
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 26 January 2022
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.805575
This article is part of the Research Topic Developmental Delay and Intellectual Disability View all 22 articles
 Tingyu Rong1,2,3†
Tingyu Rong1,2,3† Ruen Yao4†
Ruen Yao4† Yujiao Deng1,2,3
Yujiao Deng1,2,3 Qingmin Lin1,2,3
Qingmin Lin1,2,3 Guanghai Wang1,2,3
Guanghai Wang1,2,3 Jian Wang4Fan Jiang1,2,3*
Jian Wang4Fan Jiang1,2,3* Yanrui Jiang1,2,3*
Yanrui Jiang1,2,3*Mutations in the human O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthase gene (SEPSECS) are associated with progressive cerebello-cerebral atrophy (PCCA), also known as pontocerebellar hypoplasia type 2D (PCH2D). Early-onset profound developmental delay, progressive microcephaly, and hypotonia that develops toward severe spasticity have been previously reported with SEPSECS mutations. Herein we report a case with severe global developmental delay, myogenic changes in the lower limbs, and insomnia, but without progressive microcephaly and brain atrophy during infancy and toddlerhood in a child harboring the SEPSECS missense variant c.194A>G (p. Asn65Ser) and a novel splicing mutation c.701+1G>A. With these findings we communicate the first Chinese SEPSECS mutant case, and our report indicates that SEPSECS mutations can give rise to a milder phenotype.
The SEPSECS gene encodes O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthase (SepSecS), the final enzyme that catalyzes the Sep-tRNA to Sec-tRNA conversion, which is used in the synthesis of selenocysteine (1, 2). SepSecS is essential for the synthesis of selenoproteins, which are expressed unevenly and stably in the brain (3). The biallelic deletion of Trsp in mice (which impedes selenoprotein synthesis) resulted in cerebellar hypoplasia, seizure, and developmental delay (4)—phenotypes that are congruent with those described in previous clinical reports of SEPSECS mutations (5–11). Several investigators identified mutations in human SEPSECS as the cause of severe, early-onset neurological symptoms that were later characterized as causing pontocerebellar hypoplasia type 2D (PCH2D) (5–11)—with or without signs of mitochondrial deficiencies that include elevated blood lactate (7), visual impairment, and myopathy (9). Although harboring different mutant alleles, patients presented remarkably similar phenotypes typified by an autosomal recessive progressive microcephaly with profound developmental delay, progressive brain atrophy, and hypotonia (6, 7, 9). Repeated magnetic resonance imaging (MRI) of affected individuals revealed progressive cerebellar atrophy followed by cerebral atrophy involving both white and gray matter. Brain atrophy is recognized at various developmental stages, but principally within the first 18 months of life (6, 7, 9). However, SEPSECS mutations have been identified in three milder late-onset patients, with cerebellar atrophy first recognized by MRI at 9, 16, and 18 years of age (8, 10).
We hereby report on a mild phenotype without progressive microcephaly and brain atrophy up to 3 years of age in a Chinese pediatric patient who harbored biallelic SEPSECS mutations.
A 2-year-old girl was referred to the Department of Developmental and Behavioral Pediatrics due to severe global development delay and insomnia (case timeline presented in Figure 1). Clinical examination revealed left esotropia, severe muscle hypotonia, and decreased deep tendon reflexes, particularly with respect to both her lower limbs. The patient was born naturally at full-term by healthy non-consanguineous parents. Her birth weight was 3,750 g (86th percentile) and length was 52 cm (94th percentile); head circumference at birth was missing. The patient manifested low muscle tone and weak sucking upon birth, and was still unable to raise her head and turn over her body at 4 months of age. A developmental assessment at 4 months showed that the development quotients of gross motor control, fine motor control, language, reaction to objects, and reaction to people were 55, 66, 38, 28, and 28, respectively. No discomfort or malformation from heart, thorax, kidneys, genitourinary or extremities were mentioned or found. A brain MRI was performed at 5 months, which showed high signal intensities in the bilateral pallidum upon T2W1; her electroencephalography was normal. The patient was subsequently referred to the rehabilitation department for rehabilitation training.
