 Elizabeth Yen1,2*
Elizabeth Yen1,2* Jill L. Maron3,4
Jill L. Maron3,4- 1Mother Infant Research Institute, Tufts Medical Center, Boston, MA, United States
- 2Department of Pediatrics, Tufts University School of Medicine, Boston, MA, United States
- 3Department of Pediatrics, Women & Infants Hospital of Rhode Island, Providence, RI, United States
- 4Warren Alpert Medical School of Brown University, Providence, RI, United States
Opioid use disorder (OUD) among pregnant women over the last decade has led to more than a fivefold increase in the number of neonates born with withdrawal signs known as Neonatal Abstinence Syndrome (NAS) or Neonatal Opioid Withdrawal Syndrome (NOWS). The impact of prenatal opioid exposure on these neonates remains a public health and research priority due to both its short and long-term effects on offspring. Among the adverse long-term effects associated with OUD is a metabolic syndrome with accompanying cardiovascular comorbidities. The susceptibility to metabolic diseases may begin as early as conception. Neonates born in a setting of prenatal opioid exposure are known to have aberrant early growth, e.g., lower birth weight and smaller head size, and dysregulated feeding behavior that ranges from feeding difficulty to hyperphagia which may predispose these neonates to metabolic syndrome in adulthood. However, studies on this topic are lacking. In this article, we describe the reported association between OUD and metabolic syndrome in adults, animal data linking opioid receptors with the development of diet-induced obesity, the inflammatory modulation of opioids and finally, neonatal salivary transcriptomic data from our laboratory that highlighted the sex-specific impact of opioids on the hypothalamic and reward receptors that regulate feeding behavior in opioid-exposed neonates. There is a great need for future research linking opioids with epigenetic and gene expression changes, as well as neuromodulatory effects in the developing brain, that may underlie the dysregulated feeding, growth, and long-term metabolic and cardiovascular risks for these neonates.
Introduction
The rate of pregnant women misusing opioids has quadrupled between 1999 and 2014 (1), leading to a fivefold increase in Neonatal Abstinence Syndrome (NAS) or Neonatal Opioid Withdrawal Syndrome (NOWS) in the last decade (2). NAS affects multiple organs, including the gastrointestinal system, often resulting in uncoordinated feeding, excessive suck and hyperphagia, feeding intolerance, diarrhea, and poor growth (3, 4). The impact of these aberrant feeding and growth patterns is not well understood, yet they likely contributes to obesity or metabolic syndrome. Opioid use disorder (OUD) is associated with metabolic syndrome, defined as dyslipidemia, insulin resistance, obesity, and hypertension, predisposing these adults to cardiovascular disease, diabetes mellitus (DM), and death (5). Rodents exposed to opioids develop drug-induced obesity and DM type 2 (6), while administration of opioid antagonists reduces opioid craving and appetite for palatable food leading to weight loss (7, 8). The exact mechanisms by which drug addiction and metabolic syndrome are linked together are of growing interest, with some reporting no direct association (9), while others reporting disrupted reward circuitry, i.e., dopamine receptors, as a common denominator between opioid addiction, food dependence and obesity (10–12). To close this knowledge gap, our laboratory examines the expression of hypothalamic and reward genes that regulate feeding behavior in opioid-exposed neonates, with evidence of abnormal reward signaling and behavioral changes that may predispose these neonates to long-term growth and metabolic issues (13).
Metabolic Syndrome in Adults With Opioid Use Disorder
Research has demonstrated that adults with OUD undergoing treatment had increased appetite and weight several months into the program (14, 15), with a 30% prevalence of metabolic syndrome, higher than the general population (16). Methadone exposure, compared to buprenorphine, was associated with worse metabolic syndrome in individuals with heroin use disorder (17). The rate of DM in patients on methadone was much higher than in the general population, while those on buprenorphine had a similar DM risk as the general population (18). This difference could be explained by the role of μ-opioid receptor (MOR) in the metabolic pathway, regulating fatty acid oxidation and promoting fat storage (19). Further, the κ-opioid receptor (KOR) is involved in hepatic lipid metabolism and signals fat storage (20). Therefore, methadone, a full MOR and a KOR agonist, exerts stronger metabolic side effects than buprenorphine, a partial MOR agonist and a KOR antagonist (21). Methadone also antagonizes the N-methyl-D-aspartate receptors, which normally function as an appetite suppressant (22). Despite this clinical evidence, it remains unclear whether these metabolic effects result from the medications or the lifestyle changes related to being in the program. Alternatively, obesity itself is associated with chronic pain and discomfort, leading to an increased use of opioids and other pain-controlling medications (23, 24).
The activation of reward circuitry seems to underlie the propensity for both food and drug ingestion. Increasing evidence shows that common genes implicated in addictive behaviors such as dopamine (DA), proopiomelanocortin (POMC), and leptin receptor (LEPR), may also be implicated in food addiction, particularly palatable sweet and fatty foods (12). Other genes involved in opioid consumption and appetite regulation include transcription factor 7 like 2, melanocortin-4, orexin-1, and opioid receptor μ-1 (25–28). Positron emission tomography (PET) imaging of individuals with obesity showed reductions in the striatal dopamine receptor type 2 (DRD2) that were proportional to body mass index (BMI), evidence that dopamine deficiency may induce pathological eating as a mechanism to offset the reward circuitry (10). Volkow et al. demonstrated that brain imaging of subjects with obesity had reduced striatal DRD2 expression, similar in magnitude to reductions seen in subjects with cocaine addiction, further supporting a similar modulation of reward pathways in individuals with food and drug addictions (29–31).
While opioids induce signs of insulin resistance, metabolic changes, obesity, and diabetes, opioid-antagonists such as naltrexone reduce body weight and indices of metabolic syndrome (32). A clinical trial in patients with schizophrenia treated with olanzapine and randomized to naltrexone demonstrated a significant decrease in fat mass, an increase in fat-free mass, and a trend of improved insulin resistance compared to the placebo (32). These findings suggest that opioid antagonists effectively ameliorate the gain of body fat mass induced by olanzapine, an opioidergic compound. Additionally, an injection of long-acting depot naltrexone in opioid-dependent patients reduced self-reported hedonic and motivational characteristics of sucrose, which, in turn, was associated with diminished cue-induced opioid cravings (33). This study supports the link between opioid neurotransmission, hedonic signaling, and metabolic effects.
