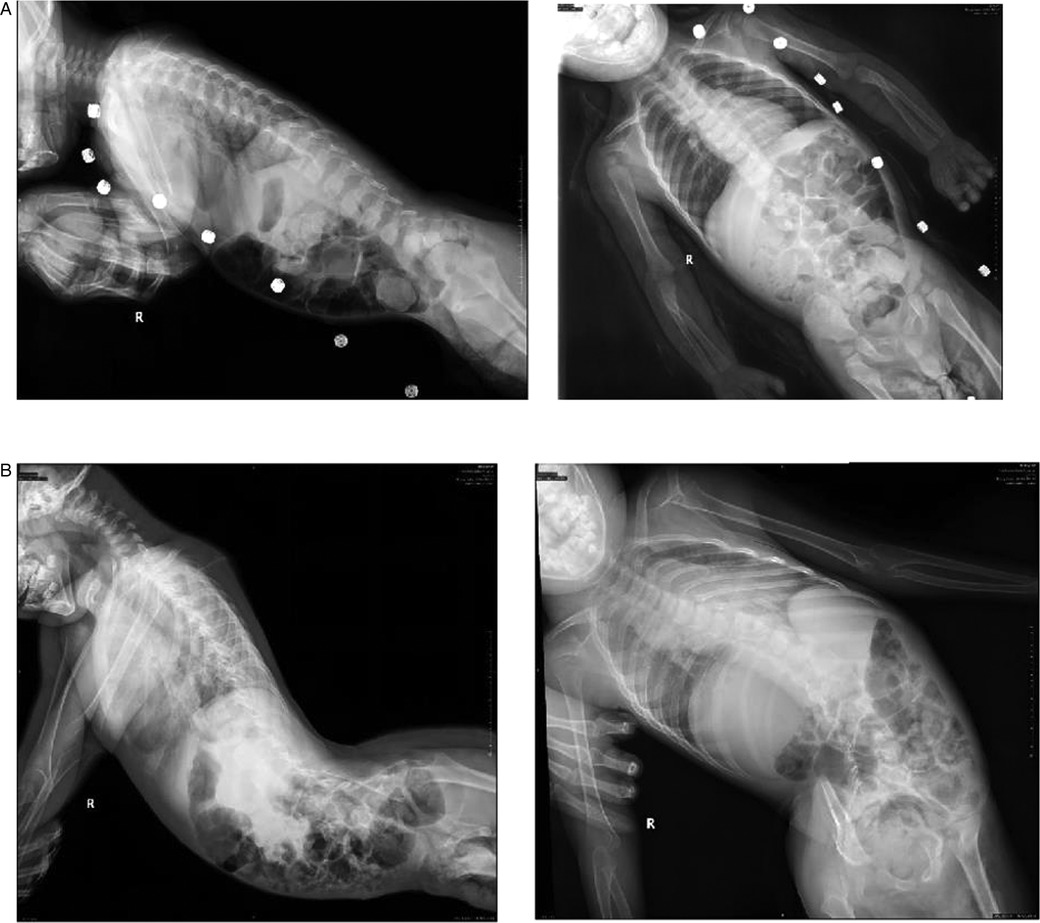
Figure 4. The spine x-rays. (A) The spine x-rays of the proband; (B) the spine x-rays of the elder brother. The spine x-rays showed that the spine of the proband was normal and the elder brother had severe scoliosis and cervical kyphosis.
The Grifths mental development scales for China (GDS-C) (8) are used to assess the development of children from birth to 8 years across six separate subscales: locomotor (A), personal-social (B), language (C), eye-hand coordination (D), performance (E), and practical reasoning (F). Developmental curves with respect to all six subscales together with the general quotient (GQ) were plotted. The proband had a GQ of 12.70 (The raw scores of the six subscales were all lower than the 1st percentile). The low score of GQ suggested that he was significantly delayed in his cognitive and physical development. The GQ of the elder brother was not available, because of his noncooperation. For this reason, genetic screening in the patient's family for inherited diseases was recommended.
