 Tao Wang
Tao Wang Yu-Xing Liu2†
Yu-Xing Liu2† Fang-Mei Luo
Fang-Mei Luo Liang-Liang Fan
Liang-Liang Fan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 29 November 2021
Sec. Pediatric Neurology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.774575
Background: Transmembrane protein 231 (TMEM231) is a component of the B9 complex that participates in the formation of the diffusion barrier between the cilia and plasma membrane. Mutations in TMEM231 gene may contribute to the Joubert syndrome (JBTS) or Meckel–Gruber syndrome (MKS). However, reports on JBTS or MKS caused by TMEM231 mutations are comparatively rare.
Method: We describe a Chinese fetus with unexplained hypoplasia of the cerebellar vermis and polydactyly, detected by ultrasound imaging. The fetus was primarily diagnosed with JBTS/MKS. The parents of this fetus were non-consanguineous and healthy. Whole-exome sequencing (WES) and bioinformatics strategies were employed to explore the genetic lesion of this family.
Results: An unknown missense variant (c.19C>T;p.R7W) of TMEM231 gene was detected. The variant was predicted as pathogenic and was absent in our 200 healthy controls.
Conclusion: WES was employed to explore the genetic lesion of a fetus with unexplained hypoplasia of the cerebellar vermis and polydactyly. A novel variant in TMEM231 gene was identified. Our study not only provided data for genetic counseling and prenatal diagnosis to this family but also broadened the spectrum of TMEM231 mutations.
Ciliopathies are multiorgan system disorders caused by the defective cilium complex (1). B9 is a cilium complex at the transition zone (TZ) and protects the cilia as a privileged membrane domain (2, 3). The B9 complex includes more than 13 proteins. Genetic deficiency of any component of the B9 complex may lead to ciliopathy (4). Transmembrane protein 231 (TMEM231) is a part of the B9 complex that localizes at the base of the ciliary axoneme at the TZ (2). Mutations in TMEM231 gene can contribute to the Joubert syndrome (JBTS, OMIM# 614970) or Meckel–Gruber syndrome (MKS, OMIM# 615397) (4, 5).
JBTS and MKS are rare but lethal ciliopathies with overlapping features (6). JBTS is an autosomal recessive (AR) disorder and is characterized by a distinctive mid-hindbrain malformation, oculomotor apraxia, abnormal breathing, intellectual impairment, and varying developmental delay. Some JBTS patients may also show retinopathy, renal cysts, and postaxial polydactyly (4). MKS is another AR disorder characterized by occipital encephalocele, polycystic kidneys, and polydactyly that is typically perinatally lethal (7). Pleiotropy is a common characteristic of ciliopathies; mutations in TMEM231 gene with different degrees of severity may cause JBTS, MKS, or oral–facial–digital syndrome (OFDS). However, owing to the genetic heterogeneity and pleiotropy, the molecular basis of this variability remains largely unknown.
In this research, we described a Chinese fetus with unexplained hypoplasia of the cerebellar vermis and polydactyly detected by ultrasound imaging. The parents of this fetus were non-consanguineous and healthy. Employing whole-exome sequencing (WES) technology and bioinformatics strategies, we identified a novel homozygous variant in TMEM231 gene.
Here, we encountered a mother from the Hebei Province in China with a 22nd-week gestation. She visited our hospital owing to an antenatal ultrasound scan that revealed the vermis of the cerebellum was indistinct in the fetus. The ultrasound examination showed the right cerebral ventricle of the fetus was widened, and Dandy–Walker malformation was suspected (Figures 1A,B). Amniocentesis revealed normal karyotype (46, XY). The following antenatal ultrasound scan, at 34th-week gestation, revealed hydrocephalus, agenesis of the cerebellar vermis, and polydactyly in the fetus (Figures 1C–F), and the parents decided to terminate the pregnancy due to the fatal deformity. The mother was 30 years old and the father was 31 years old. They were non-consanguineous and healthy. Further investigation showed the parents come from one closed and small village. Family history survey showed that proband (II-3) was the product of the third pregnancy of this family, in which all three siblings were similarly affected (Figure 1G). As the ultrasound demonstrated hydrocephalus and cerebellar dysplasia in the fetuses, the first and the second pregnancies (II-1 and II-2) were terminated (Table 1). On the other hand, 200 healthy subjects as described in our previous study were enrolled in this study to exclude polymorphisms (8).