Other studies showed a less clear linkage between OUD and metabolic syndrome. A retrospective cross-sectional study of 10,032 subjects showed that drug use was linked to a higher waist circumference but did not find a significant association between drug use and clusters of three or more cardiometabolic disease risk factors (impaired glucose tolerance, insulin resistance, hypertension, dyslipidemia, and central adiposity) (9). Complicating this linkage is that adults with OUD have multiple life stressors and psychiatric comorbidities that may contribute to the development of metabolic syndrome (34, 35). Preclinical studies allow an opportunity to mechanistically understand the impact of opioid use on the development of metabolic syndrome in a more controlled setting.
The Intersection of Food and Drug Addiction: Animal Data
Animal studies have shown that injections of morphine lead to hyperphagia and an increased intake of preferred foods (fat versus carbohydrates), resulting in weight gain. At the same time, naloxone, a MOR antagonist, decreased the intake of the preferred diet (36–39). The stimulation of MOR by injection of morphine into the lateral septum (LS) also reliably increased feeding behavior in a dose-responsive manner. In contrast, injection of high doses of MOR antagonist suppressed feeding (40). Another rodent experiment showed that administration of a highly selective MOR antagonist reduced food seeking after ingestion of palatable food, pointing to the reduction in the hedonic value of food. However, the MOR antagonist also reduced the seeking response before the delivery of palatable food. Therefore, this highly selective MOR antagonist reduced not only the hedonic/reward value of the highly palatable food but also the incentive motivational drivers that control food seeking (41), serving as evidence of direct effects of MOR on feeding behavior.
Animal research also showed that obesity, in turn, modulates the opioid system. Maternal consumption of high-fat diets and obesity was shown to upregulate the expression of MOR in offspring brain (42). Interestingly, there was an interaction between maternal obesity status and offspring sex, with pregestational obesity affecting male offspring only, while gestational obesity affecting both male and female offspring (42). Other studies also demonstrated that dams on high-fat diets produced offspring with a preference for high-fat diets, hyperphagia, and altered expression of MOR in male offspring (43). Furthermore, MOR is also susceptible to pre-pregnancy and gestational obesity, evidenced by reduced methylation in the promoter regions of MOR and the dopamine reuptake transporter, in addition to global DNA hypomethylation in the mouse brain. These behavioral and epigenetic effects were blocked with either the administration of MOR antagonist or methyl donor supplementation (44), mechanistic evidence of the intricate relationships between obesity, opioid receptor activation, and dysregulated feeding behavior.
Neural and Systemic Inflammation of Opioids
In addition to the direct opioid effects on opioid and feeding receptors, opioids also act via non-opioid receptors in the brain, particularly microglia, and induce neuroinflammation through their role as ‘foreign' agents. Microglia, which originate from yolk sac macrophages and migrate into the central nervous system (CNS) during embryogenesis, serve as the brain's innate immune system and regulators of CNS homeostasis (45, 46). Classical activation of microglia stimulates pattern recognition receptors that sense pathogen- and damage-associated molecular patterns in microorganisms, tumor cells, dying cells, or particles released in response to tissue injury and inflammation (47). Figure 1 shows that opioid binding to TLR4 on microglia activates nuclear factor-κB and the release of proinflammatory mediators (46, 48–51). TLR4 activation by opioids is similar to its classic ligand, lipopolysaccharides (52). The opioid-TLR4 signaling also induced extracellular DA increase in rodent NAc accompanied by reward-seeking behavior, e.g., conditioned place preference and opioid self-administration (53). This proinflammatory cascade is thought to actively oppose the analgesic effects of morphine and modulate the reward properties and the development of tolerance (54). Antagonism of TLR4 in animal models prevented the development of morphine tolerance in a dose-dependent manner, and injections of TLR4 agonists produced a novel tolerance to morphine (55). Together, these results confirmed the role of TLR4 in the regulation of morphine tolerance and showed that TLR4 might be a potential therapeutic target for analgesia management.
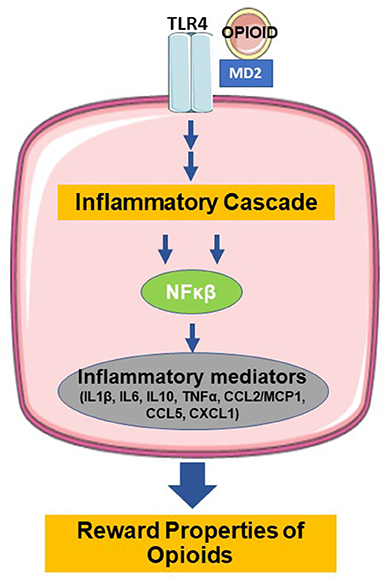
Figure 1. Binding of opioids with TLR4 induces the release of inflammatory mediators and modulation of the reward properties (TLR4=toll-like receptor type 4, MD=myeloid differentiation, NFκB=nuclear factor kappa beta, IL1β =interleukin 1 beta, IL6=interleukin 6, IL10=interleukin 10, TNFα =tumor necrosis factor alpha, CCL2=C-C motif chemokine ligand 2, MCP1=monocyte chemoattractant protein 1, CCL5=C-C motif chemokine ligand 5, CXCL1=C-X-C motif chemokine ligand 1. Figure created using derivatives of “Cellular Biology,” Servier Medical Art (https://smart.servier.com/) under the Creative Commons License Attribution 3.0 Unported License).