Whole-exome sequencing (WES)
The parents signed informed consent for genetic analysis. Our legal ethics committee approved this genetic study. Genomic DNA was extracted from the peripheral blood of the patients and phenotypically normal parent for WES. The xGen™ Exome Research Panel v2 (designed by Integrated DNA Technologies) was used for WES. Quality control (QC) of the DNA library was performed using an Agilent 2100 Bioanalyzer System. DNA nanoball (DNB) preps of clinical samples were sequenced on ultra-high throughput DNBSEQ-T7 platform (MGI, Shenzhen, China) with paired-end 150 nt strategy following manufacturer’s protocol.
Bioinformatic analysis
Sequencing data (a total of 19,136 genes) was analyzed according to our in-house (Chigene Translational Medicine Research Center) procedures. Adapters and low-quality reads were removed, and then data quantity and data quality were statistics. The trimmed reads were then mapped to UCSC GRCh37/hg19 reference genome using the software Burrows-Wheeler Aligner (BWA). GATK software was used for single nucleotide polymorphisms (SNP) and short (<50 bp) insertion/deletion (indel) calling. Samtools and Picard software packages were used to generate clean Bam data by removing duplicate data. Variants were annotated for analysis using the single nucleotide polymorphism database (dbSNP), gnomAD exomes database, and Chigene in-house minor allele frequency (MAF) database. Tools of pathogenicity prediction like SIFT, PolyPhen-2, MutPred, MutPred, and MutationTaster were used for predicting the possible impact of variants. Splice site variants were investigated with prediction programs like SpliceSiteFinder, MaxEntScan, Human Splice Finder, and SpliceAI. As a prioritized pathogenicity annotation to the American College of Medical Genetics and Genomics (ACMG) guidelines, Online Mendelian Inheritance in Man (OMIM), Human Gene Mutation Database (HGMD), and ClinVar databases were used as conferences on pathogenicity of every variant.
Variant classification
As per the guidelines of ACMG for interpreting sequence variants, variants were classified. Classification considered MAF and pathogenicity prediction of variants, disease mechanism, clinical phenotypes, literature evidence, and evolutionary conservation. Variants with MAF > 1% in population databases or predicted benign/neutral pathogenicity were excluded from further investigation. A further investigation focused on the genotype–phenotype correlations, evolutionary conservation of mutant sites, literature evidence, and disease mechanism.
Genetic analysis
Sanger sequencing (BigDye Terminator v3.1 Cycle Sequencing Kit and ABI 3730 Applied Biosystem) was used for further verification. We finally identified novel compound heterozygous FIG4 variants (c.2148delTinsAA and c.317A > G) (NM_014845.6). Cosegregation analysis was performed among family members (other available affected/unaffected relatives). The results of Sanger sequencing indicated that the father was a heterozygous carrier of the c.2148delTinsAA (p.P718Tfs*3) variant, and the mother was a heterozygous carrier of the c.317A > G (p.Y106C) variant (Figure 1; Table 1).

Table 1. Genomic findings and variants interpretation.
The allele frequency of heterozygous c.317A > G was 1/18,392 in the East Asian population of the gnomAD exome database (PM2). The position of this variant was strongly conserved (PhyloP100way score of c.317A > G was 8.99 and greater than 7.2. PhyloP100way scores are based on multiple alignments of 99 vertebrate genome sequences to the human genome). The c.317A > G (p.Y106C) variant was located in a critical and well-established functional domain (SAC phosphatase domain of FIG4 protein) (PM1). In silico predictive algorithms of pathogenicity (SIFT, PolyPhen-2, MutPred, MutPred, and MutationTaster) showed that the missense variant was damaging (PP3). A homozygous c.317A > G variant was identified in three CMT4J siblings who presented with spastic quadriplegia, epilepsy, and global developmental delay (9). Chaudhuri et al. interpreted this variant as a likely pathogenic variant (PS4; PM2; PP1; PP3; PP4) (9).
DynaMut (a web server) (10) is a well-established normal mode approach. We used it to visualize and assess the stability and interactomic interactions of the mutant protein. We put information into DynaMut as follows: wild-type structure (PDB accession code:7K1W), variant detail (Y94C, chain F) (Y106 residue mapped to Y94 in the PDB structure). The prediction outcome of stability was ΔΔG: −0.304 kcal/mol (destabilizing). The Y94 residue in the mutant site was observed to form residue interactions (colored in light green) with its surrounding residues, whereas some interactions (hydrophobic contacts, amide-amide contacts, and ionic interactions) were observed to be lost in the mutant site (Figure 5).