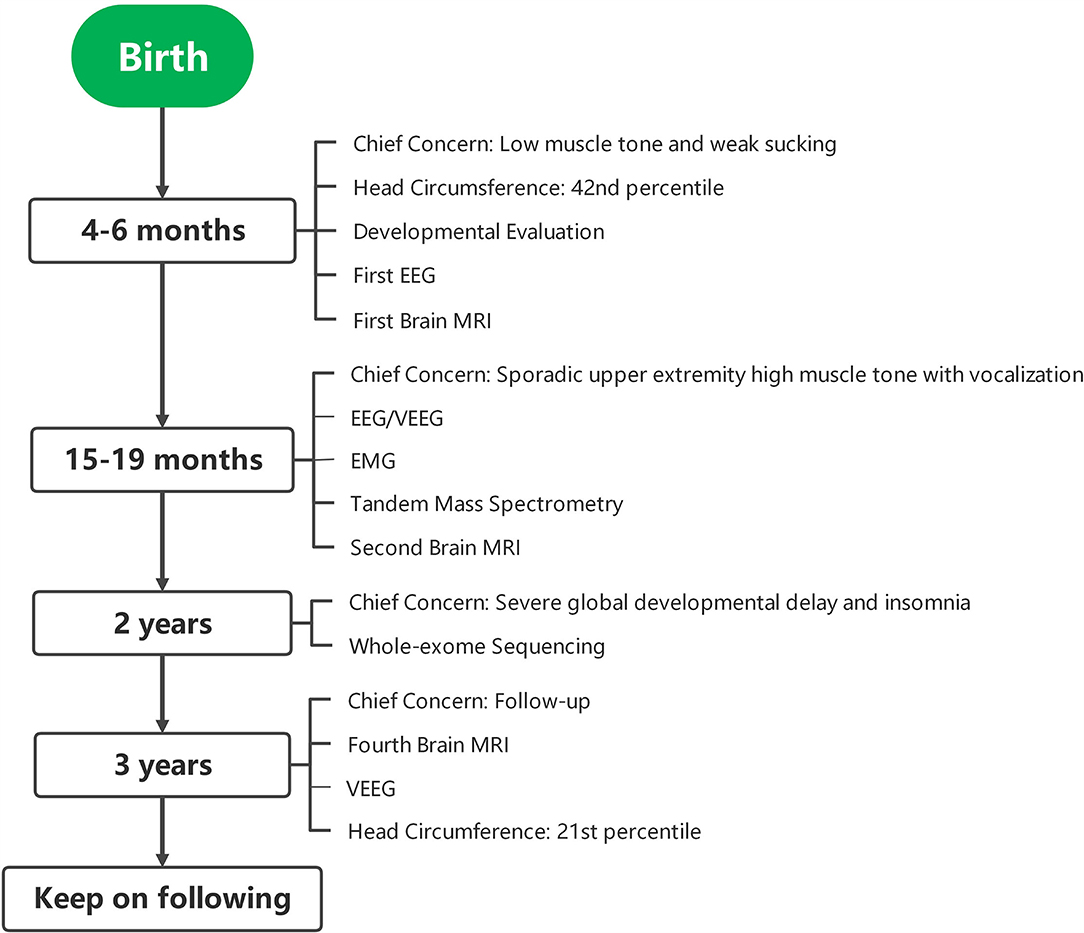
Figure 1. Case timeline. EEG, electroencephalogram; MRI, magnetic resonance imaging; VEEG, video electroencephalogram; EMG, electromyography.
At 15 months, the patient was brought to the outpatient unit for sporadic upper extremity high muscle tone with vocalization. Repeated EEG/video electroencephalogram (VEEG) at 15, 19, and 40 months exhibited no sign of epileptic seizure. The second brain MRI at 16 months revealed that the corpus callosum was slightly thin, and the lateral ventricle was plump, and we observed high-plaque signal intensity in the left frontal lobe (Figure 2). Electromyography (EMG) at 18 months showed a slight myogenic change in the tibialis anterior muscle and the peroneal muscle. Tandem mass spectrometry of blood and urine were normal. A follow-up brain MRI at 3 years of age revealed a slight enlargement of the ventricles bilaterally and a deepening of the bilateral frontotemporal sulci. Nevertheless, no obvious atrophy was found upon MRI. Electrocardiograph (ECG) result at 3 years was normal. Available head circumference data were within the normal range (42 cm at 6 months (42nd percentile) and 47.3 cm at 3 years (21st percentile)], as well as her weight [6.9 kg at 4 months (70th percentile) and 16 kg at 3 years (73rd percentile)] and lengths [68.3 cm at 4 months (>99th percentile) and 105 cm at 3 years (96th percentile)] throughout the follow-ups.