Jantzie et al. demonstrated that prenatal methadone elicits a systemic inflammatory response, leading to neuroinflammation, CNS injury, immune system dysfunction, and sustained peripheral immune hyperreactivity (56). Pups of dams implanted with mini-osmotic methadone pumps had significantly increased inflammatory cytokines in their peripheral blood mononuclear cells (PBMC) at P10, with IL1β significantly increased by P21, evidence of prolonged peripheral inflammation. Stimulation of PBMCs in the methadone-exposed pups with LPS resulted in the hypersecretion of cytokines compared to the saline-treated PBMCs, evidence of sustained immune hyperreactivity. Treatment with naloxone dissipated this cytokine response. The cortical section from methadone-exposed rats showed significantly elevated expression of Tlr4, Myd88, and key cytokines and chemokines, evidence of the opioid effects via the TLR4-dependent pathway. Furthermore, biochemical assessment and diffusion tensor imaging of the brain showed decreased myelin maturity and disrupted microstructural brain integrity in the methadone cohort, with executive function and cognitive impairment into adulthood. Prenatal opioid exposure, therefore, adversely modulates systemic inflammatory response accompanied by molecular, structural, and behavioral/cognitive consequences (56).
Neuroinflammation is associated with obesity-driven disorders through the neuroendocrine effects of opioids (57). Animal models and human conditions of stress and depression showed that the neuroimmune impact of opioids dysregulates the hypothalamic-pituitary-adrenal axis and induces the release of peptides and hormones including cortisol. These glucocorticoids, in turn, regulate the expression of inflammatory cytokines in the hypothalamus, hippocampus, and prefrontal cortex (58). Prolonged neuroinflammation may lead to imbalanced hypothalamic hormonal signaling and subsequent dysregulated energy homeostasis, impaired control of food intake, development of insulin resistance, obesity, and cardiovascular disease (59–62).
While neuroinflammation disrupts insulin sensitivity and leptin resistance (63), obesity reciprocally impairs microglia function (64). Central and peripheral inflammation processes are in constant communication and create feedback signaling that predisposes opioid-exposed individuals to adverse neurological and peripheral effects, i.e., cognitive and cardiometabolic deficits (65, 66). Obesity as a pro-inflammatory state disrupts the blood-brain barrier and reduces the expression of tight junction proteins, enhancing the permeability and recruitment of peripheral/plasma constituents with the resultant microglial inflammatory activation (66–68). Neonates with an immature blood-brain barrier are at a higher risk for both peripheral and neurological effects of prenatal opioids and are prone to the vicious cycle depicted in Figure 2.
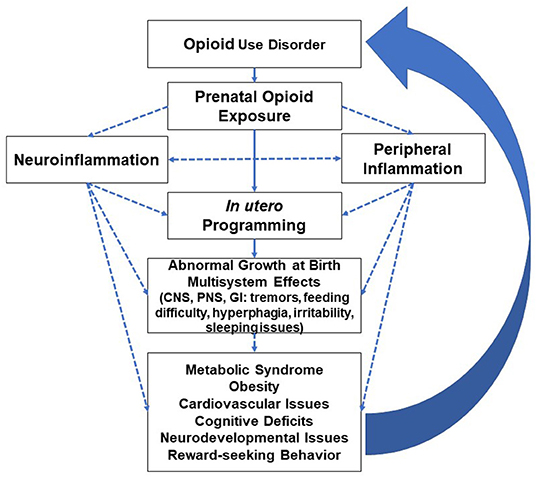
Figure 2. Inflammatory modulation of opioids and multiorgan effects with long-term adverse effects leading to increased predisposition to opioid use disorder (CNS=central nervous system, PNS=peripheral nervous system, GI=gastrointestinal system). Well-studied or known topics are indicated by solid lines; hypothetical topics or knowledge gaps are indicated by dashed lines.
Growth Dysregulation in Opioid-Exposed Neonates and Adverse Metabolic Risks
While studies increasingly found adverse neurodevelopmental outcomes and poor academic performance in children exposed to opioids in utero (69–71), the somatic growth trajectory of these neonates is poorly understood. In utero exposure to opioids and other illicit drugs is linked to prematurity, and smaller overall size, head circumference, and brain volume at birth (72–76), all postulated to be multifactorial, i.e., a poor diet during pregnancy, compromised placental transfer of nutrition, and direct effects of the drugs on fetal growth (77, 78). Exposed neonates have lower growth velocity that persists to 10 years of age (79–81). Others have reported that despite the lower growth parameters at one year, neonates demonstrate catch-up growth starting at 4 months, with a more prominent catch-up in weight by 12 months of age (82). Evidence of catch-up growth has also been reported by Shankaran et al., who reported growth discrepancies disappearing by 2 years of age (for weight and head circumference) and by 3 years of age (for height) (83).
A longitudinal study of 238 children with prenatal cocaine exposure and 323 children without exposure showed a four-fold obesity risk in cocaine-exposed children at 9 years of age (84). These exposed neonates were significantly small for gestational age (SGA) and had more rapid earlier weight gain. When analyzed based on prenatal cocaine-by-alcohol exposure, there was no difference in the prevalence of obesity from birth to 6 years of age. However, by age 7, children who were exposed to cocaine but not alcohol were more obese than those exposed to both substances, to neither substance, or to alcohol but not cocaine. This pattern continued to 9 years of age. Similarly, children with prenatal cocaine but not alcohol exposure had higher BMI Z-scores than those not exposed to either substance, with a difference in the Z-scores noted between 3 and 9 years of age, but not earlier. Obesity was associated with faster early weight gain, high caloric intake, and inadequate exercise. However, a key predictor of obesity at 9 years of age is the rate at which neonates grow from birth to one year, supporting that rapid catch-up growth in the first year of life proved to be most impactful on metabolism later in life (85, 86). A similar phenomenon has been observed in neonates with intrauterine growth restriction. The in utero nutritional deficiency compromises fetal growth and organ differentiation. quickly replaced by ample postnatal nutritional supply and a period of rapid catch-up growth. The unique fetal programming and aberrant postnatal growth trajectory predispose these neonates to a higher incidence of metabolic syndrome, early signs of atherosclerotic diseases, and increased cardiovascular risks (87, 88).
