 Scott C. Fligor
Scott C. Fligor Thomas I. Hirsch
Thomas I. Hirsch Savas T. Tsikis
Savas T. Tsikis Andrew Adeola
Andrew Adeola
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pediatr. , 14 October 2022
Sec. Pediatric Gastroenterology, Hepatology and Nutrition
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1007813
This article is part of the Research Topic Elimination of Biliary Atresia View all 8 articles
Following Kasai hepatic portoenterostomy (HPE), most patients with biliary atresia will eventually require liver transplantation due to progressive cirrhosis and liver failure. Preventing liver transplantation, or even delaying eventual liver transplantation, is the key to improving long-term outcomes. This review first examines the commonly used adjuvant therapies in post-HPE biliary atresia and the strength of the evidence supporting these therapies. Next, it examines the evolving frontiers of management through a comprehensive evaluation of both recently completed and ongoing clinical trials in biliary atresia. Promising therapies used in other cholestatic liver diseases with potential benefit in biliary atresia are discussed. Improving post-HPE management is critical to prevent complications, delay liver transplantation, and ultimately improve the long-term survival of patients with biliary atresia.
Biliary atresia is a fibro-inflammatory disease affecting both intra- and extrahepatic bile ducts and presenting with biliary obstruction (1, 2). Although rare, affecting one in 10,000–20,000 live births, biliary atresia is the most common indication for liver transplantation in childhood (2–5). The Kasai hepatic portoenterostomy (HPE) is the primary palliative procedure for restoring bile flow (6). The obliterated extrahepatic bile ducts are transected with anastomosis between the small intestine and the dissected porta hepatis to allow bile drainage via microscopic bile ducts. In modern series, half of patients survive a decade with their native liver and over 90% survive into adulthood (7, 8). However, most patients ultimately require liver transplantation as the definitive treatment for progressive native liver disease following HPE. In patients with late diagnosis and advanced liver disease at time of diagnosis, primary liver transplantation may be required.
Long-term survival depends upon delaying eventual transplantation. Patients under two years of age have higher waitlist and posttransplant mortality (9). The most important prognostic factor following HPE is successful bile drainage characterized by clearance of jaundice. Infants with successful clearance of jaundice versus those without demonstrate remarkably higher survival with their native liver at two years (86% vs. 20%) (10). Earlier diagnosis and HPE, particularly within the first 30–45 days of life, results in higher successful clearance of jaundice and improved native liver survival (11). Stool color cards are the most commonly instituted neonatal screening to facilitate early diagnosis and expeditious HPE (12).
Native liver disease in post-HPE patients includes recurrent cholangitis, cholestasis, synthetic liver failure, cirrhosis, portal hypertension, and malignancy (13, 14). Initially, native liver disease presents as a chronic cholangiopathy with bile ductular proliferation and liver fibrosis (15, 16). Molecular profiling of tissue samples obtained at HPE demonstrate distinct histologic phenotypes: inflammation-predominant, mixed, or fibrosis-predominant (17). The inflammation-predominant phenotype was found in younger patients at time of HPE and was associated with better outcomes, suggesting a pathologic progression from initial inflammation to fibrosis with implications for long-term survival of the native liver (17). While a massive inflammatory response appears to be a key part of the initial pathogenesis of biliary atresia, post-HPE inflammation may be pro-reparative (18).
The initial goals following HPE are successful bile drainage, prevention of progression of native liver disease to cirrhosis, prevention of cholangitis (which accelerates the progression to cirrhosis), and achievement of satisfactory growth and development. Thus, interventions in post-HPE patients generally focus on choleresis, preventing inflammation, preventing cholangitis, and ensuring adequate nutrition.
Few medical therapies have been rigorously investigated in biliary atresia and found to substantially alter the progression to transplantation. Here, we review the evidence for the current management of post-HPE biliary atresia and discuss emerging investigational therapies.
Glucocorticoids are among the most studied interventions following HPE for biliary atresia (19–27). The concept of “blast therapy” originated with Karrer and Lilly in 1975: a four day taper of high dose methylprednisolone (starting dose 10 mg/kg) increased bile flow and decreased serum bilirubin in post-HPE patients with cholangitis or decreased bile flow (28). Glucocorticoids also have profound anti-inflammatory effects (29). Current evidence for glucocorticoids in post-HPE biliary atresia is weak, although the data suggest a possible benefit especially in clearance of jaundice.
The highest quality evidence consists of three randomized placebo-controlled trials (19, 21, 26). Davenport et al. conducted a randomized, double-blind, placebo-controlled trial at two centers in the United Kingdom (26). In the prednisolone group, 26% underwent transplant at 12 months compared to 35% in the placebo group (P = 0.47), while 50% had normal bilirubin in the prednisolone group compared to 40% in the placebo group (P = 0.35). A subgroup analysis of infants undergoing early portoenterostomy (age < 70 days) found decreased median bilirubin at one month (64 vs. 116 umol/L, P = 0.01), but this was not sustained at six months.
The START trial was a multi-center, double-blind, randomized, placebo-controlled trial conducted at 14 United States centers (21). Infants were randomized to a 13-week steroid course or placebo (n = 70). There was no significant difference between the steroid group and control group in the primary endpoint of successful bile drainage at six months post-HPE in either the intention-to-treat (58.6% vs. 48.6%) or per-protocol analysis. In infants <70 days old at time of HPE, there was similarly no significant difference between the steroid and control groups for successful bile drainage (71.8% vs. 56.8%). There was similar survival with native liver at 24 months post-HPE (58.7% vs. 59.4%, P = 0.99) and successful bile drainage at 24 months (49.4% vs. 39.8%, P = 0.29).