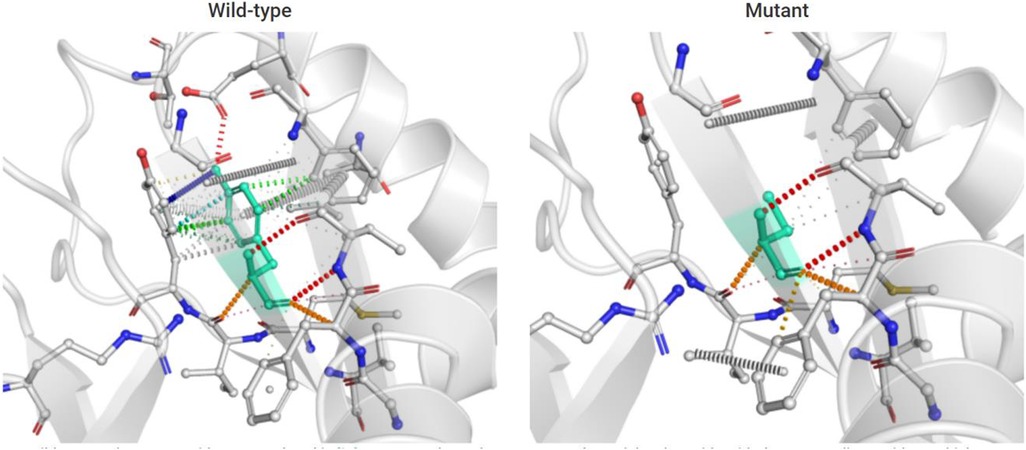
Figure 5. Prediction of interactomic interactions of the wild-type and mutant site (Y106 residue mapped to Y94 in the PDB structure). Residues in the wild-type and mutant sites were colored in light green and shown as sticks. The respective chemical interactions were labeled as dotted lines and colored as follows: hydrogen bonds—(red), weak hydrogen bonds—(orange), hydrophobic contacts—(green), amide-amide contacts—(blue), and ionic interactions—(gold). Amino acid residues were also colored according to type, namely nitrogen (blue), oxygen (red), and sulfur (yellow). In comparison to the wild-type site, some interactions (hydrophobic contacts, amide-amide contact, and ionic interactions) were observed to be lost in the mutant site.
The c.2148delTinsAA variant had not been reported in disease databases (ClinVar, HGMD, OMIM) or registered in population databases (1000 Genomes Project, gnomAD, dbSNP) (PM2). It was a null variant (frameshift mutation) in the FIG4 gene where loss-of-function (LOF) is a known mechanism of CMT4J (PVS1). As per the guidelines of ACMG for interpreting sequence variants, the compound heterozygous variants were likely pathogenic.
Conventional G-banded chromosome analysis of the patients showed a 46, XY karyotype. The results of the genetic metabolic disease screening of the patients were all normal.
Literature review
We searched PubMed, HGMD, and OMIM databases using “Charcot-Marie-Tooth disease, type 4J”, “Yunis-Varon syndrome”, and “FIG4” as keywords. The search time was from the establishment of the databases to 30 June 2022. Patients with FIG4 variants presenting with a phenotypical continuum between CMT4J and CNS involvement were included in this review. Ten documents were retrieved. Thirty CMT4J cases with homozygous or compound heterozygous FIG4 variants that were involved with CNS anomalies have been reported (Table 2). The p.I41T variant was a recurrent mutation reported in a number of unrelated families (11). It has been estimated that the population frequency of this allele was 0.001 (1).