A pilot study looking at air displacement plethysmography measurements in neonates with NAS showed a rapid increase in the mean body mass, fat-free mass, and fat percentage by 4 months of life (89). This study showed that these neonates were born smaller and remained leaner in the first few weeks of life. However, by 4 months of age, neonates with NAS had similar mean body mass and fat-free mass measurements as the non-exposed neonates, with doubling of mean fat mass percentage from 14% to 28% between weeks 4 and 16. The rapid increase in the body mass of neonates with NAS may predispose them to future obesity. Compared to non-exposed neonates, those with NAS also had a larger variability in growth in the first year of life compared to non-exposed neonates (90), evidence of a distinct growth pattern that may place them at risk for subsequent aberrant development.
Molecular and Behavioral Evidence of Sex-Specific Impact of Prenatal Opioids on Neonatal Feeding Regulation and Future Implications
In addition to the abnormal growth parameters described above, opioid-exposed neonates exhibit a unique feeding phenotype that may compound their future metabolic risk. An early withdrawal sign in NAS is poor oral coordination shortly after birth, followed by excessive eating/hyperphagia thereafter (91–93). While most neonates are discharged home without feeding difficulty, many continue to exhibit hyperphagia, consuming up to two-fold the caloric intake of healthy newborns. Using neonatal saliva, our laboratory studied the expression of select hypothalamic and reward genes that regulate feeding—neuropeptide Y2 receptor (NPY2R), POMC, LEPR, DRD2 (13). A higher proportion of opioid-exposed males required pharmacotherapy (41% males vs. 22% females). While there was no significant difference in the gene expression between opioid- and non-exposed neonates, stratification by sex showed that opioid-exposed males had significantly higher expression of DRD2 than females. Furthermore, males who required pharmacotherapy had significantly higher expression of DRD2 and LEPR than females, evidence that prenatal opioid exposure creates an imbalance between reward (DRD2) and anorexigenic (LEPR) signaling. The expression of DRD2 also correlated significantly with volume intake, evidence that hyperphagia may be a compensatory behavior to replace the reward signaling provided in utero. Males with pharmacotherapy requirement consumed an average of 180 mL/kg/day by one week of life and 210 mL/kg/day on the day of discharge, 15% above the amount ingested by females.
The expression of DRD2 in neonates requiring pharmacotherapy also showed a trend of positive correlation with percent weight change at discharge, evidence of early catch-up growth seen in other studies. Our study supports the hypothesis that opioid-exposed neonates exhibit hyperphagia early in life that continues throughout the withdrawal course, with males displaying more molecular and behavioral effects than females. While limited by the small number of subjects and duration (hospital stay), our study provided the foundation for an ongoing study examining the sex-specific impact of prenatal opioid exposure in a larger cohort and beyond the hospital stay. The higher expression of DRD2 may carry important implications for future reward-seeking behavior, albeit drugs or food. Sex-specific findings in our study may explain the predominance of males in OUD (94, 95) and male sex as a risk factor for more severe NAS and pharmacotherapy requirement (96). Additionally, serial salivary DRD2 may predict if opioid-exposed neonates are at risk for an early hyperdopaminergic followed by a hypodopaminergic state, e.g., through a desensitizing process over time. Subjects with chronic OUD and obesity showed evidence of dopamine deficiency that may induce compensatory and pathological behaviors exacerbating their drug or food dependency (30–33).
The higher expression of LEPR in our opioid-exposed male cohort may induce a hyperleptinemia state that triggers insulin resistance and cardiometabolic complications. While leptin serves as satiety signaling in the context of energy surplus (97), chronic hyperleptinemia may lead to blunted responses and obesity (97). A study in adults undergoing weight-loss treatment programs showed associations between food craving, overeating, and genetic polymorphisms involved in addiction, as well as a significant gene-gene interaction between DRD2 and LEPR, which synergistically influences the development of severe obesity (98). Our data highlighted the sex-specific impact of prenatal opioid exposure on the developing brain accompanied by anthropometric and behavioral changes, predisposing these neonates to future reward-seeking behaviors with adverse growth, neurodevelopmental, and cardiometabolic consequences. Given the intersection between food and drug dependence, future studies ought to understand the metabolic consequences of higher reward signaling, abnormal feeding phenotype, and rapid catch-up growth in these neonates.
Conclusion
Animal and human studies highlight the intricate and complex intersection between reward and feeding receptors in the brain, as well as central and peripheral inflammation that may explain the linkage between OUD and metabolic syndrome. Emerging studies in the neonatal population, including ours, indicate that the adverse effects of prenatal opioids start early on in life. However, these topics are understudied and remain significant knowledge gaps in the neonatal and pediatric population, either due to the fragile nature of the population or the limited non-invasive and validated methodologies available to this group. Additionally, the socioeconomic circumstances encountered by families with OUD pose unique challenges that may hinder robust long-term follow-up, further limiting comprehensive understanding of the cardiometabolic impact of prenatal opioids. Despite these obstacles, the field urgently needs robust research to understand the impact of prenatal opioids on brain reward signaling, inflammatory modulation of opioids, hyperphagia, and abnormal growth, including rapid catch-up growth in these neonates, and long-term cardiometabolic consequences. Animal and human adult data on the inflammatory impact of opioids provide an opportunity to develop and utilize non-opioid interventions, such as anti-inflammatory agents, in dealing with postnatal withdrawal signs and future metabolic syndrome. Furthermore, research in these topics will enable non-pharmacological interventions, such as nutritional programs and cognitive behavioral therapy, that could potentially ameliorate inflammation and prevent the development of metabolic syndrome in opioid-exposed infants and children. Well-funded public health research will enable precision and personalized medicine, such as more individualized and balanced nutrition to reduce the likelihood of early rapid catch-up growth, diligent monitoring of nutritional intake over time, exercise regimen and cognitive behavioral interventions that reduce the neuroinflammatory impact of opioids on future neurodevelopment and somatic growth, personalized health surveillance, e.g., early lipid screening, all of which may halt the vicious cycle that predisposes these growing neonates to adverse health outcomes in adulthood.
Author Contributions
EY: paper concept, writing and editing of manuscript, figure concept and creation. JM: paper concept and manuscript editing. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the Forbes Family for their generous support on the open access publication of this manuscript.