A recent randomized controlled trial at a single center in China randomized infants post-HPE (performed by a single surgeon) to steroid treatment or placebo (19). The primary outcome of clearance of jaundice with native liver at six months was significantly higher in the steroid group compared to the placebo group (54.1% vs. 31.0%, P = 0.0015) with higher native liver survival rate at 24 months (57.1% vs. 40.0%, P = 0.03). While this study is limited in generalizability due to its single center and single surgeon experience, these data are consistent in direction and magnitude with the previously described randomized controlled trials and provide weak support for post-HPE glucocorticoids in biliary atresia.
Cholangitis affects 40%–93% of post-HPE patients, most commonly in the first two years of life (30). Prophylactic antibiotics are often prescribed to prevent cholangitis and, hopefully, prevent the progression of liver disease. Surveys of primarily European and North American centers have found most utilize prophylactic antibiotics post-HPE, although there is minimal evidence to support a specific antibiotic regimen or duration of prophylaxis (30, 31).
There are no randomized placebo-controlled trials evaluating antibiotic prophylaxis. A small single center trial in Taiwan randomized patients with one episode of cholangitis post-HPE to prophylaxis with trimethoprim-sulfamethoxazole (TMP/SMZ; n = 9) or neomycin (n = 10), compared to a historical control with no prophylactic antibiotics (n = 18) (32). Patients in both antibiotic groups had decreased cholangitis recurrence compared to the historical control group (TMP/SMZ: RR 0.52 [95% CI 0.29, 0.98], neomycin: RR 0.42 [95% CI 0.22,0.82]). A recent randomized controlled trial at a single center in China randomized infants post-HPE to seven or 14 days of intravenous antibiotics (cefoperazone/sulbactam and ornidazole) prior to an oral antibiotic regimen for 6 months (alternating sulfamethoxazole and cefaclor every two weeks) (33). There was no difference in the primary outcome of cholangitis occurrence at 6 months between the short- and long-term groups (62% vs. 70%, P = 0.27), but there was decreased early onset cholangitis (within one month) in the long-term group (61% vs. 38%, P = 0.02). Older retrospective cohort studies have evaluated patients receiving antibiotic prophylaxis versus historical controls that did not receive antibiotics, with only Lally et al. (antibiotic n = 34, no antibiotic n = 7) reporting significantly decreased cholangitis incidence, while Wu et al. (antibiotic n = 16, no antibiotic n = 21) and de Vries et al. (antibiotic n = 124, no antibiotic n = 80) found no difference in cholangitis incidence (34–36).
Ursodeoxycholic acid (UDCA), a hydrophilic secondary bile acid, is used for a variety of cholestatic liver diseases (37). UDCA decreases progression to liver transplant and death in primary biliary cholangitis, and improves liver biochemistry (but not histologic progression, need for transplant, or mortality) in primary sclerosing cholangitis (38, 39). At very high doses (28–30 mg/kg/day) in an adult primary sclerosing cholangitis population, there is a doubling of severe adverse effects as well as an increase in a composite outcome of death, transplantation, or minimal listing criteria (40). This toxicity has been attributed to UDCA metabolism into lithocholic acid, which in animal models may lead to an obstructive cholangiopathy and bile duct destruction (41). UCDA has a choleretic effect through increased chloride and bicarbonate secretion, posttranscriptional modification of hepatobiliary protein levels of key ATP-binding cassette transporters, and cholangiocyte/hepatocyte protection (42–44). In vitro experiments also demonstrate that UDCA moderates the inflammatory response of peripheral blood mononuclear cells and human B cell lymphoma cells with decreased immunoglobulin and cytokine production (45).
Off-label administration of UDCA post-HPE is routine at many centers around the world, with surveys of European and Japanese centers finding near-universal use (31, 46). Despite this, a lack of high-quality literature exists to support a benefit or harm of UDCA. In a prospective single institution study of 16 children in France, after UDCA was discontinued at 18 months post-HPE, liver enzymes worsened in 13 children (47). Six months following resumption of UDCA, liver enzymes, bilirubin, and bile acids decreased, providing the most direct evidence for use of UDCA in biliary atresia. A retrospective cohort study at a single Egyptian hospital compared infants post-HPE who received UDCA (n = 108) versus those who did not (n = 33), suggesting a worse outcome in patients who received UDCA (48). A “successful” outcome, defined as absence of jaundice with aminotransferase levels below twice the upper limit of normal, was achieved in 24.2% that did not receive UDCA versus 10.2% of infants who did (P = 0.04), but there was no difference in the proportion that “failed” (75.7% vs. 77.8%, P = 0.49). The study was limited by treatment selection bias, unclear temporality of the UDCA initiation, and overall poor outcomes. The progression to transplantation as well as actual biochemical values were not reported. Given the paucity of investigation of UDCA in this population, well-designed multicenter randomized controlled trials are needed to both better understand the possible benefit or harm of this medication as well as determine the optimal dose.