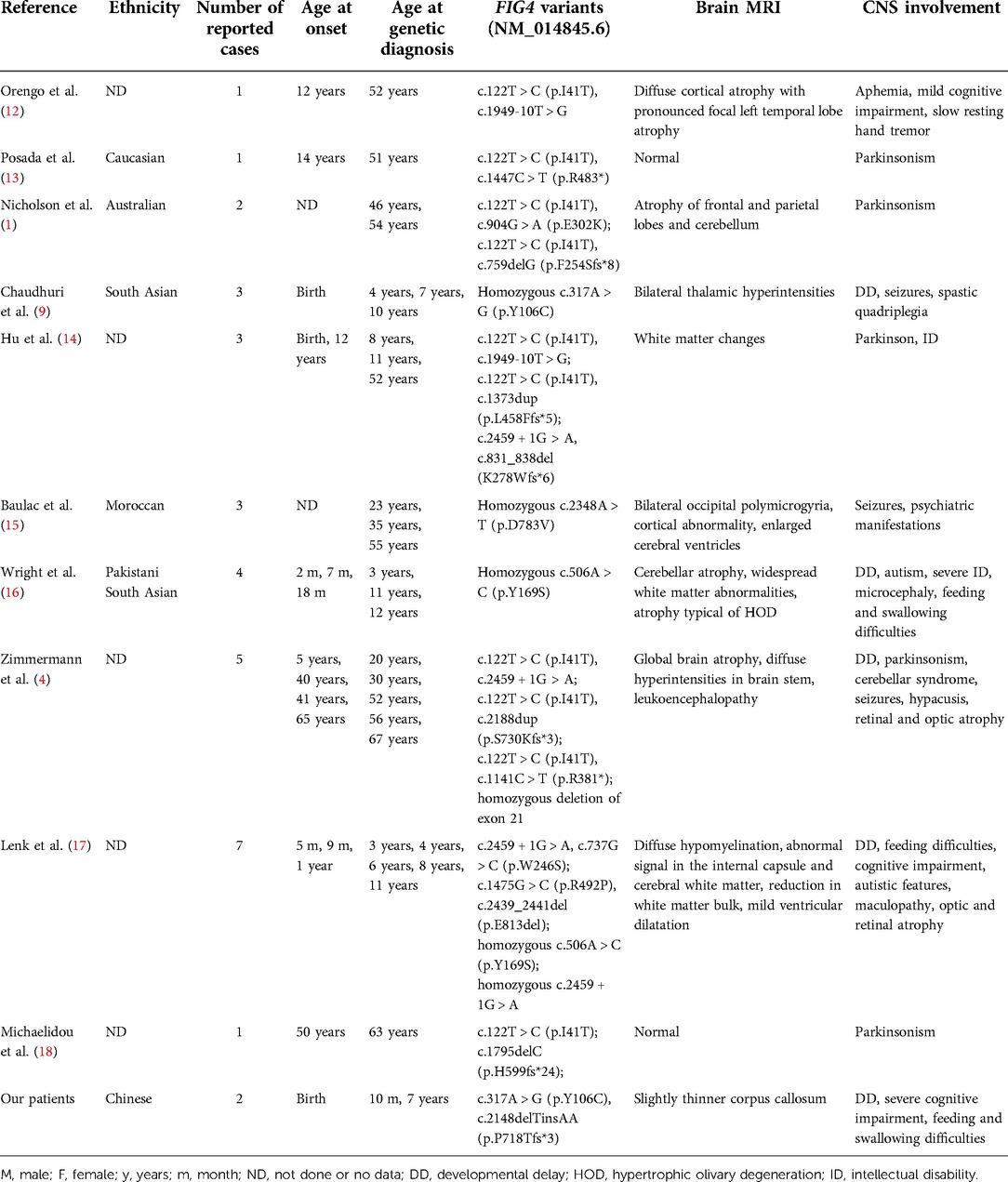
Table 2. CMT4J cases with CNS involvement.
Discussion
The human FIG4 phosphoinositide 5-phosphatase (FIG4) gene is located in chromosome 6q21. The transcript of FIG4 (NM_014845.6) has 23 exons, transcript length of 3,025 base pairs, and translation length of 907 amino acids. The polyphosphoinositide phosphatase protein (FIG4) [UniProtKB-Q92562] belongs to the SAC domain-containing protein family. Cytoplasmic expression of FIG4 is in most of the tissues. The Human Protein Atlas (HPA) shows that FIG4 is highly expressed in the brain, endocrine tissues, skin, and respiratory system. FIG4 catalyzes the dephosphorylation of PtdIns(3,5)P2 to form PtdIns3P. PtdIns3P is a phospholipid in cell membranes that helps to recruit a range of proteins. PIKfyve complex as a lipid kinase is conserved in all eukaryotes and phosphorylates PtdIns3P to PtdIns(3,5)P2 (19) (Figure 6). It is the only source of PtdIns(3,5)P2 (5). PtdIns(3,5)P2 is a low-abundance signaling lipid that maintains endomembrane homeostasis (20).