References
1. Haight SC, Ko JY, Tong VT, Bohm MK, Callaghan WM. Opioid use disorder documented at delivery hospitalization—United States. 1999-2014. MMWR Morb Mortal Wkly Rep. (2018) 67:845–9. doi: 10.15585/mmwr.mm6731a1
2. Winkelman TNA, Villapiano N, Kozhimannil KB, Daivs MM, Patrick SW. Incidence and costs of neonatal abstinence syndrome among infants with Medicaid: 2000-2014. Pediatrics. (2018) 141:e20173520. doi: 10.1542/peds.2017-3520
3. Patrick SW, Barfield WD, Poindexter B. Neonatal opioid withdrawal syndrome. Pediatrics. (2020) 146:e2020029074. doi: 10.1542/peds.2020-029074
4. Kocherlakota P. Neonatal abstinence syndrome. Pediatrics. (2004) 134:e547–61. doi: 10.1542/peds.2013-3524
5. Dydyk AM, Jain NK, Gupta M. Opioid Use Disorder. Treasure Island, FL: StatPearls Publishing (2021). Available from: https://www.ncbi.nlm.nih.gov/books/NBK553166/
6. Toorie AM, Vassoler FM, Qu F, Schonhoff CM, Bradburn S, Murgatroyd CA, et al. A history of opioid exposure in females increases the risk of metabolic disorders in their future male offspring. Addict Biol. (2021) 26:e12856. doi: 10.1111/adb.12856
7. Nogueiras R, Romero-Pic? A, Vasquez MJ, Novelle MG, L?pez M, Diéguez C. The opioid system and food intake: homeostatic and hedonic mechanisms. Obes Facts. (2012) 5:196–207. doi: 10.1159/000338163
8. Balhara YP, Kuppili PP, Shukla A, Chawla N, Gupta R. Which criteria to use to identify metabolic syndrome among patients with addictive disorders? Observations among patients with alcohol and opioid dependence syndrome. Indian J Endocrinol Metab. (2018) 22:565–8. doi: 10.4103/ijem.IJEM_617_17
9. Shah K, Joshi VV, Arinze NC, Bodicherla KP, Ghimiree S, Singh R, et al. Association of opioid use and cardiometabolic disease risk factor: evidence from the 2009-2018 National Health and Nutritional Examination Survey. Cureus. (2020) 12:e8719. doi: 10.7759/cureus.8719
10. Wang G-J, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W, et al. Brain dopamine and obesity. Lancet. (2001) 357:354–7. doi: 10.1016/S0140-6736(00)03643-6
11. Volkow ND, Wang G-J, Tomasi D, Baler R. The addictive dimensionality of obesity. Biol Psychiatry. (2013) 73:811–8. doi: 10.1016/j.biopsych.2012.12.020
12. Heber D, Carpenter CL. Addictive genes and the relationship to obesity and inflammation. Mol Neurobiol. (2011) 44:160–5. doi: 10.1007/s12035-011-8180-6
13. Yen E, Kaneko-Tarui T, Ruthazer R, Harvey-Wilkes H, Hassaneen M, Maron JL. Sex-dependent gene expression in neonates with neonatal opioid withdrawal syndrome. J Pediatr. (2019) 214:60–5.e2. doi: 10.1016/j.jpeds.2019.07.032
14. Baykara S, Alban K. The effects of buprenorphine/naloxone maintenance treatment on sexual dysfunction, sleep and weight gain in opioid use disorder patients. Psych Res. (2019) 272:450–3. doi: 10.1016/j.psychres.2018.12.153
15. Parvaresh N, Sabani AR, Mazari S, Gilani H. A study of sexual function, sleep, and weight gain status of patients after 6 months of methadone maintenance treatment. Addict Health. (2015) 7:24–9.
16. Mattoo SK, Chakraborty K, Basu D, Gosh A, Kumar V, Kulhara P. Prevalence and correlates of metabolic syndrome in alcohol and opioid dependent inpatients. Indian J Med Res. (2011) 134:341–8.
17. Vallecillo G, J?se Robles M, Torrens M, Samos P, Roquer A, Martires PK, et al. Metabolic syndrome among individuals with heroin use disorders on methadone therapy: Prevalence, characteristics, and related factors. Substance Abuse J. (2018) 39:46–51. doi: 10.1080/08897077.2017.1363122
18. Nolan LJ, Scagnelli LM. Preference for sweet foods and higher body mass index in patients being treated in long-term methadone maintenance. Subst Use Misuse. (2007) 42:1555–66. doi: 10.1080/10826080701517727
19. Tabarin A, Diz-Chaves Y. del Carmen Carmona M, Catargi B, Zorrilla EP, Roberts AJ, et al. Resistance to diet-induced obesity in μ-opioid receptor-deficient mice. Diabetes. (2005) 54:3510–6. doi: 10.2337/diabetes.54.12.3510
20. Czyzyk TA, Nogueiras R, Lockwood JF, McKinzie JH, Coskun T, Pintar JE, et al. Kappa-opioid receptors control the metabolic response to a high-energy diet in mice. FASEB J. (2010) 24:1151–9. doi: 10.1096/fj.09-143610
21. Jordan CJ, Cao J, Newman AH Xi ZX. Progress in agonist therapy for substance use disorders: lessons learned from methadone and buprenorphine. Neuropharmacology. (2019) 158:107609. doi: 10.1016/j.neuropharm.2019.04.015
22. Guard DB, Swartz TD, Ritter RC, Burns GA, Covasa M. Blockade of hindbrain NMDA receptors containing NR2 subunits increases sucrose intake. Am J Physiol Regul Integr Comp Physiol. (2009) 296:R921–8. doi: 10.1152/ajpregu.90456.2008
23. Stokes A, Berry KM, Collins JM, Hsiao C-W, Waggoner JR, Johnston SS, et al. The contribution of obesity to prescription opioid use in the United States. Pain. (2019) 160:2255–62. doi: 10.1097/j.pain.0000000000001612
24. Stokes A, Lundberg DJ, Sheridan B, Hempstead K, Moron NE, Lasser KE, et al. Association of obesity with prescription opioids for painful conditions in patients seeking primary care in the US. JAMA Netw Open. (2020) 3:e202012. doi: 10.1001/jamanetworkopen.2020.2012
25. Ip W, Chiang YT, Jin T. The involvement of the wnt signaling pathway and TCF7L2 in diabetes mellitus: the current understanding, dispute, and perspective. Cell Biosci. (2012) 2:28. doi: 10.1186/2045-3701-2-28
26. Garfield AS Li C, Madara JC, Shah BP, Webber E, Steger JS, et al. A neural basis for melanocortin-4 receptor-regulated appetite. Nat Neurosci. (2015) 18:863–71. doi: 10.1038/nn.4011
27. Kasai S, Ikeda K. Pharmacogenomics of the human μ-opioid receptor. Pharmacogenomics. (2011) 12:1305–20. doi: 10.2217/pgs.11.68
28. Fragale JE, Pantazis CB, James MH, Aston-Jones G. The role of orexin-1 receptor signaling in demand for the opioid fentanyl. Neuropsychopharmacol. (2019) 44:1690–7. doi: 10.1038/s41386-019-0420-x
29. Volkow ND, Wise RA. How can drug addiction help us understand obesity? Nature Neurosci. (2005) 8:555–60. doi: 10.1038/nn1452
30. Koob GF, Volkow ND. Neurobiology of addiction. Lancet Psychiatr. (2016) 3:760–83. doi: 10.1016/S2215-0366(16)00104-8
31. Volkow ND, Michalides M, Baler R. The neuroscience of drug reward and addiction. Physiol Rev. (2019) 99:2015–40. doi: 10.1152/physrev.00014.2018
32. Taveira TH, Wu WC, Tschibelu E, Borsook D, Simonson DC, Yamamoto R, et al. The effect of naltrexone on body fat mass in olanzapine-treated schizophrenic or schizoaffective patients: a randomized double-blind placebo-controlled pilot study. J Psychopharmacol. (2014) 28:395–400. doi: 10.1177/0269881113509904
33. Langleben DD, Busch EL, O'Brien CP, Elman I. Depot naltrexone decreases rewarding properties of sugar in patients with opioid dependence. Psychopharmacol. (2012) 220:559–64. doi: 10.1007/s00213-011-2503-1
34. Wiss DA. A biopsychosocial overview of the opioid crisis: considering nutrition and gastrointestinal health. Front Public Health. (2019) 7:193. doi: 10.3389/fpubh.2019.00193
35. Osadchiy V, Mayer EA, Bhatt R, Labus JS, Gao L, Kilpatrick LA, et al. History of early life adversity is associated with increased food addiction and sex-specific alterations in reward network connectivity in obesity. Obes Sci Pract. (2019) 5:416–36. doi: 10.1002/osp4.362
36. Gosnell BA, Krahn DD, Majchrzak MJ. The effects of morphine on diet selection are dependent upon baseline diet preferences. Pharmacol Biochem Behav. (1990) 37:207–12. doi: 10.1016/0091-3057(90)90322-9
37. Glass MJ, Grace M, Cleary JP, Billington CJ, Levine AS. Potency of naloxone's anorectic effect in rats is dependent on diet preference. Am J Physiol. (1996) 271:R217–21. doi: 10.1152/ajpregu.1996.271.1.R217
38. Mena JD, Sadeghian K, Baldo BA. Induction of hyperphagia and carbohydrate intake by μ-oioid receptor stimulation in circumscribed regions of frontal cortex. J Neurosci. (2011) 31:3249–60. doi: 10.1523/JNEUROSCI.2050-10.2011
39. Zhang M, Kelley AE. Enhanced intake of high-fat food following striatal mu-opioid stimulation: microinjection mapping and fos expression. Neurosci. (2000) 99:267–77. doi: 10.1016/S0306-4522(00)00198-6
40. Calderwood MT, Tseng A, Stanley BG. Lateral septum mu opioid receptors in stimulation of feeding. Brain Res. (2020) 1734:146648. doi: 10.1016/j.brainres.2020.146648
41. Giuliano C, Robbins TW, Nathan PDJ, Bullmore ET, Everitt BJ. Inhibition of opioid transmission at the μ-opioid receptor prevents both food seeking and binge-like eating. Neuropsychopharm. (2012) 37:2643–52. doi: 10.1038/npp.2012.128
42. Grissom NM, Lyde R, Christ L, Sasson IE, Carlin JL, Vitins AP, et al. Obesity at conception programs the opioid system in the offspring brain. Neuropsychopharmacol. (2014) 39:801–10. doi: 10.1038/npp.2013.193
43. Ong ZY, Wanasuria AF, Lin MZ, Hiscock J, Muhlhausler BS. Chronic intake of a cafeteria diet and subsequent abstinence. Sex-specific effects on gene expression in the mesolimbic reward system. Appetite. (2013) 65:189–99. doi: 10.1016/j.appet.2013.01.014
44. Carlin J, George R, Reyes TM. Methyl donor supplementation blocks the adverse effects of maternal high fat diet on offspring physiology. PLoS ONE. (2013) 85:e63549. doi: 10.1371/journal.pone.0063549
45. Ginhoux F, Prinz M. Origins of microglia: current concepts and past controversies. Cold Spring Harbor Perspect Biol. (2015) 7:a020537. doi: 10.1101/cshperspect.a020537
46. Saijo K, Glass CK. Microglia cell origin and phenotypes in health and disease. Nat Rev Immunol. (2011) 11:775–87. doi: 10.1038/nri3086
47. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. (2012) 249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x
48. Buchanan MM, Hutchinson M, Watkins LR, Yin H. Toll-like receptor 4 in CNS pathologies. J Neurochem. (2010) 114:13–27. doi: 10.1111/j.1471-4159.2010.06736.x
49. Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. (2011) 25:181–213. doi: 10.1016/j.bbi.2010.10.015
50. Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun. (2010) 24:83–95. doi: 10.1016/j.bbi.2009.08.004
51. Hutchinson MR, Northcutt AL Hiranita T, Wang X, Lewis SS, Thomas J, et al. Opioid activation of toll-like receptor 4 contributes to drug reinforcement. J Neurosci. (2012) l32:11187–200. doi: 10.1523/JNEUROSCI.0684-12.2012
52. Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci USA. (2012) 109:6325–30. doi: 10.1073/pnas.1200130109
53. Hutchinson MR, Lewis SS, Coats BD, Rezvani N, Zhang Y, Wiseler JL, et al. Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neurosci. (2010) 167:880–93. doi: 10.1016/j.neuroscience.2010.02.011
54. Eidson LN, Murphy AZ. Persistent peripheral inflammation attenuates morphine-induced periaqueductal gray glial cell activation and analgesic tolerance in the male rat. J Pain. (2013) 14:393–405. doi: 10.1016/j.jpain.2012.12.010
55. Cui C, Shurtleff D, Harris RA. Neuroimmune mechanisms of alcohol and drug addiction. Int Rev Neurobiol. (2014) 118:1–12. doi: 10.1016/B978-0-12-801284-0.00001-4
56. Jantzie LL, Maxwell JR, Newville JC, Yellowhair TY, Kitase Y, Madurai N, et al. Prenatal opioid exposure: the next neonatal neuroinflammatory disease. Brain Behav Immun. (2020) 84:45–58. doi: 10.1016/j.bbi.2019.11.007
57. Hueston CM, Deak T. The inflamed axis: the interaction between stress, hormones, and the expression of inflammatory-related genes within key structures comprising the hypothalamic-pituitary-adrenal axis. Physiol Behav. (2014) 124:77–91. doi: 10.1016/j.physbeh.2013.10.035
58. Munhoz CD, Lepsch LB, Kawamoto EM, Malta MB, de Sá Lima L, Avellar MC, et al. Chronic unpredictable stress exacerbates lipopolysaccharide-induced activation of nuclear factor-κB in the frontal cortex and hippocampus via glucocorticoid secretion. J Neurosci. (2006) 26:3813–20. doi: 10.1523/JNEUROSCI.4398-05.2006
59. Cai D, Liu T. Hypothalamic inflammation: a double-edged sword to nutritional diseases. Ann NY Acad Sci. (2011) 1243:E1–39. doi: 10.1111/j.1749-6632.2011.06388.x
60. Thaler JP, Schwartz MW. Inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrionol. (2010) 151:4109–15. doi: 10.1210/en.2010-0336
61. Jais A, Brunning JC. Hypothalamic inflammation in obesity and metabolic disease. J Clin Investig. (2017) 127:24–32. doi: 10.1172/JCI88878
62. Purkayastha S, Zhang H, Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nat Med. (2011) 17:883–7. doi: 10.1038/nm.2372
63. Kleinridders A, Schenten D, Konner AC, Belgardt BF, Mauer J, Okamura T, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. (2009) 10:249–59. doi: 10.1016/j.cmet.2009.08.013
64. Alexaki VI. The impact of obesity on microglial function: immune, metabolic and endocrine perspectives. Cells. (2021) 10:1584. doi: 10.3390/cells10071584
65. Dantzer R. Neuroimmune interactions: from the brain to the immune system and vice versa. Physiol Rev. (2018) 8:477–504. doi: 10.1152/physrev.00039.2016
66. Dionysopoulou S, Charmandari E, Bargioat A, Vlahos N, Mastorakos G, Valsamkis G. The role of hypothalamic inflammation in diet-induced obesity and its association with cognitive and mood disorder. Nutrients. (2021) 13:498. doi: 10.3390/nu13020498
67. Balasubramanian P, Kiss T, Tarantini S, Nyúl-T?th A, Ahire C, Yabluchanskiy A, et al. Obesity-induced cognitive impairment in older adults: a microvascular perspective. Am J Physiol Heart Circ Physiol. (2021) 320:H740–61. doi: 10.1152/ajpheart.00736.2020
68. Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. (2012) 3:1227. doi: 10.1038/ncomms2230
69. Yeoh SL, Eastwood J, Wright IM, Morton R, Melhuish E, Ward M, et al. Cognitive and motor outcomes of children with prenatal opioid exposure: a systematic review and meta-analysis. JAMA Netw Open. (2019) 2:e197025. doi: 10.1001/jamanetworkopen.2019.7025
70. Larson JJ, Graham DL, Singer LT, Beckwith AM, Terplan M, Davis JM, et al. Cognitive and behavioral impact on children exposed to opioids during pregnancy. Pediatrics. (2019) 144:e20190514. doi: 10.1542/peds.2019-0514
71. Oei JL, Melhuish E, Uebel H, Azzam N, Breen C, Burns L, et al. Neonatal abstinence syndrome and high school performance. Pediatrics. (2017) 139:e20162651. doi: 10.1542/peds.2016-2651
72. Brogly S. Maternal and child health after prenatal opioid exposure. JAMA Netw Open. (2019) 2:e196428. doi: 10.1001/jamanetworkopen.2019.6428
73. Merhar SL, Kline JE, Braimah A, Kline-Fath BM, Tkach JA, Altaye M, et al. Prenatal opioid exposure is associated with smaller brain volumes in multiple regions. Pediatr Res. (2021) 90:397–402. doi: 10.1038/s41390-020-01265-w
74. Towers CV, Hyatt BW, Visconti KC, Chernicky L, Chattin K, Fortner KB. Neonatal head circumference in newborns with neonatal abstinence syndrome. Pediatrics. (2019) 143:e20180541. doi: 10.1542/peds.2018-0541
75. Yuan Q, Rubic M, Seah J, Rae C, Wright IMR, Kaltenbach K, et al. Do maternal opioids reduce neonatal regional brain volumes? A pilot study J Perinatol. (2014) 34:909–13. doi: 10.1038/jp.2014.111
76. Monnelly VJ, Anblagan D, Quigley A, Cabez MB, Cooper ES, Mactier H, et al. Prenatal methadone exposure is associated with altered neonatal brain development. Neuroimage Clin. (2018) 18:9–14. doi: 10.1016/j.nicl.2017.12.033
77. Serra AE, Lemon L, Mokhtari NB, Parks WT, Catov JM, Venkataram R, et al. Delayed villous maturation in term placentas exposed to opioid maintenance therapy: a retrospective cohort study. Am J Obstet Gynecol. (2017) 216:418.e1–5. doi: 10.1016/j.ajog.2016.12.016