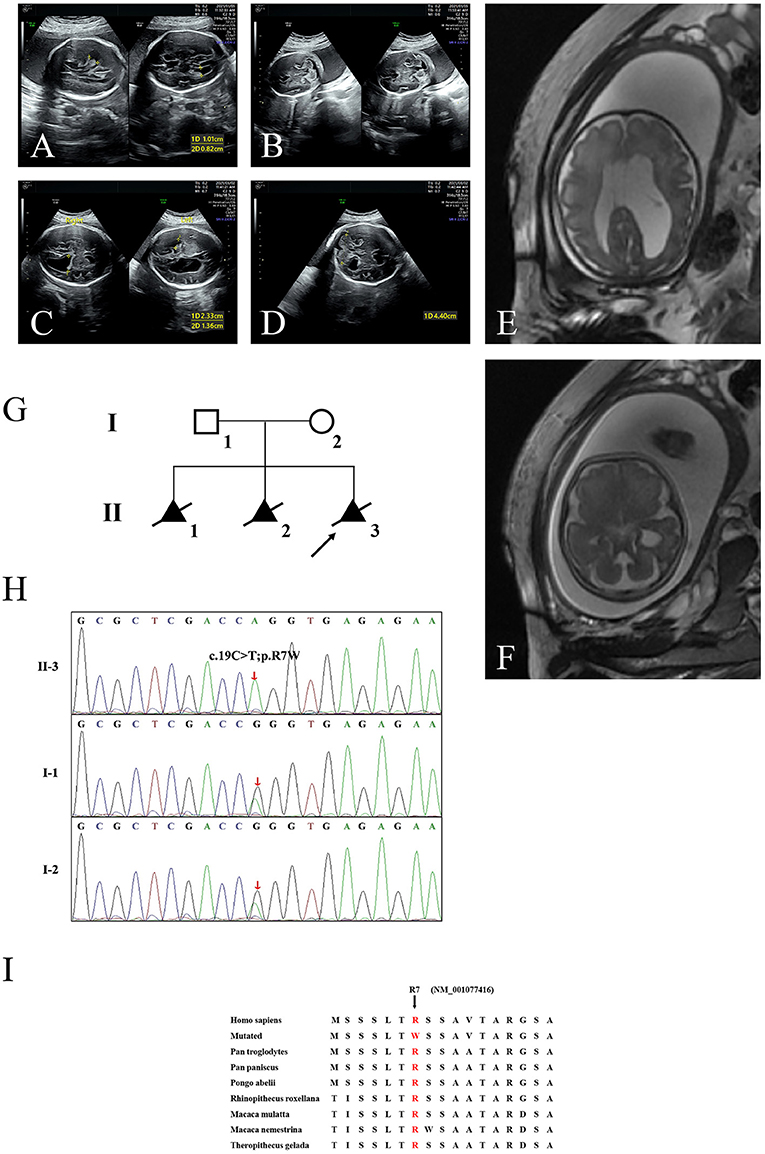
Figure 1. The clinic and sequencing data of this JBTS/MKS family. Ultrasound images show lateral ventricle (A) and cerebellum in transverse section (B) of the proband (II-3) at 22nd-week gestation. Lateral ventricle (C) and cerebellum (D) ultrasound images at 34th-week gestation. 3D FIESTA shows lateral ventricle (E) and cerebellum in transverse section (F) at 34th-week gestation. (G) The genealogy of this JBTS/MKS family. Squares indicate male family members; circles, female members; triangles, fetus; black symbols, the affected members; white symbols, unaffected members; arrow, proband. (H) Sanger DNA sequencing chromatogram detected a homozygosity mutation (c.19C>T;p.R7W) of TMEM231 gene in the proband. (I) Alignment of multiple TMEM231 protein sequences across species. Letters in red show the R7 site (NM_001077416) are evolutionarily conserved. FIESTA, Fast Imaging Employing Steady-state Acquisition.
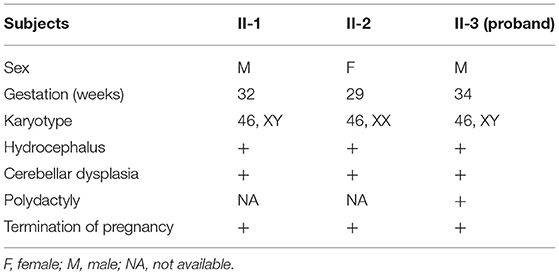
Table 1. Clinical data of three fetuses in this family.
This study was approved by the Review Board of the HeBei General Hospital, ShiJiaZhuang, China. Written informed consent was obtained from all adult participants and legal guardians of the minor participants.
Genomic DNA was extracted from peripheral blood lymphocytes of all the participants using the DNeasy Blood &Tissue Kit (Qiagen, Valencia, CA, USA.). The central part of the WES services and the necessary bioinformatics analyses were provided by the Novogene Bioinformatics Institute (Beijing, China). Exomes were captured using SureSelect Human All Exon V6 kits (Agilent, Santa Clara, CA, USA), and next-generation sequencing (NGS) was conducted with a HiSeq X-10 system (Illumina, San Diego, CA, USA). The strategies of data filtering refer to a previous study published by our laboratory group (9).
Sanger sequencing was used to validate the candidate variants identified in WES. Segregation analysis was performed in the family members of this study. Primer pairs were designed employing the PrimerQuest Tool IDT (http://sg.idtdna.com/Primerquest/Home/Index), and sequences of the polymerase chain reaction (PCR) products were determined using the ABI 3100 Genetic Analyzer (ABI, Foster City, CA).
The Polyphen-2, SIFT, and MutationTaster programs were used to predict the effects of mutations on the function of the protein. Swiss-Model software (https://swissmodel.expasy.org/interactive) was used to identify the function of the mutation. Local hydrophobicity was predicted by ProtScale (https://web.expasy.org/protscale/). The conservation analysis was performed by comparing amino acid sequences among different species.
WES yielded 11.9 Gb of data with 99.1% coverage of the target region and 98.98% of the target covered over 10 ×. After data filtering, the variants were further filtered by ciliopathy-related genes as described in the previous study (10) (Supplementary Table 1). A set of six variants in five genes were detected and further analyzed. Information related to inheritance pattern, Online Mendelian Inheritance in Man (OMIM) clinical phenotypes (https://omim.org/), ToppGene gene function (11), and American College of Medical Genetics and Genomics (ACMG) classification (12) of these six variants are shown in Table 2. Sanger sequencing was performed in the family members (I-1, I-2, and II-3) and showed that a novel homozygous variant (c.19C>T;p.R7W) of the TEME231 gene (NM_001077416) may underlie the genetic factor of this fetus with hypoplasia of the cerebellar vermis and polydactyly (Figure 1H). Each parent of this fetus carried an allele of this homozygous variant (Figure 1H). The (c.19C>T;p.R7W) variant targets exon 1 of the canonical isoform of TMEM231 (NM_001077416), as well as the other predicted protein-coding isoforms (NM_001077418). In NM_001077416, the variant results in the substitution of arginine acid by tryptophan at code 7, while in NM_001077418, it leads to the substitution of proline acid by leucine at code 10 (c.29C>T;p.P10L). This variant was not present in our 200 healthy controls group. Alignment of TMEM231 (NM_001077416) amino acid sequences revealed that R7 is conserved in primates (Figure 1I). Furthermore, modeling of proteins before and after missense variant were performed by SWISS-MODEL software (https://swissmodel.expasy.org/) and revealed that the missense variant at R7W may lead to the change of TMEM231 protein structure as marked by the red frame in the figure (Figure 2A). ProtScale software (https://web.expasy.org/protscale/) analyses revealed that the R7W variant raised local hydrophobicity compared with wild-type (WT) protein (Figure 2B).