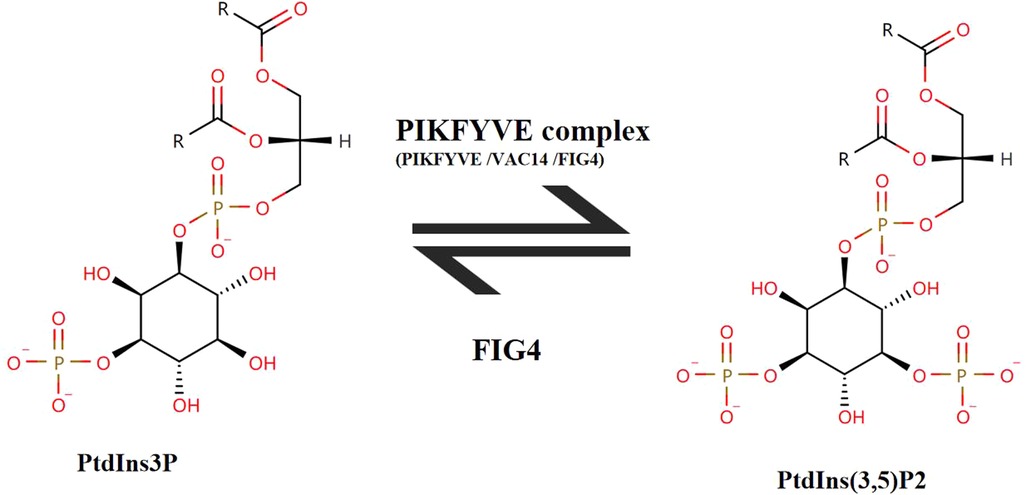
Figure 6. The transition between PtdIns(3,5)P2 and PtdIns3P. FIG4 catalyzes the dephosphorylation of PtdIns(3,5)P2 to form PtdIns3P. PIKFYVE complex consists of three subunits (PIKFYVE, VAC14, FIG4). PIKFYVE is the sole enzyme that catalyzes the phosphorylation of PtdIns3P on the fifth hydroxyl of the myo-inositol ring to form PtdIns(3,5)P2.
FIG4 contains an N-terminal SAC phosphatase domain of 331 amino acids (residues 93–423). Most disease-causing FIG4 variants occurred within the SAC phosphatase domain. Axonal degeneration of motor neurons and demyelination of Schwann cells were observed in Fig4-null mice (21). Neonatal Fig4−/− mice exhibited hypomyelination, spongiform degeneration of neural tissues, intention tremor, and juvenile lethality (6). Neurons are particularly sensitive to reduced abundance of PtdIns(3,5)P2 that is localized to the cytoplasmic surface of endolysosomal vesicles. The kinase PIKFYVE complex regulates the abundance of PtdIns(3,5)P2. The other two components of the PIKFYVE complex are the PIKFYVE activator VAC14 and the phosphoinositide 5-phosphatase FIG4 (Figure 6). The phosphatase FIG4 is bound to the scaffold protein VAC14. A 50% reduction in the level of PtdIns(3,5)P2 was observed in Fig4−/− fibroblasts (22). Thus, FIG4 variants may lead to reduce the activity of FIG4 protein, and then LOF of FIG4 protein damages the PIKFYVE complex. FIG4 variants were associated with dominant ALS11. Dorrity et al. (23) identified the conserved position of the “dominant negative fragment” in yeast. Chow et al. suggested that heterozygous missense variants of the FIG4 gene could exert their effects by a dominant negative mechanism via competition with the wild-type protein for incorporation into the multimeric PtdIns(3,5)P2-regulatory complex (24). But in literature review of ALS11 cases, the variants (Q403X and R183X) were identified in both familial ALS11 cases and CMT4J cases (25). We also did not find “dominant negative fragment” or specific/hot mutation regions/domains for ALS11, CMT4J, and YVS-related FIG4 variants in literature review of Liu et al. (26) and Umair et al. (27). Thus, LOF is a known mechanism of FIG4 gene-related recessive diseases.