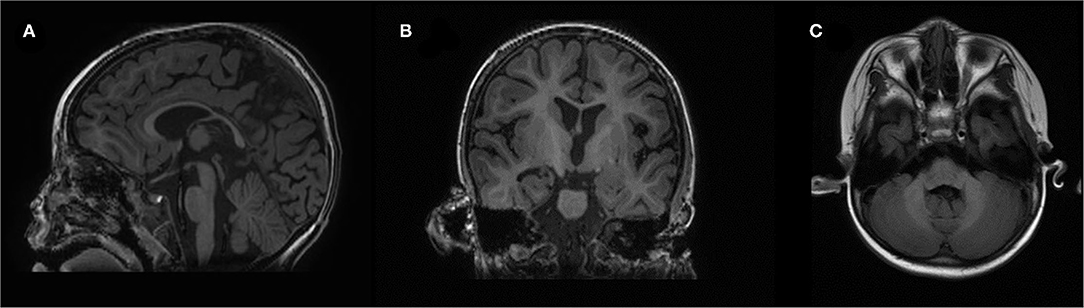
Figure 2. Brain magnetic resonance imaging (MRI): (A) sagittal, (B) coronal, and (C) OAx planes.
Whole-exome sequencing was executed to ascertain a possible molecular cause for the patient's condition. Two variants in SEPSECS were subsequently identified: c.194A>G leading to the missense variant p.Asn65Ser, and a novel splice variant c.701+1G>A. Sanger sequencing confirmed these variants and a parental test revealed a compound heterozygous state for the pedigree. To determine the impact of the splice variant, mRNA was extracted from the patient's blood sample for reverse transcription polymerase chain reaction (RT-PCR) assay. We designed the PCR primers as follows: F, 5′-ATCACTGCAGGTTTTGAGCC-3′; R, 5′-ACGCAGACAATGACAACCAC-3′; amplification produced an abnormal cDNA fragment, proving the presence of an alternative splice product with retention of the fifth intron (Figure 3). According to the American College of Medical Genetics and Genomics (ACMG) guidelines for interpreting sequence variants, the c.701+1G>A variant was categorized as pathogenic.
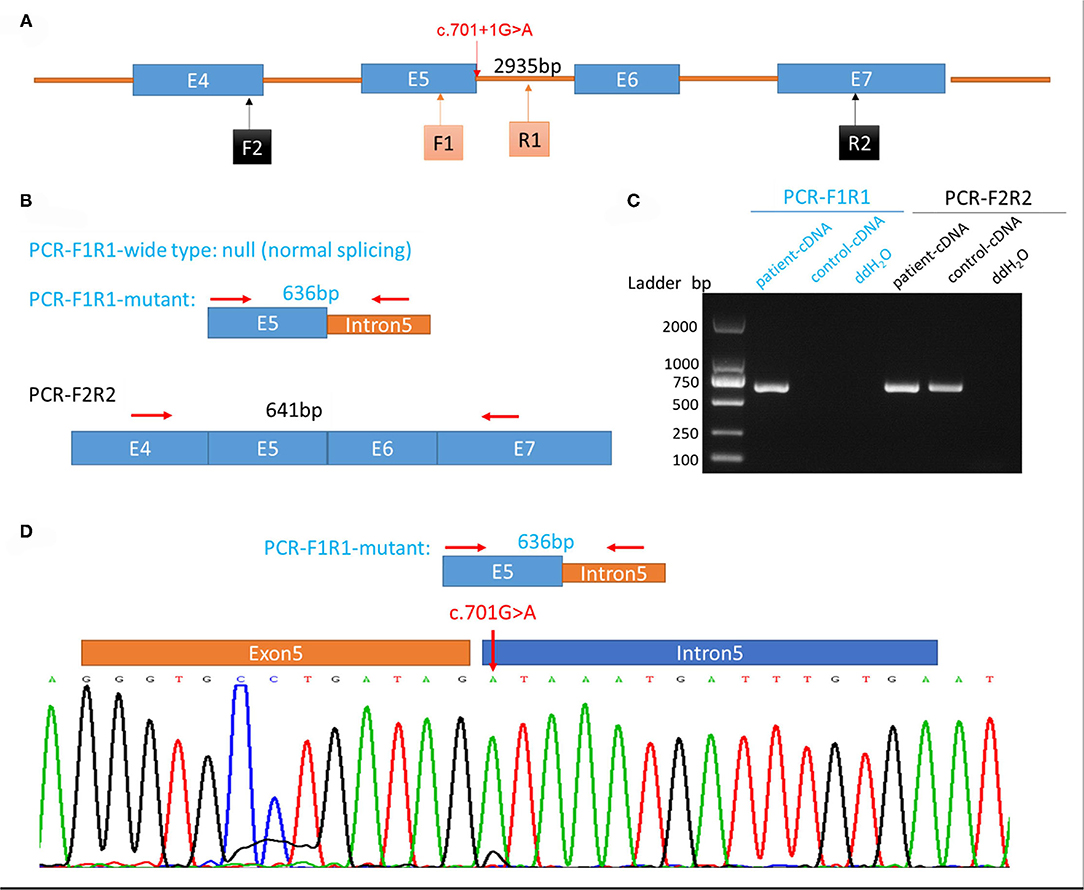
Figure 3. Genetic testing of the patient and analysis of aberrant splicing. (A) Schematic diagram of primer design. (B) Schematic diagram of expected polymerase chain reaction (PCR) product. (C) Electrophoretogram of PCR product from the patient's and control's blood samples. (D) Sequence of aberrant splicing product with intron 5 retention (red arrow indicates the mutation site).