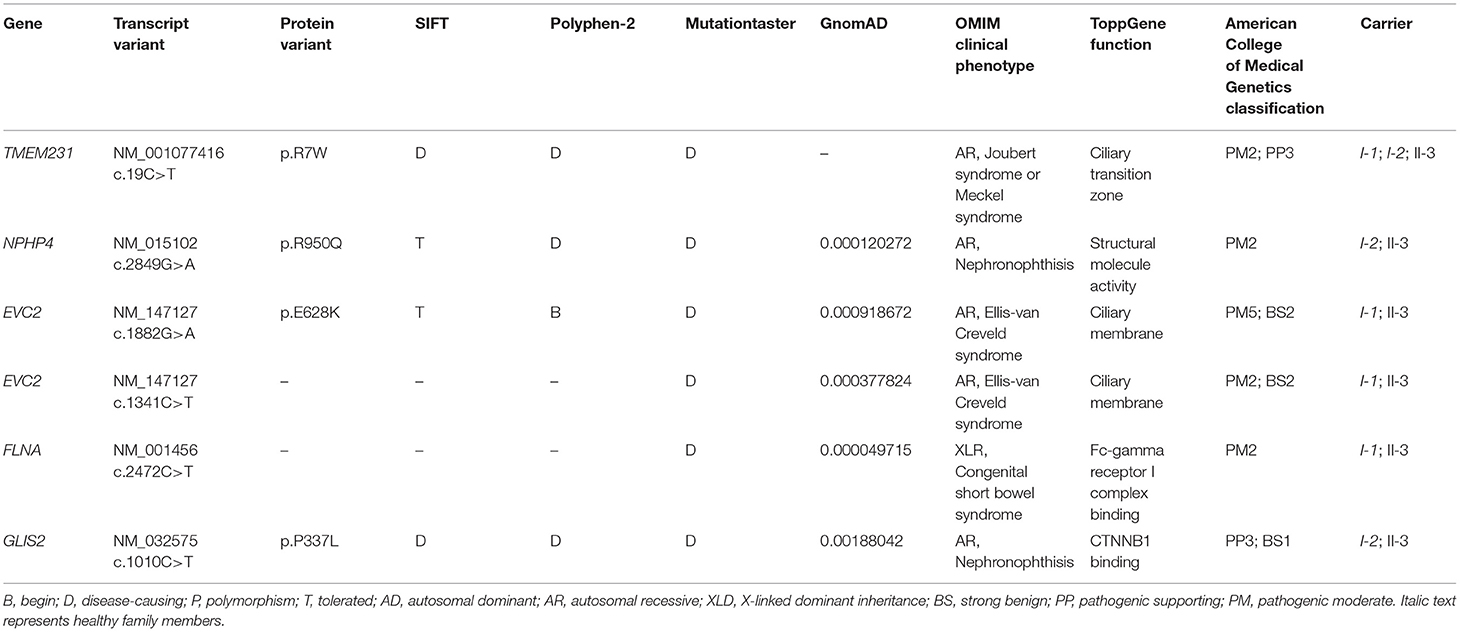
Table 2. Variants identified by WES in this family.
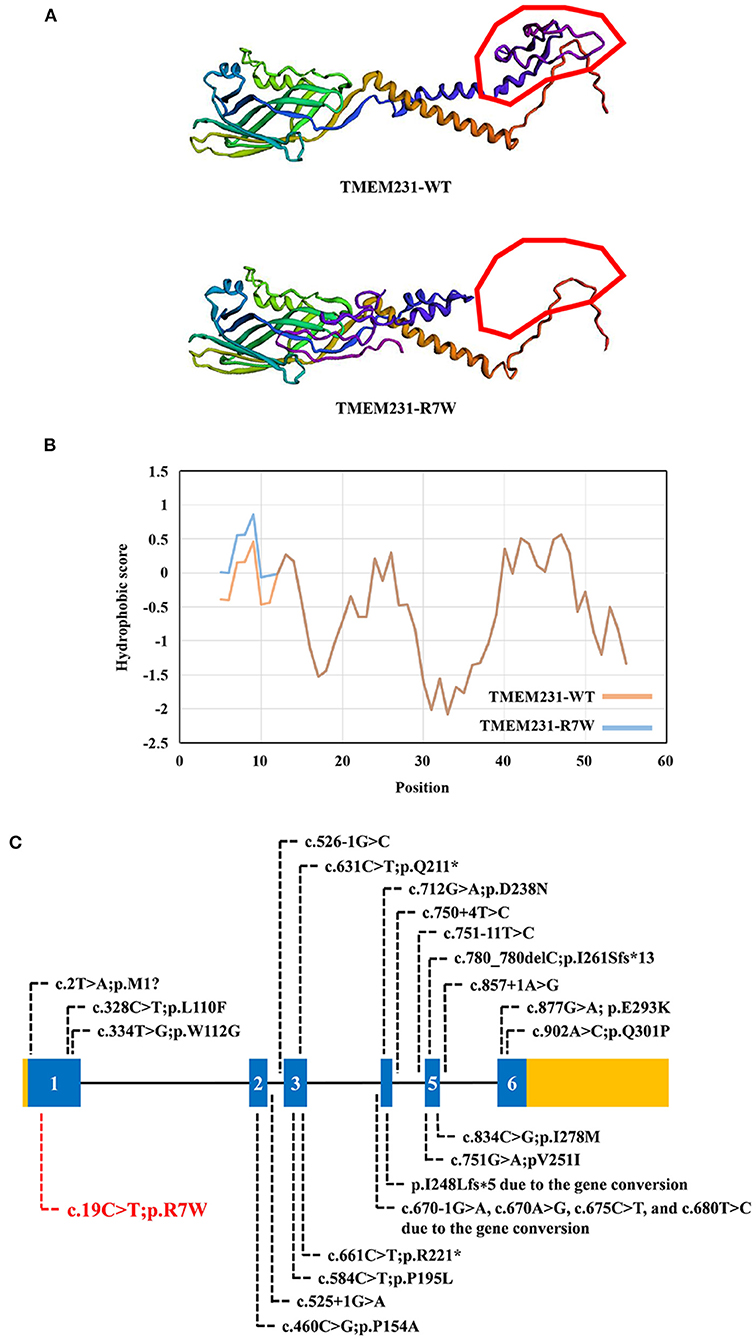
Figure 2. The bioinformatics analysis of mutations. (A) Structure prediction of the mutant protein. The wild-type TMEM231 (TMEM231-WT) protein structure and the p.R7W mutant TMEM231 (TMEM231-p.R7W) protein structure were predicted by SWISS-MODEL online software. (B) The Protscale online software predicts the hydrophobicity of wild-type and p.R7W mutant TMEM231 protein. The yellow curve shows the hydrophobicity score of each amino acid of wild-type TMEM231. The blue curve shows the p.R7W mutant TMEM231. (C) Schematic of TMEM231 mutations identified in JBTS- and MKS-affected individuals. The TMEM231 gene is showed, with all currently known TMEM231 mutations (black letters) and novel mutation (red letters).
In this study, we reported a Chinese fetus with unexplained hypoplasia of the cerebellar vermis and polydactyly detected by ultrasound imaging. The fetus was primarily diagnosed with JBTS/MKS. The parents of this fetus were non-consanguineous and healthy. A new homozygous variant (c.19C>T, p.R7W) of TMEM231 gene was detected by employing WES in combination with ciliopathy-related gene-filtering. Sanger sequencing confirmed that this fetus (II-3) harbored the homozygous missense variant in TMEM231, and each parent of this fetus carried an allele of this homozygous variant. DNA from the first (II-1) and the second fetal demises (II-2) were not available. Given that both the parents carried the TMEM231 c.19C>T;p.R7W heterozygous variant and all three siblings had similar phenotypes, we suspected that their fetuses (II-1 and II-2) with hydrocephalus and cerebellar dysplasia also carried this homozygous variant.
JBTS and MKS are rare ciliopathies. JBTS patients manifest a characteristic “molar tooth sign” on brain imaging in conjunction with retinopathy, nephropathy, and polydactyly. Although some individuals with JBTS die in infancy, most of the patients survive with variable developmental outcomes. MKS is a much more deadly perinatal syndrome. As the severe part of the ciliopathy phenotypic spectrum, MKS-affected individuals present with polycystic kidneys, occipital encephalocele, and polydactyly (13). To date, mutations in more than 27 genes are associated with JBTS, and pathogenic variants in at least 17 genes have been identified in MKS patients. Importantly, mutations in 12 of these genes, including TMEM231, TMEM67, and TEME237, are reported to cause both conditions (13, 14). As JBTS and MKS share overlapping features and are genetically heterogeneous, some laboratories routinely deploy a gene panel diagnostic approach that examines all JBTS- or MKS- associated disease genes, irrespective of which of the two diagnostic categories the clinical features suggest (13). Additionally, TMEM231 mutations have also been reported in OFDS patients. OFDS are rare disorders characterized by facial, oral, and digital abnormalities associated with a broad range of additional features. TMEM231, initially implicated in JBTS and MKS, also caused unclassified OFDS, with cerebellar hypoplasia, severe microcephaly, or polycystic kidney disease. The OFDS phenotype was clinically heterogeneous, and recent researches have confirmed the clinical and genetic overlap between OFDS and other ciliopathies, such as JBTS and MKS (15). A more comprehensive review of previously reported cases with TMEM231 variants is presented in Table 3 and may help to establish a genotype–phenotype correlation (18–21). Consistent with the previous study, the present study was also based on a non-consanguineous family. The proband was a product of the third pregnancy of this family and was primarily diagnosed as JBTS/MKS. A homozygote variant (c.19C>T;p.R7W) in TMEM231 was identified as a possible cause of the genetic lesion in this family. Besides, Dandy–Walker malformation is also one of the symptoms associated with MKS (22). The same was observed in the proband of the present study.
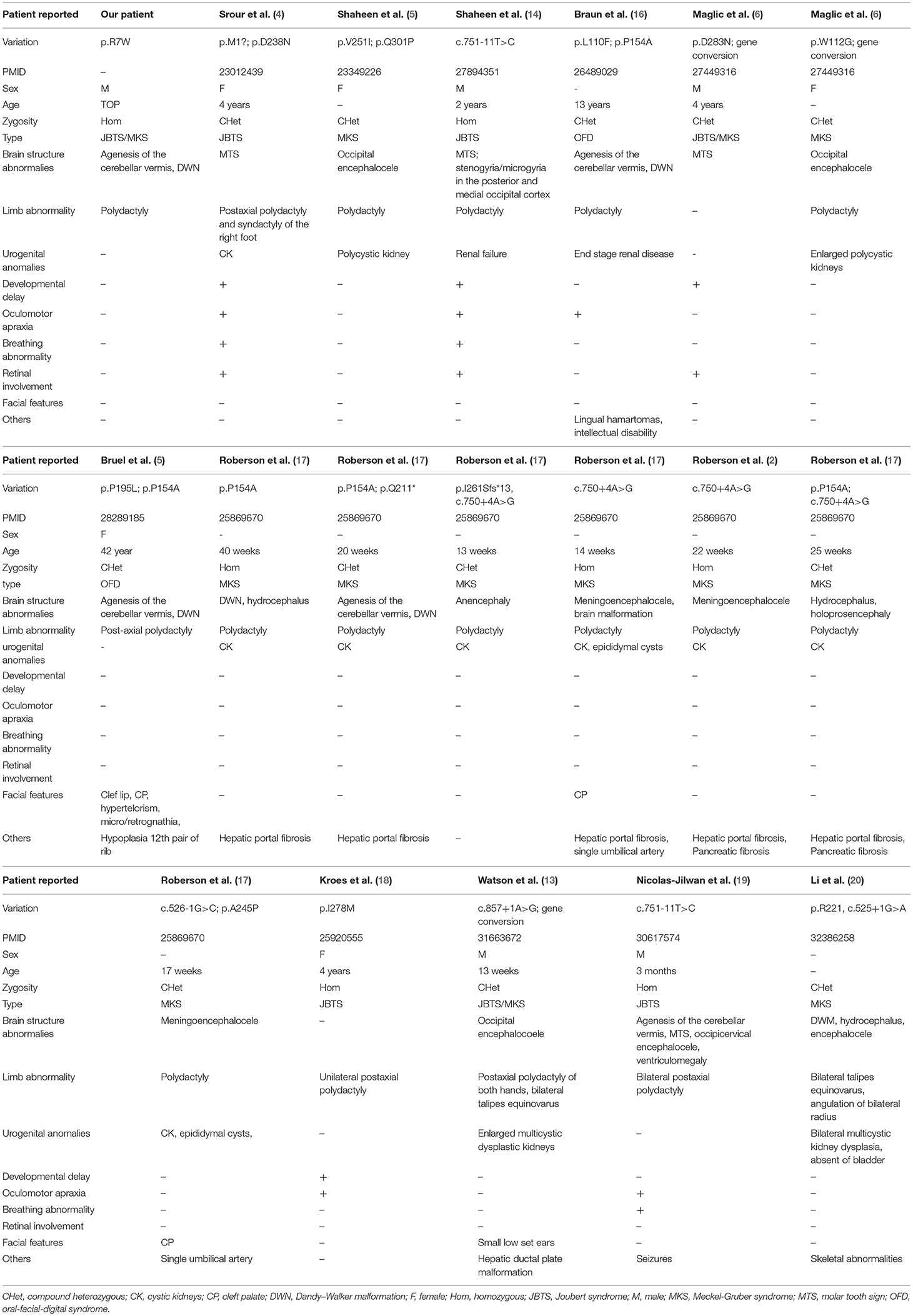
Table 3. The summary of reported patients with TMEM231 variant.
Tmem231 is a 36-kD two-pass transmembrane protein that locates at the base of the ciliary axoneme at the TZ (2). Tmem231 has a role in mammalian development (23). In C57BL/6 mice, Tmem231−/− embryos die before birth with distinct features of ciliopathy, including abrogated hedgehog signaling and polydactyly (2). Tmem231 mutant mouse embryos also exhibit ciliopathy hallmarks such as polydactyly, microphthalmia, and dorsalization of the neural tube (17). As a part of the B9 complex, TMEM231 is crucial for the constitution and functions of the cilia and physically interacts with many JBTS- or MKS-related genes (2, 17). Therefore, mutations in TMEM231 may lead to JBTS/MKS. In our research, we detected a homozygous variant (c.19C>T;p.R7W) of TMEM231 in a fetus with unexplained hypoplasia of the cerebellar vermis and polydactyly. This missense variant (p. R7W) locates in the exon 1 of TMEM231 gene that alters the arginine codon at position 7 to a tryptophan codon. As predicted by the SWISS-MODEL software, the missense variant at R7W may lead to the change of TMEM231 protein structure (Figure 2A). ProtScale software analyses revealed that the local hydrophobicity at and near the altered amino acid was increased compared with the WT TMEM231 protein (Figure 2B). In this case, the structural change might affect the transmembrane and localization of TMEM231 protein. And increased hydrophobicity at the R7W variant site might affect protein–protein interactions. No other TMEM231 coding or splicing variants were detected in the exome of this fetus. Furthermore, gene conversion in TEME231 may also lead to JBTS/MKS. As WES does not apply to detecting large deletions, ciliopathy patients who were only detected one heterozygous TMEM231 variant should be further examined (6).
By far, ~20 variants of TMEM231, including two gene conversions, have been identified in JBTS-, MKS-, and OFDS-affected individuals (Figure 2C). Although the pathogenic mechanism involved requires further investigation, our findings offer more evidence that TMEM231 gene variation is significant in JBTS/MKS. Notably, the variant (c.19C>T;p.R7W) detected in this research has not been published earlier and, therefore, is considered novel.
We used WES to explore the genetic lesion in a Chinese fetus with unexplained hypoplasia of the cerebellar vermis and polydactyly. A novel homozygous variant (c.19C>T;p.R7W) of TMEM231 was detected. Our study not only provided data for genetic counseling and prenatal diagnosis to this family but also broadened the spectrum of TMEM231 mutations.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI BioSample; PRJNA773747.
The studies involving human participants were reviewed and approved by the Review Board of the HeBei General Hospital, ShiJiaZhuang, China. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
TW and Y-XL enrolled the family members. F-ML performed DNA isolation and Sanger sequencing. YD performed genetic analysis and bioinformatics analysis. Y-XL and L-LF wrote the manuscript. Y-LL and L-LF supported the project. All authors reviewed the manuscript.
This study was supported by National Natural Science Foundation of China (82000427), National Natural Science Foundation of Hunan province (2020JJ5785), the Fundamental Research Funds for the Central Universities of Central South University (2021zzts0079), Hunan Provincial Innovation Foundation for Postgraduate (CX20210177), and the clinical medical personnel training program of Hebei Provincial Health Commission (Y-LL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all subjects for participating in this study. We also thank to Dr. Chen-yu Wang for her help in the bioinformatics analysis during the process of revision. And we would like to thank Editage (www.editage.cn) for English language editing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.774575/full#supplementary-material
1. Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. (2017) 18:533–47. doi: 10.1038/nrm.2017.60
2. Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. (2011) 14:61–72. doi: 10.1038/ncb2410
3. Czarnecki PG, Shah JV. The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol. (2012) 22:201–10. doi: 10.1016/j.tcb.2012.02.001
4. Srour M, Hamdan FF, Schwartzentruber JA, Patry L, Ospina LH, Shevell MI, et al. Mutations in TMEM231 cause Joubert syndrome in French Canadians. J Med Genet. (2012) 49:636–41. doi: 10.1136/jmedgenet-2012-101132
5. Shaheen R, Ansari S, Mardawi EA, Alshammari MJ, Alkuraya FS. Mutations in TMEM231 cause Meckel-Gruber syndrome. J Med Genet. (2013) 50:160–2. doi: 10.1136/jmedgenet-2012-101431
6. Maglic D, Stephen J, Malicdan MC, Guo J, Fischer R, Konzman D, et al. TMEM231 gene conversion associated with joubert and meckel-gruber syndromes in the same family. Hum Mutat. (2016) 37:1144–148. doi: 10.1002/humu.23054
8. Xiang R, Fan LL, Huang H, Cao BB, Li XP, Peng DQ, et al. A novel mutation of GATA4 (K319E) is responsible for familial atrial septal defect and pulmonary valve stenosis. Gene. (2014) 534:320–3. doi: 10.1016/j.gene.2013.10.028
9. Fan LL, Liu JS, Huang H, Du R, Xiang R. Whole exome sequencing identified a novel mutation (p.Ala1884Pro) of beta-spectrin in a Chinese family with hereditary spherocytosis. J Gene Med. (2019) 21:e3073. doi: 10.1002/jgm.3073
10. Zhang M, Chang Z, Tian Y, Wang L, Lu Y. Two novel TCTN2 mutations cause Meckel-Gruber syndrome. J Hum Genet. (2020) 65:1039–43. doi: 10.1038/s10038-020-0804-0
11. Kaimal V, Bardes EE, Tabar SC, Jegga AG, Aronow BJ. ToppCluster: a multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. (2010) 38:W96–102. doi: 10.1093/nar/gkq418
12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
13. Watson CM, Dean P, Camm N, Bates J, Carr IM, Gardiner CA, et al. Long-read nanopore sequencing resolves a TMEM231 gene conversion event causing Meckel-Gruber syndrome. Hum Mutat. (2020) 41:525–31. doi: 10.1002/humu.23940
14. Shaheen R, Szymanska K, Basu B, Patel N, Ewida N, Faqeih E, et al. Characterizing the morbid genome of ciliopathies. Genome Biol. (2016) 17:242. doi: 10.1186/s13059-016-1099-5
15. Bruel AL, Franco B, Duffourd Y, Thevenon J, Jego L, Lopez E, et al. Fifteen years of research on oral-facial-digital syndromes: from 1 to 16 causal genes. J Med Genet. (2017) 54:371–80. doi: 10.1136/jmedgenet-2016-104436
16. Braun DA, Schueler M, Halbritter J, Gee HY, Porath JD, Lawson JA, et al. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int. (2016) 89:468–75. doi: 10.1038/ki.2015.317
17. Roberson EC, Dowdle WE, Ozanturk A, Garcia-Gonzalo FR, Li C, Halbritter J, et al. TMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone. J Cell Biol. (2015) 209:129–42. doi: 10.1083/jcb.201411087
18. Kroes HY, Monroe GR, van der Zwaag B, Duran KJ, de Kovel CG, van Roosmalen MJ, et al. Joubert syndrome: genotyping a Northern European patient cohort. Eur J Hum Genet. (2016) 24:214–0. doi: 10.1038/ejhg.2015.84
19. Nicolas-Jilwan M, Al-Ahmari AN, Alowain MA, Altuhaini KS, Alshail EA. Tectocerebellar dysraphia with occipital encephalocele: a phenotypic variant of the TMEM231 gene mutation induced Joubert syndrome. Childs Nerv Syst. (2019) 35:1257–61. doi: 10.1007/s00381-019-04048-9
20. Li L, Fu F, Li R, Xiao W, Yu Q, Wang D, et al. Genetic tests aid in counseling of fetuses with cerebellar vermis defects. Prenat Diagn. (2020) 40:1228–38. doi: 10.1002/pd.5732
21. Hartill V, Szymanska K, Sharif SM, Wheway G, Johnson CA. Meckel-gruber syndrome: an update on diagnosis, clinical management, and research advances. Front Pediatr. (2017) 5:244. doi: 10.3389/fped.2017.00244
22. Herriot R, Hallam LA, Gray ES. Dandy-Walker malformation in the Meckel syndrome. Am J Med Genet. (1991) 39:207–10. doi: 10.1002/ajmg.1320390218
Keywords: JBTS, MKS, TMEM231, mutation, homozygote, whole-exome sequencing
Citation: Wang T, Liu Y-X, Luo F-M, Dong Y, Li Y-L and Fan L-L (2021) A Novel Homozygous Variant of TMEM231 in a Case With Hypoplasia of the Cerebellar Vermis and Polydactyly. Front. Pediatr. 9:774575. doi: 10.3389/fped.2021.774575
Received: 12 September 2021; Accepted: 22 October 2021;
Published: 29 November 2021.
Edited by:
Kette D. Valente, Universidade de São Paulo, BrazilReviewed by:
Andrea Accogli, Istituto Giannina Gaslini, ItalyCopyright © 2021 Wang, Liu, Luo, Dong, Li and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ya-Li Li, bHlsODcwM0BzaW5hLmNvbQ==; Liang-Liang Fan, c3dmYW5saWFuZ2xpYW5nQGNzdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.