As we could see from Table 2, 58% (18/31) of cases were affected with CTM4J and CNS abnormalities during the infant period. The age of onset of CTM4J varied from early childhood to the sixth decade. The congenital or infantile onset of CMT4J has been rarely reported. YVS was caused by null variants in FIG4, which resulted in complete LOF (28). Compared with CTM4J, YVS was a more severe disorder characterized by CNS problems and skeletal abnormalities (cleidocranial dysplasia, digital anomalies). Enlarged cytoplasmic vacuoles were found in neurons, muscles, and cartilage. YVS patients most frequently died during the neonatal period or in early childhood (27). Our patients had global developmental delays, but they were unlike classical YVS. They did not have cleidocranial dysplasia, absent thumbs, dysmorphic features, aphalangia of fingers and toes, or hearing loss, but they presented with partial symptoms of YVS like severe developmental delay, intellectual disability, and feeding and swallowing difficulties. Besides, the symptoms (parkinsonism, autistic features, maculopathy, optic and retinal atrophy, microcephaly, aphemia, spastic quadriplegia, and psychiatric manifestations) have been reported in CMT4J patients. Thus, it is should be noted for clinicians to recognize the phenotypic variability of CMT4J. In the adult PNS, FIG4 was required to protect myelinated axons from Wallerian degeneration; in the CNS, FIG4 was required for myelin repair but not maintenance. The greater vulnerability of the PNS to FIG4 deficiency in the mouse was consistent with clinical observations in patients with CMT4J (29). Thus, CMT4J patients with CNS defects have been rarely reported.
Reducing the intralysosomal Ca2+ by application of TRPML1 synthetic ligand rescued abnormal lysosomal storage in Fig4−/− culture cells. It might be a potential therapy against diseases with FIG4 deficiency (30). Now therapies for CMT4J like gene silencing, gene replacement therapies, and small molecule treatments are in preclinical testing, some therapies reached the clinical trial stage (31).
In conclusion, we described that two Chinese siblings with compound heterozygous deleterious FIG4 variants presented with abnormal PNS and CNS features. Then we reviewed related literature and helped clinicians to be aware of the wide phenotypic spectrum of FIG4-related diseases. We hoped that this study could be helpful to early diagnosis and treatment.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Materials.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethical Committee of the Children’s Hospital, Zhejiang University School of Medicine. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
YLY and HWY contributed to the manuscript equally; KW wrote the main manuscript text; CLM, XYJ, and WCL carried out the molecular genetic experiment; YLY, HWY, and HFL prepared the clinical data and imaging data; KW contributed to the checking of revision, genetic evaluation, and gene database analysis; KW critically revised the manuscript. All authors reviewed the manuscript. The authors read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
We are grateful to the patient and their families for participation in this study, as well as for the help of all the physicians in the course of the medical treatment. We wish to thank the staff of Chigene (Beijing) Translational Medical Research Center Co. Ltd. for assisting with whole-genome sequencing and Sanger sequencing.