Children with biliary atresia are at high risk for malnutrition, growth failure, and vitamin deficiencies (especially fat-soluble vitamins) due to malabsorption, anorexia, and altered nutrient metabolism (49). Growth failure is associated with increased pre- and post-transplant mortality and graft failure (50, 51). Close surveillance combined with aggressive nutritional supplementation is critical. Initial recommendations include high protein intake (3–4 g/kg/day) and up to 130%–150% of recommended calories for age, with sequential stepwise escalation of nutritional support to nasogastric feeds and ultimately parenteral nutrition (49). Medium chain triglyceride supplementation is under study as a source of caloric dense, easily absorbable lipids (NCT05072626). Critically, medium chain triglycerides do not contain the essential polyunsaturated long-chain fatty acids necessary for growth and development. As growth failure is common in biliary atresia and often leads to early liver transplantation, further research is critical in this area to optimize nutrition, as well as further investigating detoxification genomics in biliary atresia (52).
Various viral infections have been proposed as possible inciting events in a subset of biliary atresia pathogenesis, with the greatest investigation of cytomegalovirus (CMV). Between 10% and 74% of biliary atresia patients have positive CMV serology depending on series, and CMV-positive patients have worse outcomes (53). Limited investigation has evaluated the role of antiviral therapy in this population. One retrospective single center trial found that in 36 CMV IgM+ patients, treatment with ganciclovir and/or valganciclovir in the early postoperative period until negative CMV serology resulted in greatly improved clearance of jaundice (75 vs. 21%, P = 0.04) (54). Prospective investigation is critical to demonstrate efficacy of targeted antiviral therapy.
Cholestyramine and phenobarbital are used at some centers despite minimal evidence of effectiveness (31). Cholestyramine is a bile acid sequestrant, while phenobarbital is a choleretic that, in rats, increases the bile acid-independent portion of bile flow (55). The primary trial investigating these agents (from 1986) randomized infants with biliary atresia following surgical repair (primarily Kasai HPE) to no choleretic (n = 12), phenobarbital (n = 21), or cholestyramine (n = 23) for three months (56). There was no difference in total bilirubin or bile acid levels at three months in any group.
There are several emerging therapies under investigation for the treatment of patients with biliary atresia. Ongoing late-stage clinical trials of repurposed drugs that are FDA-approved for non-biliary atresia indications are shown in Table 1.
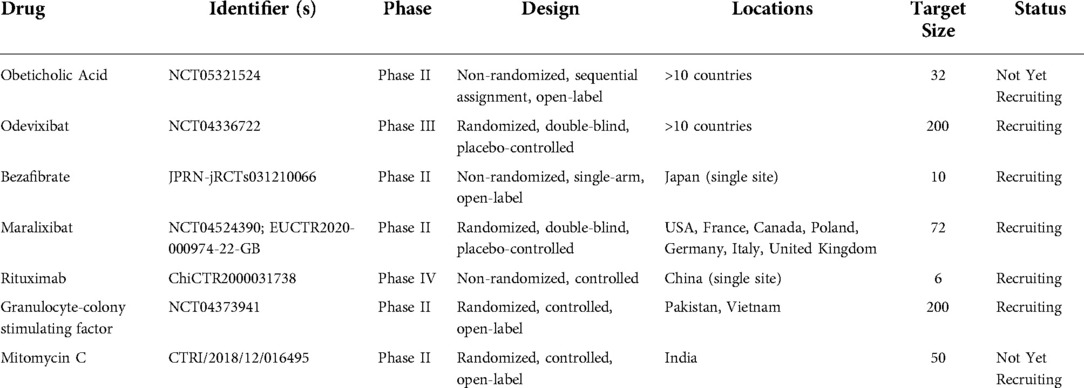
Table 1. Selected ongoing clinical trials investigating repurposed therapeutics for biliary atresia.
Odevixibat is an oral small molecule ileal bile acid transporter inhibitor FDA-approved for pruritus progressive familial intrahepatic cholestasis (PFIC). Inhibition of the ileal bile acid transporter prevents bile acid absorption in the terminal ileum, increasing fecal bile acid excretion. Importantly, this may lead to deficiency in fat-soluble vitamin absorption and the impact on growth and development is unknown. In the PEDFIC 1 trial, daily administration of odevixibat reduced serum bile acids and pruritis in children compared to placebo, and was well tolerated (57). Notably, the PEDFIC 1 trial only included type I and type II PFIC, which are distinct from type III and IV PFIC due to their low-GGT cholestasis. Odevixibat is currently under study in a randomized, double-blind, placebo-controlled phase III trial for biliary atresia (NCT04336722).
Maralixibat (Mirum Pharmaceuticals) is a second oral small molecule ileal bile acid transporter inhibitor FDA-approved for treatment of cholestatic pruritis in Alagille Syndrome (58). In the phase IIb randomized placebo-controlled ICONIC trial, maralixibat treatment reduced serum bile acids and pruritis in children with Alagille syndrome complicated by cholestasis and intractable pruritis (59). A multinational, randomized, double-blind, placebo-controlled phase II trial is currently recruiting patients with biliary atresia to evaluate maralixibat (NCT04524390).
Obeticholic acid is a semi-synthetic bile acid that is a potent Farsenoid X receptor (FXR) agonist FDA-approved to treat UDCA-resistant primary biliary cholangitis (60). FXR is a key ligand-modulated nuclear regulator of hepatic lipogenesis, insulin resistance, bile metabolism, and inflammation (61). In a phase III trial for UDCA-resistant primary biliary cholangitis, obeticholic acid decreased alkaline phosphatase compared to placebo, although pruritis was a common side effect (62). A phase I multicenter open-label single/multiple dose study investigating the safety and tolerability of obeticholic acid in biliary atresia patients is underway in Europe (NCT05321524).