78. Bandstra ES, Morrow CE, Mansoor E, Accornero VH. Prenatal drug exposure: infant and toddler outcomes. J Addictive Dis. (2010) 29:245–58. doi: 10.1080/10550881003684871
79. Favara MT, Smith J, Friedman D, Lafferty M, Carola D, Adeniyi-Jones S, et al. Growth failure in infants with neonatal abstinence syndrome in the neonatal intensive care unit. J Perinatol. (2021). doi: 10.1038/s41372-021-01183-7
80. Covington CY, Nordstrom-Klee B, Ager J, Sokol R, Delaney-Black V. Birth to age 7 growth of children prenatally exposed to drugs: a prospective cohort study. Neurotoxicol Teratol. (2002) 24:489–96. doi: 10.1016/S0892-0362(02)00233-7
81. Richardson GA, Goldschmidt L, Larkby C. Effects of prenatal cocaine exposure on growth: a longitudinal analysis. Pediatrics. (2007) 120:1017–27. doi: 10.1542/peds.2006-3482
82. Vance JC, Chant DC, Tudehope DI, Gray PH, Hayes AJ. Infants born to narcotic dependent mothers: physical growth patterns in the first 12 months of life. J Paediatr Child Health. (1997) 33:504–8. doi: 10.1111/j.1440-1754.1997.tb01659.x
83. Shankaran S, Lester BM, Das B, Bauer CR, Bada H, LaGlasse L, et al. Impact of maternal substance use during pregnancy on childhood outcome. Semin Fetal Neonatal Med. (2007) 12:143–50. doi: 10.1016/j.siny.2007.01.002
84. LaGlasse L, Gaskins RB, Bada HS, Shankaran S, Liu J, Lester BM, et al. Prenatal cocaine exposure and childhood obesity at nine years. Neurotoxicol Teratol. (2011) 33:188–97. doi: 10.1016/j.ntt.2010.11.002
85. Silveira PP, Pokhvisneva I, Gaudreau H, Rifkin-Graboi A, Broekman BFP, Steiner M, et al. Birth weight and catch up growth are associated with childhood impulsivity in two independent cohorts. Sci Rep. (2018) 8:13705. doi: 10.1038/s41598-018-31816-5
86. Fabricius-Bjerre S, Jensen RB, Færcj K, Larsen T, Mølgaard C, Michaelsen KF, et al. Impact of birth weight and early infant weight gain on insulin resistance and associated cardiovascular risk factors in adolescence. PLoS ONE. (2011) 6:e20595. doi: 10.1371/journal.pone.0020595
87. Menendez-Castro C, Rascher W, Hartner A. Intrauterine growth restriction—impact on cardiovascular disease later in life. Mol Cell Pediatr. (2018) 5:4. doi: 10.1186/s40348-018-0082-5
88. Hwang IT. Long-term care, from neonatal period to adulthood, of children born small for gestational age. Clin Ped Endocrinol. (2019) 28:97–103. doi: 10.1297/cpe.28.97
89. Corr TE, Schaefer EW, Paul IM. Body composition during the first 4 months in infants affected by neonatal abstinence syndrome: a pilot study. J Dev Orig Health Dis. (2021) 1–8. doi: 10.1017/S2040174421000052
90. Corr TE, Schaefer EW, Paul IM. Growth during the first year in infants affected by neonatal abstinence syndrome. BMC Pediatr. (2018) 18:343. doi: 10.1186/s12887-018-1327-0
91. Taylor K, Maguire D. A review of feeding practices in infants with neonatal abstinence syndrome. Adv Neonatal Care. (2020) 20:430–9. doi: 10.1097/ANC.0000000000000780
92. Shephard R, Greenough A, Johnson K, Gerada C. Hyperphagia, weight gain, and neonatal drug withdrawal. Acta Paediatr. (2002) 91:951–3. doi: 10.1111/j.1651-2227.2002.tb02883.x
93. Martinez A, Kastner B, Taeusch HW. Hyperphagia in neonates withdrawing from methadone. Arch Dis Child Fetal Neonatal Ed. (1999) 80:F178–82. doi: 10.1136/fn.80.3.F178
94. Blanco C, Volkow ND. Management of opioid use disorder in the USA: present status and future directions. Lancet. (2019) 10182:1760–72. doi: 10.1016/S0140-6736(18)33078-2
95. Gao W, Leighton C, Chen YM, Jones J, Mistry P. Predicting opioid use disorder and associated risk factors in a Medicaid managed care population. Am J Manag Care. (2021) 27:148–54. doi: 10.37765/ajmc.2021.88617
96. Charles MK, Cooper WO, Jansson LM, Dudley J, Slaughter JC, Patrick SW. Male sex associated with increased risk of neonatal abstinence syndrome. Hosp Pediatr. (2017) 7:328–34. doi: 10.1542/hpeds.2016-0218
97. Knight ZA, Hannan KS, Greenberg ML, Friedman JM. Hyperleptinemia is required for the development of leptin resistance. PLoS ONE. (2010) 5:e11376. doi: 10.1371/journal.pone.0011376
98. Carpenter CL, Wong AM Li Z, Noble EP, Heber D. Association of dopamine D2 receptor and leptin receptor genes with clinically severe obesity. Obesity. (2013) 21:E467–73. doi: 10.1002/oby.20202
Keywords: opioid epidemic, Neonatal Abstinence Syndrome, Inflammation, Neuromodulation, Feeding dysregulation, metabolic syndrome
Citation: Yen E and Maron JL (2022) Aberrant Feeding and Growth in Neonates With Prenatal Opioid Exposure: Evidence of Neuromodulation and Behavioral Changes. Front. Pediatr. 9:805763. doi: 10.3389/fped.2021.805763
Received: 30 October 2021; Accepted: 28 December 2021;
Published: 21 January 2022.
Edited by:
Lauren Jantzie, Johns Hopkins University, United StatesReviewed by:
Sinno Simons, Erasmus Medical Center, NetherlandsCopyright © 2022 Yen and Maron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth Yen, ZXllbkB0dWZ0c21lZGljYWxjZW50ZXIub3Jn