Conflict of interest
Authors Changli Ma and Ke Wu were employed by company Chigene (Beijing) Translational Medical Research Center Co Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nicholson G, Lenk GM, Reddel SW, Grant AE, Towne CF, Ferguson CJ, et al. Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P2 phosphatase FIG4. Brain. (2011) 134(Pt 7):1959–71. doi: 10.1093/brain/awr148
2. DiVincenzo C, Elzinga CD, Medeiros AC, Karbassi I, Jones JR, Evans MC, et al. The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med. (2014) 2(6):522–9. doi: 10.1002/mgg3.106
3. Umair M, Alkharfy TM, Sajjad S, Alfadhel M. FIG4-associated Yunis-Varon syndrome: identification of a novel missense variant. Mol Syndromol. (2021) 12(6):386–92. doi: 10.1159/000516971
4. Zimmermann M, Schuster S, Boesch S, Korenke GC, Mohr J, Reichbauer J, et al. FIG4 mutations leading to parkinsonism and a phenotypical continuum between CMT4J and Yunis-Varon syndrome. Parkinsonism Relat Disord. (2020) 74:6–11. doi: 10.1016/j.parkreldis.2020.03.021
5. Lees JA, Li P, Kumar N, Weisman LS, Reinisch KM Insights into lysosomal PI(3,5)P2 homeostasis from a structural-biochemical analysis of the PIKfyve lipid Kinase complex. Mol Cell. (2020) 80(4):736–43.e4. doi: 10.1016/j.molcel.2020.10.003
6. Mironova YA, Lin JP, Kalinski AL, Huffman LD, Lenk GM, Havton LA, et al. Protective role of the lipid phosphatase Fig4 in the adult nervous system. Hum Mol Genet. (2018) 27(14):2443–53. doi: 10.1093/hmg/ddy145
7. Meseguer-Henarejos AB, Sánchez-Meca J, López-Pina JA, Carles-Hernández R Inter- and intra-rater reliability of the Modified Ashworth Scale: a systematic review and meta-analysis. Eur J Phys Rehabil Med. (2018) 54(4):576–90. doi: 10.23736/S1973-9087.17.04796-7
8. Tso WWY, Wong VCN, Xia X, Faragher B, Li M, Xu X, et al. The Griffiths Development Scales-Chinese (GDS-C): a cross-cultural comparison of developmental trajectories between Chinese and British children. Child Care Health Dev. (2018) 44(3):378–83. doi: 10.1111/cch.12548
9. Chaudhuri J, Dutta AK, Biswas T, Biswas A, Ray BK, Ganguly G Charcot-Marie-Tooth disease type 4J with spastic quadriplegia, epilepsy and global developmental delay: a tale of three siblings. Int J Neurosci. (2020):1–4. doi: 10.1080/00207454.2020.1840373
10. Rodrigues CH, Pires DE, Ascher DB. Dynamut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. (2018) 46(W1):W350–5. doi: 10.1093/nar/gky300
11. Lafontaine M, Lia AS, Bourthoumieu S, Beauvais-Dzugan H, Derouault P, Arné-Bes MC, et al. Clinical features of homozygous FIG4-p.Ile41Thr Charcot-Marie-Tooth 4J patients. Ann Clin Transl Neurol. (2021) 8(2):471–6. doi: 10.1002/acn3.51175
12. Orengo JP, Khemani P, Day JW, Li J, Siskind CE, et al. Charcot-Marie-Tooth disease type 4J with complex central nervous system features. Ann Clin Transl Neurol. (2018) 5(2):222–5. doi: 10.1002/acn3.525
13. Posada IJ, Domínguez-González C. CMT4J, Parkinsonism and a new FIG4 mutation. Parkinsonism Relat Disord. (2020) 81:82–3. doi: 10.1016/j.parkreldis.2020.10.011
14. Hu B, McCollum M, Ravi V, Arpag S, Moiseev D, Castoro R, et al. Myelin abnormality in Charcot-Marie-Tooth type 4J recapitulates features of acquired demyelination. Ann Neurol. (2018) 83(4):756–70. doi: 10.1002/ana.25198
15. Baulac S, Lenk GM, Dufresnois B, Ouled Amar Bencheikh B, Couarch P, Renard J, et al. Role of the phosphoinositide phosphatase FIG4 gene in familial epilepsy with polymicrogyria. Neurology. (2014) 82(12):1068–75. doi: 10.1212/WNL.0000000000000241
16. Wright GC, Brown R, Grayton H, Livingston JH, Park SM, Parker APJ, et al. Clinical and radiological characterization of novel FIG4-related combined system disease with neuropathy. Clin Genet. (2020) 98(2):147–54. doi: 10.1111/cge.13771