Mutations in SEPSECS have now been found in individuals of several races and ethnicities—including Iraqi, Moroccan, Finnish, Arabic, Jordanian, and Japanese (6–9). Herein we reported the first case of pathogenic SEPSECS mutations in the Chinese population, which included the pathogenic splice variant c.701+1G>A.
Unlike previously reported cases, the patient in our study did not manifest the typical characteristics of PCH2D, and brain MRI results did not support obvious atrophy. There have been three cases reported of late-onset brain atrophy, either homozygous or heterozygous, and their mutation types are shown in Table 1 (8, 10). The molecular structure of SepSecS includes three domains, two insertions, one pyridoxal phosphate-binding site, and one non-canonical N terminal (1). Van Dijk et al. reported a female patient who exhibited a mild phenotype, with cerebellar atrophy detected at 16 years of age (10). The authors argued that the late-onset phenotype might be the result of a mutation site in the last exon, and this may have allowed partial resumption of enzyme activity (10). Iwama et al. reported two patients with compound heterozygous mutations who shared the same c.356A>G mutation located in the 4th α-helix of SepSecS (8). However, our patient's mutations were located at the 1st insertion and between the 6th β-fold and 5th α-helix. Mutations related to milder phenotypes are located at various sites, and an in vitro test of enzymatic activity was not available to us due to technical reasons. Without sufficient evidence to infer prognosis, regular brain imaging and developmental evaluations are still required to determine whether cerebello-cerebral atrophy would occur in the future.
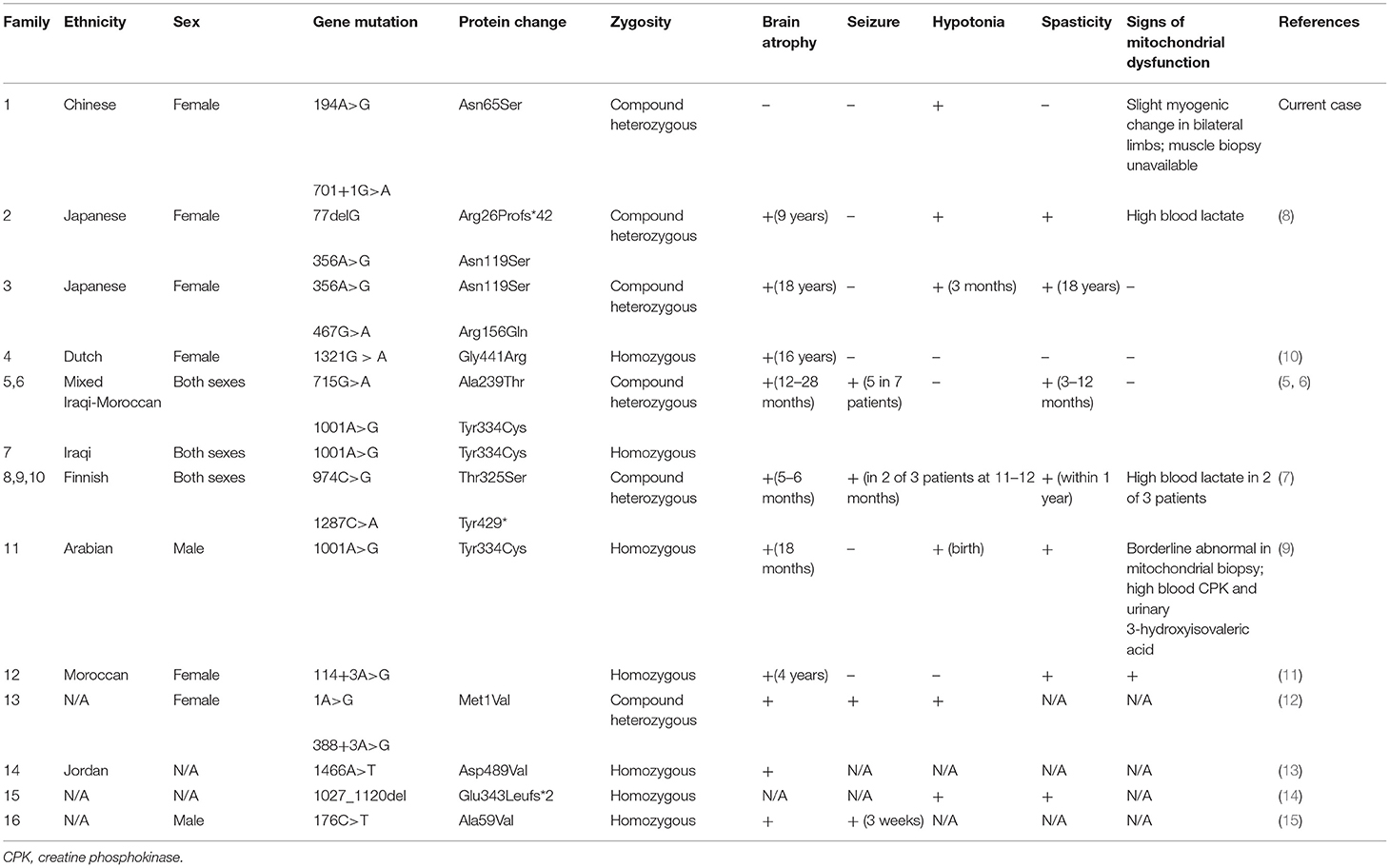
Table 1. Summary of SEPSECS mutations and related phenotypes.
Epilepsy is a common comorbidity of encephalopathy, and evidence shows that it could exacerbate neurocognitive dysfunction (16). As a result, early diagnosis and treatment of epilepsy are required. Myoclonic or generalized tonic-clonic seizures were previously reported in most SEPSECS cases, primarily occurring during the first or second year of life (5, 7, 17). Absence of epilepsy or seizures has also been reported (8–11), especially for milder phenotypes where cerebello-cerebral atrophy was detected in children older than 9 years of age (8, 10). Our patient exhibited seizure-like symptoms at 15 months, as she occasionally manifested upper-extremity high muscle tone together with monotone vocalizations; however, repeated EEG/VEEGs showed no epileptiform discharges at 15, 19, and 40 months. Ruling out epilepsy in our patient was congruent with her milder phenotype. However, with the potential of progressive encephalopathy, additional repeated measurements are essential once symptoms of epilepsy, such as focal or generalized seizures emerge.
Heterogeneous manifestations have been reported in cases with mutations of tRNA synthetase. For example, mutations in HARS2 (which encodes the histidyl-tRNA synthetase) result in deafness in both genders and infertility in females (18); while mutations in WARS (which encodes the tryptophanyl-tRNA synthetase) result in distal hereditary motor neuropathy (19). The heterogeneity in different gene mutations is attributed to the complicated catalytic processes inherent to each tRNA, and the diverse functions of and mechanisms underlying the gene-encoded proteins. SEPSECS is essential for selenoprotein synthesis (1); most selenoproteins are oxidoreductases and participate in cellular redox regulation and antioxidant activities (20). Elevated blood lactate levels and visual impairment have been previously reported, suggesting potential mitochondrial impairment due to SEPSECS mutations (7–9). Our patient did manifest hypotonia and the EMG showed a slight myogenic change in the lower limbs, although metabolic products in blood and urine remained normal. Muscle biopsy is still an option if further diagnosis is needed.
In conclusion, we have identified a novel pathogenic mutation in the SEPSECS gene, which is the first case reported in China, and it is associated with a mild phenotype that encompasses developmental delay, myogenic changes in the lower limbs, and sleep disorder but without progressive microcephaly and brain atrophy. According to other reported cases, disease progression in the future is not precluded, and thus a targeted and detailed follow-up is needed.
The original contributions presented in the study are included in the article/supplementary files, further inquiries can be directed to the corresponding author/s.
Ethical approval for this study was obtained from the Ethics Committee of Shanghai Children's Medical Center, School of Medicine, Shanghai Jiaotong University (SCMCIRB-W2021030). Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
TR collected the clinical data and wrote the initial draft of the manuscript. RY determined the mutant gene and participated in writing and reviewing of the manuscript. YD, QL, GW, and JW participated in the diagnosis and critically reviewed the manuscript. FJ and YJ identified and followed up the patient, supervised data collection, and critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
The study was supported by National Natural Science Foundation of China (81773443; 81602868; 82073568; 82071493); Ministry of Science and Technology of China (2016YFC1305203); Science and Technology Commission of Shanghai Municipality (2018SHZDZX05; 18JC1420305; 21Y11907400; 19QA1405800; 19411968800).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to the patient and her parents to participate in the study and consent us to report on the case.
1. Ganichkin OM, Xu XM, Carlson BA, Mix H, Hatfield DL, Gladyshev VN, et al. Structure and catalytic mechanism of eukaryotic selenocysteine synthase. J Biol Chem. (2008) 283:5849–65. doi: 10.1074/jbc.M709342200
2. Palioura S, Sherrer RL, Steitz TA, Soll D, Simonovic M. The human SEPSECS-tRNASec complex reveals the mechanism of selenocysteine formation. Science. (2009) 325:321–5. doi: 10.1126/science.1173755
3. Zhang Y, Zhou Y, Schweizer U, Savaskan NE, Hua D, Kipnis J, et al. Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. J Biol Chem. (2008) 283:2427–38. doi: 10.1074/jbc.M707951200
4. Wirth EK, Bharathi BS, Hatfield D, Conrad M, Brielmeier M, Schweizer U. Cerebellar hypoplasia in mice lacking selenoprotein biosynthesis in neurons. Biol Trace Elem Res. (2014) 158:203–10. doi: 10.1007/s12011-014-9920-z
5. Ben-Zeev B, Hoffman C, Lev D, Watemberg N, Malinger G, Brand N, et al. Progressive cerebellocerebral atrophy: a new syndrome with microcephaly, developmental relay, spastic quadriplegia. J Med Genet. (2003) 40:e96. doi: 10.1136/jmg.40.8.e96
6. Agamy O, Ben Zeev B, Lev D, Marcus B, Fine D, Su D, et al. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am J Hum Genet. (2010) 87:538–44. doi: 10.1016/j.ajhg.2010.09.007
7. Anttonen AK, Hilander T, Linnankivi T, Isohanni P, French RL, Liu Y, et al. Selenoprotein biosynthesis defect causes progressive encephalopathy with elevated lactate. Neurology. (2015) 85:306–15. doi: 10.1212/WNL.0000000000001787
8. Iwama K, Sasaki M, Hirabayashi S, Ohba C, Iwabuchi E, Miyatake S, et al. Milder progressive cerebellar atrophy caused by biallelic SEPSECS mutations. J Hum Genet. (2016) 61:527–31. doi: 10.1038/jhg.2016.9
9. Pavlidou E, Salpietro V, Phadke R, Hargreaves IP, Batten L, McElreavy K, et al. Pontocerebellar hypoplasia type 2D and optic nerve atrophy further expand the spectrum associated with selenoprotein biosynthesis deficiency. Eur J Paediatr Neurol. (2016) 20:483–8. doi: 10.1016/j.ejpn.2015.12.016
10. van Dijk T, Vermeij JD, van Koningsbruggen S, Lakeman P, Baas F. A SEPSECS mutation in a 23-year-old woman with microcephaly and progressive cerebellar ataxia. J Inherit Metab Dis. (2018) 41:897–8. doi: 10.1007/s10545-018-0151-x
11. Arrudi-Moreno M, Fernández-Gómez A, Peña-Segura JL. A new mutation in the SEPSECS gene related to pontocerebellar hypoplasia type 2D. Med Clin. (2021) 156:94–5. doi: 10.1016/j.medcli.2019.10.005
12. Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. (2015) 17:774–81. doi: 10.1038/gim.2014.191
13. Makrythanasis P, Nelis M, Santoni FA, Guipponi M, Vannier A, Béna F, et al. Diagnostic exome sequencing to elucidate the genetic basis of likely recessive disorders in consanguineous families. Hum Mutat. (2014) 35:1203–10. doi: 10.1002/humu.22617
14. Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. (2015) 10:148–61. doi: 10.1016/j.celrep.2014.12.015
15. Olson HE, Kelly M, Lacoursiere CM, Pinsky R, Tambunan D, Shain C, et al. Genetics and genotype-phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann Neurol. (2017) 81:419–29. doi: 10.1002/ana.24883
16. Nickels KC, Zaccariello MJ, Hamiwka LD, Wirrell EC. Cognitive and neurodevelopmental comorbidities in paediatric epilepsy. Nat Rev Neurol. (2016) 12:465–76. doi: 10.1038/nrneurol.2016.98
17. Hady-Cohen R, Ben-Pazi H, Adir V, Yosovich K, Blumkin L, Lerman-Sagie T, et al. Progressive cerebello-cerebral atrophy and progressive encephalopathy with edema, hypsarrhythmia and optic atrophy may be allelic syndromes. Eur J Paediatr Neurol. (2018) 22:1133–8. doi: 10.1016/j.ejpn.2018.07.003
18. Tiosano D, Mears JA, Buchner DA. Mitochondrial dysfunction in primary ovarian insufficiency. Endocrinology. (2019) 160:2353–66. doi: 10.1210/en.2019-00441
19. Wang B, Li X, Huang S, Zhao H, Liu J, Hu Z, et al. A novel WARS mutation (p.Asp314Gly) identified in a Chinese distal hereditary motor neuropathy family. Clin Genet. (2019) 96:176–82. doi: 10.1111/cge.13563
Keywords: SEPSECS mutation, PCCA, PCH2D, milder phenotype, developmental delay
Citation: Rong T, Yao R, Deng Y, Lin Q, Wang G, Wang J, Jiang F and Jiang Y (2022) Case Report: A Relatively Mild Phenotype Produced by Novel Mutations in the SEPSECS Gene. Front. Pediatr. 9:805575. doi: 10.3389/fped.2021.805575
Received: 30 October 2021; Accepted: 20 December 2021;
Published: 26 January 2022.
Edited by:
Chen Li, Zhejiang University, ChinaCopyright © 2022 Rong, Yao, Deng, Lin, Wang, Wang, Jiang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fan Jiang, ZmFuamlhbmdAc2hzbXUuZWR1LmNu; Yanrui Jiang, eWFucnVpMTAyN0AxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.