N-acetylcysteine is a hepatoprotective and antioxidant agent used for acetaminophen overdose that drives glutathione synthesis and increases bile flow (63, 64). Previous investigations of N-acetylcysteine in pediatric diseases such as parenteral nutrition-associated cholestasis and bronchopulmonary dysplasia demonstrated excellent safety, although it did not demonstrate efficacy (65–67). A phase II open-label single center trial (NCT03499249) is underway in the United States to evaluate if seven days intravenous infusion of N-acetylcysteine following HPE improves bile flow (68).
Intravenous immunoglobulin (IVIG) is a preparation of immunoglobulin G purified from donor plasma (69). Gamma globulins were first used for primary humoral immunodeficiency; IVIG was later introduced for autoimmune disease in idiopathic thrombocytopenic purpura (70, 71). The immunomodulatory and anti-inflammatory actions of IVIG involve both innate and adaptive immune cells, with effects extending beyond the half-life due to poorly understood mechanisms (72). Due to the proposed centrality of autoimmunity and inflammation in the pathogenesis of biliary atresia, IVIG has been investigated as a potential therapy.
The PRIME trial was a multicenter phase I/IIa trial performed at eight North American centers. Twenty-nine patients with biliary atresia received IVIG and were compared to a historical “placebo” control cohort (n = 64) from the START trial (73). Subjects received three 1 g/kg infusions of IVIG at days 3–5, 30, and 60 post-HPE. The primary outcome was safety: 89.7% of IVIG patients had a serious adverse event compared to 75% in the historical cohort, although no events were directly attributable to IVIG infusion. There was no difference in serum bilirubin, but there was a non-significant trend towards decreased transplant-free survival at 360 days in the IVIG group (58.6% vs. 70.5%, P > 0.05). Randomized controlled trials comparing IVIG and glucocorticoids are necessary.
Several small trials are underway to investigate other repurposed therapies for biliary atresia. Granulocyte-colony stimulating factor (GCSF) stimulates hematopoietic stem cell (HSC) and neutrophil production and mobilization. Typically used in myelosuppression, it recently has been investigated in various liver diseases due to the potential for HSCs to modulate hepatic inflammation and promote regeneration (74). GCSF is currently being investigated in post-HPE type 3 biliary atresia (NCT04373941). Autologous bone marrow mononuclear cell infusion has also been investigated in a small phase I/IIa open-label trial in Vietnam, although the primary outcomes assessed were safety-related and efficacy cannot be determined (75).
Rituximab is an anti-CD20 human/murine chimeric monoclonal antibody that targets B cells. Initially developed for B cell lymphoma, it under investigation for several inflammatory liver diseases. A small non-randomized clinical trial is currently enrolling patients in China (ChiCTR2000031738).
Bezafibrate is a peroxisome proliferator-activated receptor agonist that was found in a randomized, double-blind, placebo-controlled trial to decrease cholestatic pruritus in a combined population of primary sclerosing cholangitis and primary biliary cholangitis patients (76). A study at a single center in Japan for biliary atresia is underway (jRCTs031210066).
The installation of mitomycin C at the portal plate at time of HPE is being investigated in a randomized phase II trial due to its antifibrotic properties (CTRI/2018/12/016495).
The gut microbiome is altered in children with biliary atresia (77). Two small trials have investigated Lactobacillus casei rhamnosus (LGG) supplementation. A small randomized controlled trial found no difference in jaundice, cholangitis, or need for transplantation at two years between LGG (n = 14) and placebo (n = 16) (78). A second trial randomized patients to LGG (n = 10) or neomycin (n = 10) for cholangitis prophylaxis, finding a similar frequency of cholangitis episodes (although less than a small historical control with no prophylaxis) (79). Further research is necessary to evaluate the efficacy of probiotics for cholangitis prophylaxis and other outcomes.
Current adjuvant therapy for biliary atresia post-HPE varies depending on center and is often based upon expert opinion, with few high-quality randomized controlled trials to support individual interventions. Given the rarity of biliary atresia and variation in management, studies are often both underpowered and not necessarily generalizable. The development of multicenter research networks with the infrastructure to conduct rigorous investigations is an encouraging trend.
The challenge in finding novel therapies for biliary atresia is compounded by a poor understanding of its underlying pathogenesis and a lack of representative animal models (80). Investigating emerging therapies found to be beneficial in other cholestatic and inflammatory liver diseases is perhaps the most promising approach to improve the outcomes of post-HPE biliary atresia.
A substantial need exists for well-designed trials to both optimize current therapies and investigate candidate therapeutics for post-HPE biliary atresia. Until then, the single most effective evidence-supported intervention to improve native liver survival is younger age at time of HPE, highlighting the importance of screening and early diagnosis.
SF and MP contributed to the conception and design of the study. SF, TH, AA compiled and reviewed relevant literature. SF, TH, and ST wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
This study is funded in part by NIH grants 5T32HL007734 (SF, TH), 5T32DK007754 (ST), and 5T35HL11084 (AA); the Richard and Sandra Cummings Research Fellowship of the Beth Israel Deaconess Medical Center Department of Surgery (SF); The Boston Children's Hospital Vascular Biology Program; and The Boston Children's Hospital Surgical Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Haber BA, Russo P. Biliary atresia. Gastroenterol Clin North Am. (2003) 32(3):891–911. doi: 10.1016/S0889-8553(03)00049-9
2. Bezerra JA, Wells RG, Mack CL, Karpen SJ, Hoofnagle JH, Doo E, et al. Biliary atresia: clinical and research challenges for the twenty-first century. Hepatology. (2018) 68(3):1163–73. doi: 10.1002/hep.29905
3. Lupo PJ, Isenburg JL, Salemi JL, Mai CT, Liberman RF, Canfield MA, et al. Population-based birth defects data in the United States, 2010-2014: a focus on gastrointestinal defects. Birth Defects Res. (2017) 109(18):1504–14. doi: 10.1002/bdr2.1145
4. Hopkins PC, Yazigi N, Nylund CM. Incidence of biliary atresia and timing of hepatoportoenterostomy in the United States. J Pediatr. (2017) 187:253–7. doi: 10.1016/j.jpeds.2017.05.006
5. Chardot C, Buet C, Serinet MO, Golmard JL, Lachaux A, Roquelaure B, et al. Improving outcomes of biliary atresia: French national series 1986-2009. J Hepatol. (2013) 58(6):1209–17. doi: 10.1016/j.jhep.2013.01.040
6. Ohi R. A history of the Kasai operation: hepatic portoenterostomy for biliary atresia. World J Surg. (1988) 12(6):871–4. doi: 10.1007/BF01655504
7. Fanna M, Masson G, Capito C, Girard M, Guerin F, Hermeziu B, et al. Management of biliary atresia in France 1986 to 2015: long-term results. J Pediatr Gastroenterol Nutr. (2019) 69(4):416–24. doi: 10.1097/MPG.0000000000002446
8. Witt M, van Wessel DBE, de Kleine RHJ, Bruggink JLM, Hulscher JBF, Verkade HJ, et al. Prognosis of biliary atresia after 2-year survival with native liver: a nationwide cohort analysis. J Pediatr Gastroenterol Nutr. (2018) 67(6):689–94. doi: 10.1097/MPG.0000000000002130
9. Arnon R, Annunziato RA, D’Amelio G, Chu J, Shneider BL. Liver transplantation for biliary atresia: is there a difference in outcome for infants? J Pediatr Gastroenterol Nutr. (2016) 62(2):220–5. doi: 10.1097/MPG.0000000000000986
10. Shneider BL, Magee JC, Karpen SJ, Rand EB, Narkewicz MR, Bass LM, et al. Total serum bilirubin within 3 months of hepatoportoenterostomy predicts short-term outcomes in biliary atresia. J Pediatr. (2016) 170:211–217.e1-2. doi: 10.1016/j.jpeds.2015.11.058
11. Sokol RJ, Shepherd RW, Superina R, Bezerra JA, Robuck P, Hoofnagle JH. Screening and outcomes in biliary atresia: summary of a National Institutes of Health workshop. Hepatology. (2007) 46(2):566–81. doi: 10.1002/hep.21790
12. Rabbani T, Guthery SL, Himes R, Shneider BL, Harpavat S. Newborn screening for biliary atresia: a review of current methods. Curr Gastroenterol Rep. (2021) 23(12):28. doi: 10.1007/s11894-021-00825-2
13. Bessho K. Complications and quality of life in long-term survivors of biliary atresia with their native livers. J Pediatr. (2015) 167(6):1202–6. doi: 10.1016/j.jpeds.2015.08.041
14. Kelay A, Davenport M. Long-term outlook in biliary atresia. Semin Pediatr Surg. (2017) 26(5):295–300. doi: 10.1053/j.sempedsurg.2017.09.003
15. Lampela H, Kosola S, Heikkilä P, Lohi J, Jalanko H, Pakarinen MP. Native liver histology after successful portoenterostomy in biliary atresia. J Clin Gastroenterol. (2014) 48(8):721–8. doi: 10.1097/MCG.0000000000000013
16. Kerola A, Lampela H, Lohi J, Heikkilä P, Mutanen A, Jalanko H, et al. Molecular signature of active fibrogenesis prevails in biliary atresia after successful portoenterostomy. Surgery. (2017) 162(3):548–56. doi: 10.1016/j.surg.2017.04.013
17. Moyer K, Kaimal V, Pacheco C, Mourya R, Xu H, Shivakumar P, et al. Staging of biliary atresia at diagnosis by molecular profiling of the liver. Genome Med. (2010) 2(5):33. doi: 10.1186/gm154
18. Nijagal A, Perito ER. Treating biliary atresia: the challenge continues. J Pediatr Gastroenterol Nutr. (2019) 68(4):464–5. doi: 10.1097/MPG.0000000000002302
19. Lu X, Jiang J, Shen Z, Chen G, Wu Y, Xiao X, et al. Effect of adjuvant steroid therapy in type 3 biliary atresia: a single-center, open-label, randomized controlled trial. Ann Surg. (2022). doi: 10.1097/SLA.0000000000005407. [Online ahead of print].
20. Alonso EM, Ye W, Hawthorne K, Venkat V, Loomes KM, Mack CL, et al. Impact of steroid therapy on early growth in infants with biliary atresia: the multicenter steroids in biliary atresia randomized trial. J Pediatr. (2018) 202:179–185.e4. doi: 10.1016/j.jpeds.2018.07.002
21. Bezerra JA, Spino C, Magee JC, Shneider BL, Rosenthal P, Wang KS, et al. Use of corticosteroids after hepatoportoenterostomy for bile drainage in infants with biliary atresia: the START randomized clinical trial. JAMA. (2014) 311(17):1750–9. doi: 10.1001/jama.2014.2623
22. Japanese Biliary Atresia Society, Nio M, Muraji T. Multicenter randomized trial of postoperative corticosteroid therapy for biliary atresia. Pediatr Surg Int. (2013) 29(11):1091–5. doi: 10.1007/s00383-013-3377-6
23. Davenport M, Parsons C, Tizzard S, Hadzic N. Steroids in biliary atresia: single surgeon, single centre, prospective study. J Hepatol. (2013) 59(5):1054–8. doi: 10.1016/j.jhep.2013.06.012
24. Chung HY, Kak Yuen Wong K, Cheun Leung Lan L, Kwong Hang Tam P. Evaluation of a standardized protocol in the use of steroids after Kasai operation. Pediatr Surg Int. (2008) 24(9):1001–4. doi: 10.1007/s00383-008-2200-2
25. Petersen C, Harder D, Melter M, Becker T, Wasielewski RV, Leonhardt J, et al. Postoperative high-dose steroids do not improve mid-term survival with native liver in biliary atresia. Am J Gastroenterol. (2008) 103(3):712–9. doi: 10.1111/j.1572-0241.2007.01721.x
26. Davenport M, Stringer MD, Tizzard SA, McClean P, Mieli-Vergani G, Hadzic N. Randomized, double-blind, placebo-controlled trial of corticosteroids after Kasai portoenterostomy for biliary atresia. Hepatology. (2007) 46(6):1821–7. doi: 10.1002/hep.21873
27. Escobar MA, Jay CL, Brooks RM, West KW, Rescorla FJ, Molleston JP, et al. Effect of corticosteroid therapy on outcomes in biliary atresia after Kasai portoenterostomy. J Pediatr Surg. (2006) 41(1):99–103; discussion 99-103. doi: 10.1016/j.jpedsurg.2005.10.072
28. Karrer FM, Lilly JR. Corticosteroid therapy in biliary atresia. J Pediatr Surg. (1985) 20(6):693–5. doi: 10.1016/S0022-3468(85)80026-9
29. Tyraskis A, Parsons C, Davenport M. Glucocorticosteroids for infants with biliary atresia following Kasai portoenterostomy. Cochrane Database Syst Rev. (2018) 5:CD008735. doi: 10.1002/14651858.CD008735.pub3
30. Calinescu AM, Madadi-Sanjani O, Mack C, Schreiber RA, Superina R, Kelly D, et al. Cholangitis definition and treatment after Kasai hepatoportoenterostomy for biliary atresia: a delphi process and international expert panel. J Clin Med. (2022) 11(3):494. doi: 10.3390/jcm11030494
31. Wong ZH, Davenport M. What happens after Kasai for biliary atresia? A European multicenter survey. Eur J Pediatr Surg. (2019) 29(1):1–6. doi: 10.1055/s-0038-1668146
32. Bu LN, Chen HL, Chang CJ, Ni YH, Hsu HY, Lai HS, et al. Prophylactic oral antibiotics in prevention of recurrent cholangitis after the Kasai portoenterostomy. J Pediatr Surg. (2003) 38(4):590–3. doi: 10.1053/jpsu.2003.50128
33. Chen G, Liu J, Huang Y, Wu Y, Lu X, Dong R, et al. Preventive effect of prophylactic intravenous antibiotics against cholangitis in biliary atresia: a randomized controlled trial. Pediatr Surg Int. (2021) 37(8):1089–97. doi: 10.1007/s00383-021-04916-z
34. Lally KP, Kanegaye J, Matsumura M, Rosenthal P, Sinatra F, Atkinson JB. Perioperative factors affecting the outcome following repair of biliary atresia. Pediatrics. (1989) 83(5):723–6. doi: 10.1542/peds.83.5.723
35. Wu ET, Chen HL, Ni YH, Lee PI, Hsu HY, Lai HS, et al. Bacterial cholangitis in patients with biliary atresia: impact on short-term outcome. Pediatr Surg Int. (2001) 17(5–6):390–5. doi: 10.1007/s003830000573
36. de Vries W, de Langen ZJ, Groen H, Scheenstra R, Peeters PMJG, Hulscher JBF, et al. Biliary atresia in The Netherlands: outcome of patients diagnosed between 1987 and 2008. J Pediatr. (2012) 160(4):638–644.e2. doi: 10.1016/j.jpeds.2011.09.061
37. Kriegermeier A, Green R. Pediatric cholestatic liver disease: review of bile acid metabolism and discussion of current and emerging therapies. Front Med. (2020) 7:149. doi: 10.3389/fmed.2020.00149
38. Harms MH, van Buuren HR, Corpechot C, Thorburn D, Janssen HLA, Lindor KD, et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J Hepatol. (2019) 71(2):357–65. doi: 10.1016/j.jhep.2019.04.001
39. Poropat G, Giljaca V, Stimac D, Gluud C. Bile acids for primary sclerosing cholangitis. Cochrane Database Syst Rev. (2011) 2011(1):CD003626. doi: 10.1002/14651858.CD003626.pub2
40. Lindor KD, Kowdley KV, Luketic VAC, Harrison ME, McCashland T, Befeler AS, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. (2009) 50(3):808–14. doi: 10.1002/hep.23082
41. Sinakos E, Marschall HU, Kowdley KV, Befeler A, Keach J, Lindor K. Bile acid changes after high-dose ursodeoxycholic acid treatment in primary sclerosing cholangitis: relation to disease progression. Hepatology. (2010) 52(1):197–203. doi: 10.1002/hep.23631
42. Beuers U, Spengler U, Zwiebel FM, Pauletzki J, Fischer S, Paumgartner G. Effect of ursodeoxycholic acid on the kinetics of the major hydrophobic bile acids in health and in chronic cholestatic liver disease. Hepatology. (1992) 15(4):603–8. doi: 10.1002/hep.1840150409
43. Marschall HU, Wagner M, Zollner G, Fickert P, Diczfalusy U, Gumhold J, et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology. (2005) 129(2):476–85. doi: 10.1016/j.gastro.2005.05.009
44. Beuers U, Trauner M, Jansen P, Poupon R. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol. (2015) 62(1 Suppl):S25–37. doi: 10.1016/j.jhep.2015.02.023
45. Yoshikawa M, Tsujii T, Matsumura K, Yamao J, Matsumura Y, Kubo R, et al. Immunomodulatory effects of ursodeoxycholic acid on immune responses. Hepatology. (1992) 16(2):358–64. doi: 10.1002/hep.1840160213
46. Sakamoto S, Hashizume N, Yagi M, Sasaki H, Nio M. Japanese biliary atresia society. Postoperative pharmacotherapy for patients with biliary atresia in Japan. Pediatr Int Off J Jpn Pediatr Soc. (2022) 64(1):e14990. doi: 10.1111/ped.14990
47. Willot S, Uhlen S, Michaud L, Briand G, Bonnevalle M, Sfeir R, et al. Effect of ursodeoxycholic acid on liver function in children after successful surgery for biliary atresia. Pediatrics. (2008) 122(6):e1236–1241. doi: 10.1542/peds.2008-0986
48. Kotb MA. Review of historical cohort: ursodeoxycholic acid in extrahepatic biliary atresia. J Pediatr Surg. (2008) 43(7):1321–7. doi: 10.1016/j.jpedsurg.2007.11.043
49. Boster JM, Feldman AG, Mack CL, Sokol RJ, Sundaram SS. Malnutrition in biliary atresia: assessment, management, and outcomes. Liver Transpl. (2022) 28(3):483–92. doi: 10.1002/lt.26339
50. Utterson EC, Shepherd RW, Sokol RJ, Bucuvalas J, Magee JC, McDiarmid SV, et al. Biliary atresia: clinical profiles, risk factors, and outcomes of 755 patients listed for liver transplantation. J Pediatr. (2005) 147(2):180–5. doi: 10.1016/j.jpeds.2005.04.073
51. DeRusso PA, Ye W, Shepherd R, Haber BA, Shneider BL, Whitington PF, et al. Growth failure and outcomes in infants with biliary atresia: a report from the biliary atresia research consortium. Hepatology. (2007) 46(5):1632–8. doi: 10.1002/hep.21923
52. Hausman-Cohen SR, Hausman-Cohen LJ, Williams GE, Bilich CE. Genomics of detoxification: how genomics can be used for targeting potential intervention and prevention strategies including nutrition for environmentally acquired illness. J Am Coll Nutr. (2020) 39(2):94–102. doi: 10.1080/07315724.2020.1713654
53. Fischler B, Czubkowski P, Dezsofi A, Liliemark U, Socha P, Sokol RJ, et al. Incidence, impact and treatment of ongoing CMV infection in patients with biliary atresia in four European centres. J Clin Med. (2022) 11(4):945. doi: 10.3390/jcm11040945
54. Parolini F, Hadzic N, Davenport M. Adjuvant therapy of cytomegalovirus IgM + ve associated biliary atresia: prima facie evidence of effect. J Pediatr Surg. (2019) 54(9):1941–5. doi: 10.1016/j.jpedsurg.2018.12.014
55. Okuda H, Sorrentino D, Alpini G, Tavoloni N, Jones MJ, Kiang CL, et al. Bile acid secretion and pool size during phenobarbital induced hypercholeresis. Proc Soc Exp Biol Med. (1988) 187(2):202–8. doi: 10.3181/00379727-187-42655
56. Vajro P, Couturier M, Lemonnier F, Odièvre M. Effects of postoperative cholestyramine and phenobarbital administration on bile flow restoration in infants with extrahepatic biliary atresia. J Pediatr Surg. (1986) 21(4):362–5. doi: 10.1016/S0022-3468(86)80205-6
57. Thompson RJ, Arnell H, Artan R, Baumann U, Calvo PL, Czubkowski P, et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: a randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. (2022) 7(9):830–42. doi: 10.1016/S2468-1253(22)00093-0
58. Shirley M. Maralixibat: first approval. Drugs. (2022) 82(1):71–6. doi: 10.1007/s40265-021-01649-0
59. Gonzales E, Hardikar W, Stormon M, Baker A, Hierro L, Gliwicz D, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Lancet. (2021) 398(10311):1581–92. doi: 10.1016/S0140-6736(21)01256-3
60. Tian SY, Chen SM, Pan CX, Li Y. FXR: structures, biology, and drug development for NASH and fibrosis diseases. Acta Pharmacol Sin. (2022) 43(5):1120–32. doi: 10.1038/s41401-021-00849-4
61. Jonker JW, Liddle C, Downes M. FXR And PXR: potential therapeutic targets in cholestasis. J Steroid Biochem Mol Biol. (2012) 130(0):147–58. doi: 10.1016/j.jsbmb.2011.06.012
62. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. (2016) 375(7):631–43. doi: 10.1056/NEJMoa1509840
63. Tahan G, Tarcin O, Tahan V, Eren F, Gedik N, Sahan E, et al. The effects of N-Acetylcysteine on bile duct ligation–induced liver fibrosis in rats. Dig Dis Sci. (2007) 52(12):3348–54. doi: 10.1007/s10620-006-9717-9
64. Shattuck KE, Rassin DK, Grinnell CD. N-acetylcysteine (NAC) protects bile flow during amino acid infusion. Pediatr Res. (1999) 45(7):117–117. doi: 10.1203/00006450-199904020-00698
65. Ahola T, Lapatto R, Raivio KO, Selander B, Stigson L, Jonsson B, et al. N-acetylcysteine does not prevent bronchopulmonary dysplasia in immature infants: a randomized controlled trial. J Pediatr. (2003) 143(6):713–9. doi: 10.1067/S0022-3476(03)00419-0
66. Mager DR, Marcon M, Wales P, Pencharz PB. Use of N-acetyl cysteine for the treatment of parenteral nutrition-induced liver disease in children receiving home parenteral nutrition. J Pediatr Gastroenterol Nutr. (2008) 46(2):220–3. doi: 10.1097/MPG.0b013e3180653ce6
67. Soghier LM, Brion LP. Cysteine, cystine or N-acetylcysteine supplementation in parenterally fed neonates. Cochrane Database Syst Rev. (2006) 2006(4):CD004869. doi: 10.1002/14651858.CD004869.pub2
68. Tessier MEM, Shneider BL, Brandt ML, Cerminara DN, Harpavat S. A phase 2 trial of N-acetylcysteine in biliary atresia after Kasai portoenterostomy. Contemp Clin Trials Commun. (2019) 15:100370. doi: 10.1016/j.conctc.2019.100370
69. Berger M. A history of immune globulin therapy, from the harvard crash program to monoclonal antibodies. Curr Allergy Asthma Rep. (2002) 2(5):368–78. doi: 10.1007/s11882-002-0069-z
71. Imbach P, Barandun S, d’Apuzzo V, Baumgartner C, Hirt A, Morell A, et al. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet. (1981) 1(8232):1228–31. doi: 10.1016/S0140-6736(81)92400-4
72. Tha-In T, Bayry J, Metselaar HJ, Kaveri SV, Kwekkeboom J. Modulation of the cellular immune system by intravenous immunoglobulin. Trends Immunol. (2008) 29(12):608–15. doi: 10.1016/j.it.2008.08.004
73. Mack CL, Spino C, Alonso EM, Bezerra JA, Moore J, Goodhue C, et al. A phase I/IIa trial of intravenous immunoglobulin following portoenterostomy in biliary atresia. J Pediatr Gastroenterol Nutr. (2019) 68(4):495–501. doi: 10.1097/MPG.0000000000002256
74. Engelmann C, Herber A, Franke A, Bruns T, Reuken P, Schiefke I, et al. Granulocyte-colony stimulating factor (G-CSF) to treat acute-on-chronic liver failure: a multicenter randomized trial (GRAFT study). J Hepatol. (2021) 75(6):1346–54. doi: 10.1016/j.jhep.2021.07.033
75. Nguyen TL, Nguyen HP, Ngo DM, Ha THT, Mai KA, Bui TH, et al. Autologous bone marrow mononuclear cell infusion for liver cirrhosis after the Kasai operation in children with biliary atresia. Stem Cell Res Ther. (2022) 13(1):108. doi: 10.1186/s13287-022-02762-x
76. de Vries E, Bolier R, Goet J, Parés A, Verbeek J, de Vree M, et al. Fibrates for itch (FITCH) in fibrosing cholangiopathies: a double-blind, randomized, placebo-controlled trial. Gastroenterology. (2021) 160(3):734–743.e6. doi: 10.1053/j.gastro.2020.10.001
77. Song W, Sun LY, Zhu ZJ, Wei L, Qu W, Zeng ZG, et al. Association of gut microbiota and metabolites with disease progression in children with biliary atresia. Front Immunol. (2021) 12:698900. doi: 10.3389/fimmu.2021.698900
78. Orłowska E, Czubkowski P, Wołochowska K, Jarzębicka D, Motyl I, Socha P. Assessment of lactobacillus casei rhamnosus (LGG) therapy in children with biliary atresia - randomized placebo controlled trial. Clin Res Hepatol Gastroenterol. (2021) 45(6):101753. doi: 10.1016/j.clinre.2021.101753
79. Lien TH, Bu LN, Wu JF, Chen HL, Chen AC, Lai MW, et al. Use of lactobacillus casei rhamnosus to prevent cholangitis in biliary atresia after kasai operation. J Pediatr Gastroenterol Nutr. (2015) 60(5):654–8. doi: 10.1097/MPG.0000000000000676
Keywords: biliary atresia, therapeutics, cholestasis, cholangitis, liver transplant, glucocorticoids, survival, children
Citation: Fligor SC, Hirsch TI, Tsikis ST, Adeola A and Puder M (2022) Current and emerging adjuvant therapies in biliary atresia. Front. Pediatr. 10:1007813. doi: 10.3389/fped.2022.1007813
Received: 31 July 2022; Accepted: 26 September 2022;
Published: 14 October 2022.
Edited by:
Satoshi Ieiri, Kagoshima University, JapanReviewed by:
Magd Kotb, Cairo University, Egypt© 2022 Fligor, Hirsch, Tsikis, Adeola and Puder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark Puder TWFyay5QdWRlckBjaGlsZHJlbnMuaGFydmFyZC5lZHU=
Specialty Section: This article was submitted to Pediatric Gastroenterology, Hepatology and Nutrition, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.