17. Lenk GM, Berry IR, Stutterd CA, Blyth M, Green L, Vadlamani G, et al. Cerebral hypomyelination associated with biallelic variants of FIG4. Hum Mutat. (2019) 40(5):619–30. doi: 10.1002/humu.23720
18. Michaelidou K, Tsiverdis I, Erimaki S, Papadimitriou D, Amoiridis G, Papadimitriou A, et al. Whole exome sequencing establishes diagnosis of Charcot-Marie-Tooth 4J, 1C, and X1 subtypes. Mol Genet Genomic Med. (2020) 8(4):e1141. doi: 10.1002/mgg3.1141
19. Jin N, Lang MJ, Weisman LS. Phosphatidylinositol 3,5-bisphosphate: regulation of cellular events in space and time. Biochem Soc Trans. (2016) 44(1):177–84. doi: 10.1042/BST20150174
20. McCartney AJ, Zhang Y, Weisman LS. Phosphatidylinositol 3,5-bisphosphate: low abundance, high significance. Bioessays. (2014) 36(1):52–64. doi: 10.1002/bies.201300012
21. Vaccari I, Carbone A, Previtali SC, Mironova YA, Alberizzi V, Noseda R, et al. Loss of Fig4 in both Schwann cells and motor neurons contributes to CMT4J neuropathy. Hum Mol Genet. (2015) 24(2):383–96. doi: 10.1093/hmg/ddu451
22. Zolov SN, Bridges D, Zhang Y, Lee WW, Riehle E, Verma R, et al. In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc Natl Acad Sci U S A. (2012) 109(43):17472–7. doi: 10.1073/pnas.1203106109
23. Dorrity MW, Queitsch C, Fields S. High-throughput identification of dominant negative polypeptides in yeast. Nat Methods. (2018) 16(5):413–6. doi: 10.1038/s41592-019-0368-0
24. Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. (2008) 84(1):85–8. doi: 10.1016/j.ajhg.2008.12.010
25. Osmanovic A, Rangnau I, Kosfeld A, Abdulla S, Janssen C, Auber B, et al. FIG4 Variants in central European patients with amyotrophic lateral sclerosis: a whole-exome and targeted sequencing study. Eur J Hum Genet. (2021) 25(3):324–31. doi: 10.1038/ejhg.2016.186
26. Liu CY, Lin JL, Feng SY, Che CH, Huang HP, Zou ZY Novel variants in the FIG4 gene associated with Chinese sporadic amyotrophic lateral sclerosis with slow progression. J Clin Neurol. (2022) 18(1):41–7. doi: 10.3988/jcn.2022.18.1.41
27. Umair M, Alkharfy TM, Sajjad S, Alfadhel M FIG4-associated Yunis-Varon syndrome: identification of a novel missense variant. Mol Syndromol. (2021) 12(6):386–92. doi: 10.1159/000516971
28. Raghu P, Joseph A, Krishnan H, Singh P, Saha S Phosphoinositides: regulators of nervous system function in health and disease. Front Mol Neurosci. (2019) 12:208. doi: 10.3389/fnmol.2019.00208
29. Mironova YA, Lenk GM, Lin JP, Lee SJ, Twiss JL, Vaccari I, et al. PI(3,5)P2 biosynthesis regulates oligodendrocyte differentiation by intrinsic and extrinsic mechanisms. Elife. (2016) 5:e13023. doi: 10.7554/eLife.13023.27008179
30. Zou J, Hu B, Arpag S, Yan Q, Hamilton A, Zeng YS, et al. Reactivation of lysosomal Ca2+ efflux rescues abnormal lysosomal storage in FIG4-deficient cells. J Neurosci. (2015) 35(17):6801–12. doi: 10.1523/JNEUROSCI.4442-14.2015
Keywords: FIG4 gene, Charcot-Marie-Tooth disease, central nervous system, cognitive deficits, compound heterozygous variants
Citation: Yu Y, Yin H, Ma C, Jia X, Chen W, Li H and Wu K (2022) Case report and literature review: Novel compound heterozygous FIG4 variants causing both of peripheral and central nervous system defects. Front. Pediatr. 10:1008251. doi: 10.3389/fped.2022.1008251
Received: 31 July 2022; Accepted: 26 September 2022;
Published: 21 October 2022.
Edited by:
Jun Mitsui, The University of Tokyo, JapanReviewed by:
Abdul Wahab Siddique, College of Physicians and Surgeons Pakistan, PakistanIoannis Zaganas, University of Crete, Greece
© 2022 Yu, Yin, Ma, Jia, Chen, Li and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ke Wu NzU0Mjk5MDU4QHFxLmNvbQ==
†These authors have contributed equally to this work
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics