 Linda Gaillard1*Anne Goverde2Quincy C. C. van den Bosch2Fernanda S. Jehee2Erwin Brosens2Danielle Veenma2Frank Magielsen2Annelies de Klein2Irene M. J. Mathijssen1†Marieke F. van Dooren2†
Linda Gaillard1*Anne Goverde2Quincy C. C. van den Bosch2Fernanda S. Jehee2Erwin Brosens2Danielle Veenma2Frank Magielsen2Annelies de Klein2Irene M. J. Mathijssen1†Marieke F. van Dooren2†- 1Department of Plastic, Reconstructive and Hand Surgery, Erasmus Medical Center—Sophia Children's Hospital, University Medical Center Rotterdam, Rotterdam, Netherlands
- 2Department of Clinical Genetics, Erasmus Medical Center—Sophia Children's Hospital, University Medical Center Rotterdam, Rotterdam, Netherlands
Congenital diaphragmatic hernia (CDH) is a life-threatening birth defect that presents as either an isolated diaphragm defect or as part of a complex disorder with a wide array of anomalies (complex CDH). Some patients with complex CDH display distinct craniofacial anomalies such as craniofrontonasal dysplasia or craniosynostosis, defined by the premature closure of cranial sutures. Using clinical whole exome sequencing (WES), we found a BCL11B missense variant in a patient with a left-sided congenital diaphragmatic hernia as well as sagittal suture craniosynostosis. We applied targeted sequencing of BCL11B in patients with craniosynostosis or with a combination of craniosynostosis and CDH. This resulted in three additional BCL11B missense mutations in patients with craniosynostosis. The phenotype of the patient with both CDH as well as craniosynostosis was similar to the phenotype of previously reported patients with BCL11B missense mutations. Although these findings imply that both craniosynostosis as well as CDH may be associated with BCL11B mutations, further studies are required to establish whether BCL11B variants are causative mutations for both conditions or if our finding was coincidental.
Introduction
The genetic etiology of congenital diaphragmatic hernia (CDH) is complex. Structural variants, small and large insertions or deletions and single-nucleotide variants (SNVs) in over a 100 genes have been associated with CDH (1). Only a few of these genes are mutated recurrently in multiple individuals and even then the phenotype can differ largely due to incomplete penetrance. CDH has a prevalence of 2.3–2.7 per 10.000 live births (2–4) and can present as isolated CDH or in association with additional congenital anomalies (non-isolated CDH or complex CDH), as seen in ~40 (5) to 49% (6) of the cases. Complex CDH may present as part of a recognizable genetic syndrome, chromosome abnormality, or a collection of major congenital malformations. One of the less common malformations in patients with CDH are craniofacial anomalies and in particular craniosynostosis.
Although less common in syndromes associated with complex CDH, several studies have described syndromes such as Apert's syndrome and Craniofrontonasal syndrome, that include both CDH as well as craniosynostosis as cardinal or relatively common features (7–22). Craniosynostosis, a developmental disorder defined by the premature fusion of one or more sutures, has a prevalence of 7.2 per 10,000 live births (23). It can be divided into non-syndromic and syndromic craniosynostosis, with syndromic craniosynostosis being characterized by additional congenital anomalies, such as limb anomalies and neurodevelopmental delays (24, 25). As part of our standard clinical care, all patients diagnosed with craniosynostosis are offered targeted genetic analysis (26). In a female patient with both CDH as well as craniosynostosis, we observed a genetic variant of the B cell leukemia 11b gene (BCL11B; OMIM 606558) resulting in an amino acid change at position 667 [p.(Gly667Glu)].
Searching the literature for BCL11B mutations we found several case reports. First, a case report described a male patient with a heterozygous de novo missense BCL11B mutation (p.Asn441Lys), who presented with severe combined immunodeficiency as well as neurological, dermal and facial dysmorphisms including hypertelorism, short palpebral fissures and micrognathia (27). Second, a study reported on 13 patients with heterozygous germline mutations in BCL11B (28). Most of these patients presented with neurodevelopmental disorders and immunodeficiency with reduced type 2 innate lymphoid cell and were carriers of loss of function mutations in BCL11B. However, in the most severely affected patient (patient EII-I) a missense BCL11B mutation p.(Asn807Lys) was found. This patient had a similar phenotype as compared to the first patient described by Punwani et al. (27) including a myopathic facial appearance, hypertelorism and small palpebral fissures. Neither study reported on the presence of craniosynostosis or CDH although craniofacial anomalies were described for patient EII-I and are apparent for the patient described by Punwani et al. (27). However, we previously discovered a de novo BCL11B missense mutation in exon 1 which encodes for [p.(Arg3Ser)] in a male patient with unilateral coronal suture craniosynostosis (29). In addition, a de novo BCL11B missense mutation in exon 4 [p.(Arg350Cys)] was reported in a patient who presented with CDH, an abnormal optic nerve, increased intraocular pressure and scoliosis (30). These findings suggest that both craniosynostosis as well as CDH may be associated with BCL11B missense mutations.
For this report, we selected patients who had undergone genetic testing with craniosynostosis or combined CDH and craniosynostosis from the Erasmus MC- Sophia Children's hospital. This search resulted in three additional BCL11B missense variants. In this report we present the clinical reports of these four new patients with BCL11B variants and a brief review of disorders that are characterized by both CDH as well as craniosynostosis. Informed consent to publish case descriptions and images was obtained from patients, and/or their parents if applicable. All genetic information is provided according to the HGNC guidelines (31).
Clinical Reports
Patient A
Patient A [NM_138576.3(BCL11B):c.2000G>A, p.(Gly667Glu)], a 30 year old female, is the third child of healthy non-consanguineous parents, born at 41 weeks of gestation with a birth weight of ~2,200 g, following an uncomplicated pregnancy. Shortly after birth, the patient developed severe respiratory insufficiency, resulting in apnea and asystole. After resuscitation, the patient was transferred to our tertiary care center. She presented with left-sided CDH, resulting in respiratory insufficiency and pulmonary hypertension, for which she underwent surgery the second day after birth. She was weaned off respiratory support after 2 weeks. She showed several dysmorphic features including, slight ocular proptosis, hypertelorism, down slanting palpebral fissures, ptosis, arched eyebrows, and syndactyly of the second and third toes of both feet (Figures 1A–C). In addition, she suffered from feeding difficulties and gastroesophageal reflux due to pyloric stenosis, which was surgically corrected at the age of 2 months. She remained hypotonic and developed psychomotor delays during the first year of life. At the age of 11 months she was diagnosed with progressive sagittal suture craniosynostosis, for which she underwent surgery at the ages of 14 and 16 months. During surgery part of the calvarium looked abnormal and was sent for pathological investigation, revealing a capillary and cavernous hemangioma. She developed divergent strabismus, latent nystagmus, hypermetropia and astigmatism, and suffered from recurrent episodes of pneumonia, sinusitis and rhinitis. She showed no signs of severe cognitive impairment.
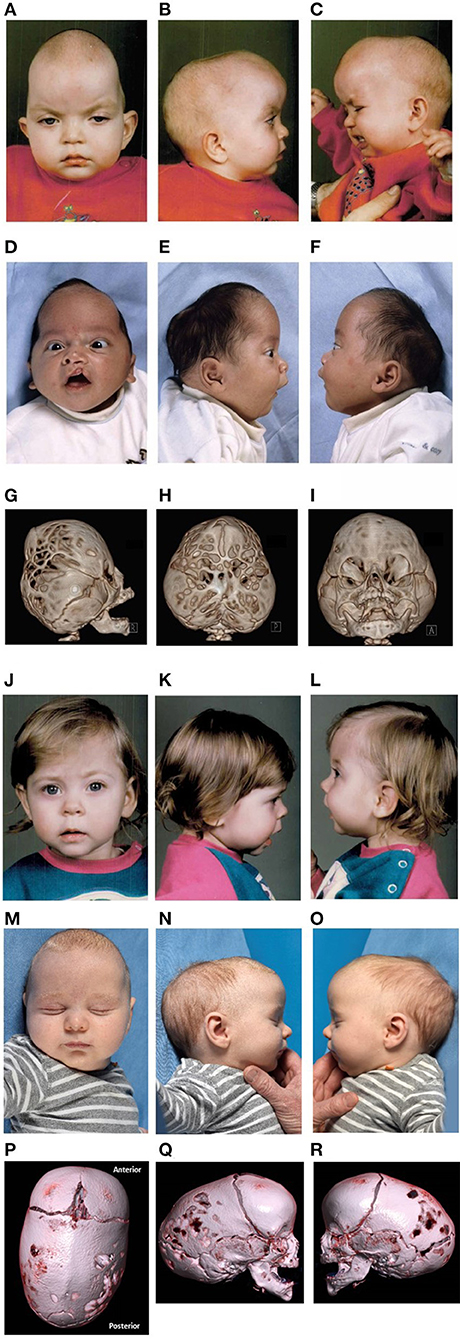
Figure 1. Clinical features. Photos published with consent. (A–C) Pre-operative features of patient A at the age of 14 months. Historic pre-operative radiological imaging could not be retrieved of the CDH and craniosynostosis. (D–I) Pre-operative features and 3DCT-scan imaging of patient B at the age of 1 and 6 months, respectively. (J–L) Pre-operative features of patient C at the age of 12 months. Historic pre-operative radiological imaging of the craniosynostosis could not be retrieved. (M–R) Pre-operative features and 3DCT-scan imaging of patient D at the age of 2 months.
Patient B
Patient B [NM_138576.3(BCL11B): c.1744G>A, p.(Gly582Ser)], a 16 year old male, is the second child of unaffected non-consanguineous parents. He was born at 40 weeks of gestation, with a birth weight of 3,710 g. He presented with craniosynostosis of the sagittal suture and both lambdoid sutures, for which he underwent surgery at the age of 6 months. In addition, he suffered from an incomplete left-sided unilateral cleft lip including the alveolar arch, which was surgically corrected in three stages (at the ages of 6 months, 6, and 11 years). He showed several dysmorphic features, including frontal bossing, down-slanting palpebral fissures, hypotelorism, mild webbing of the neck, and hyperpigmentation on the left shoulder (Figures 1D–I). Magnetic Resonance Imaging (MRI) of the brain at the age of 8 years showed a Chiari I malformation. Ophthalmological assessment revealed astigmatism and myopia. He developed a proportionate short stature of −2.5 to 2 SD with delayed bone age, for which puberty was postponed using leuprorelin and letrozole. He had no neurodevelopmental delays. Motor and speech development were within normal range although he required speech therapy and physical therapy. He developed Kawasaki disease at the age of 7 years, which responded well to intravenous immunoglobulins. At the age of 16 years he underwent a second cranial vault surgery including biparietal expansion and occipital decompression. He did not suffer from frequent infections.
Patient C
Patient C [NM_138576.3(BCL11B):c.2018C>G, p.(Pro673Arg)], a 30 year old female, is the child of non-consanguineous phenotypically normal parents. A sibling and paternal half-sibling are both healthy. She presented with left-sided unicoronal craniosynostosis, for which she underwent surgery at the age of 14 months. She developed no neurodevelopmental delays and did not suffer from recurrent infections to our knowledge, although the cranial vault operation was postponed twice due to upper airway infections. She required glasses from the age of 8 years onwards. She did not display any evident dysmorphic features with the exception of mild vertical orbital dystopia (Figures 1J–L).
Patient D
Patient D [NM_138576.3(BCL11B):c.1265C>T, p.(Pro422Leu)], a 2 year old male, is the second child of unaffected non-consanguineous parents. In addition, the patient has two unaffected paternal half-siblings. He was born with a birth weight of 4,230 g at 41.3 weeks of gestation. He was born with the umbilical cord wrapped around his neck but recovered well after stimulation and did not require oxygen support. The pregnancy was otherwise uncomplicated. He presented with craniosynostosis of the sagittal suture at the outpatient clinic at the age of 3 months for which he underwent spring-assisted surgery at the age of 6 months. He had no neurodevelopmental delays and did not suffer from recurrent infections. Dysmorphic features were mild and included thick alae nasi and mild retrognathia with an overbite (Figures 1M–R).
Overview of Patients With BCL11B Missense Mutations and Variants
An overview of all patients described above, including genetic and phenotypic information of each patient can be found in Table 1. In addition, it shows the previously described patients with BCL11B missense mutations. Phenotypical features vary heavily among patients. The patient, reported by Goos et al. (29), displayed a similar phenotype to our Patient C, with both patients presenting with coronal suture synostosis without neurodevelopmental delays or other severe associated features. Patient A and patient B, are more similar to patients reported by Punwani et al. (27) and Lessel et al. (28). They have a more severe phenotype, presenting with a combination of multi-sutural craniosynostosis, CDH and cleft. In an additional study, one patient with a de novo BCL11B missense mutation [p.(Arg350Cys)] was reported to have presented with complex CDH (30). All cases summarized above have missense variants in exon 4 with the exception of the patient reported by Goos et al. (29), who had a de novo missense mutation of exon 1.
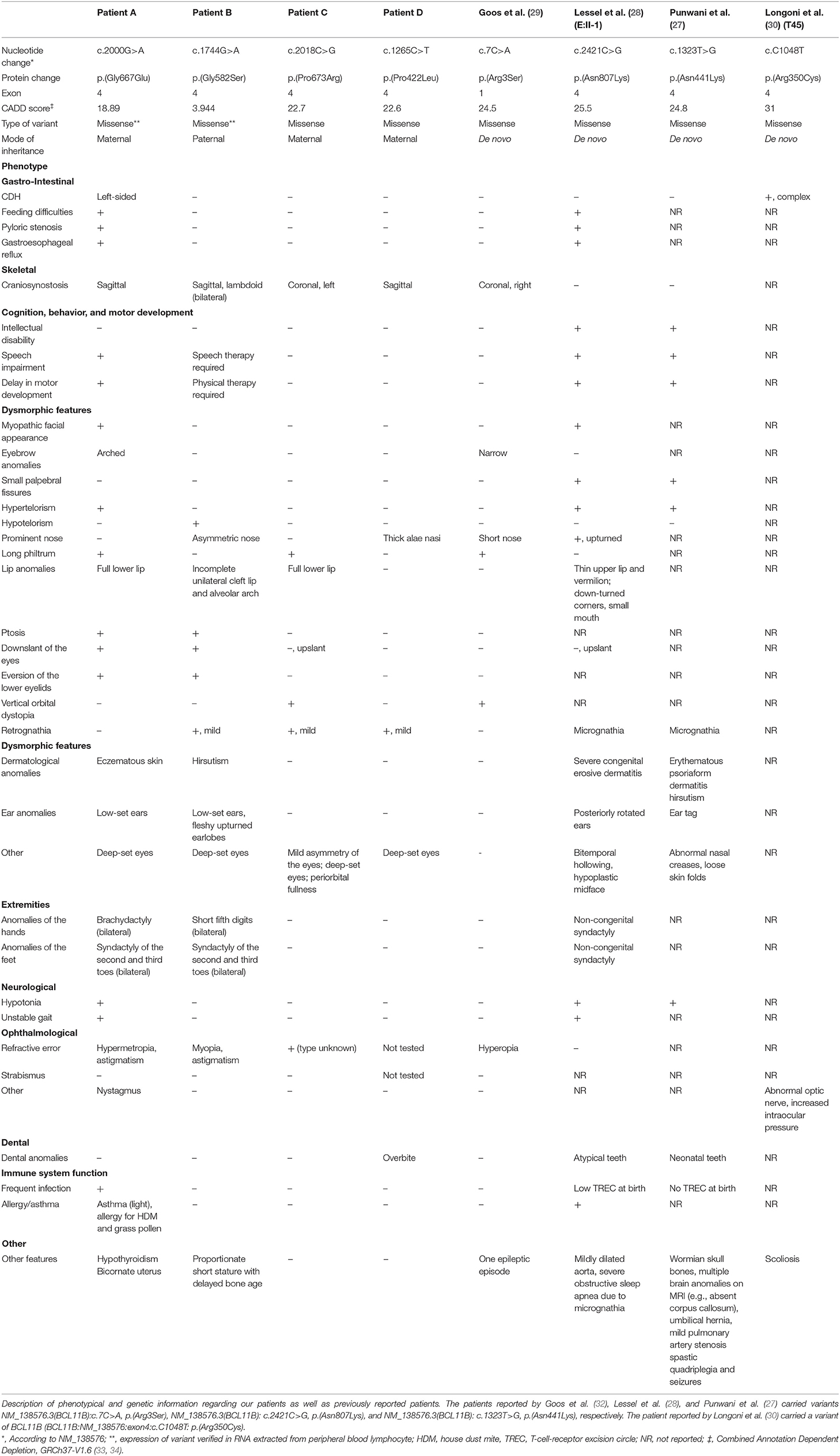
Table 1. Case descriptions of patients with BCL11B missense variants: genetic and phenotypical features.
Discussion
In this study, we present four new patients with missense variants in BCL11B (Patient A: p.(Gly667Glu); Patient B: p. (Gly582Ser); Patient C: p. (Pro673Arg); Patient D: p.(Pro422Leu). Patient A displayed both CDH as well as craniosynostosis of the sagittal suture. The other three patients were diagnosed with craniosynostosis without a diaphragm defect. To our knowledge, this is the second study to report on craniosynostosis in patients with BCL11B mutations and the first to report on the co-occurrence of CDH and craniosynostosis in a patient with a missense BCL11B variant. Based on our current cohort of patients with BCL11B missense variants, the phenotype associated with BCL11B mutations is highly variable. Clinical features range from isolated craniosynostosis to CDH, severe immunological deficiencies and neurodevelopmental delays (Table 1).
BCL11B has a key function in fetal development and is involved in a multitude of systems and pathways (35). In line with this, many patients with BCL11B missense mutations are reported to suffer from a wide array of clinical anomalies (27, 28). In addition to altered craniofacial development, mutations, mostly loss of function mutations in BCL11B, have been reported to affect neurodevelopment as well as the development of the immune system, skin and the teeth (27–29, 36–49). In mice models, BCL11B is key in regulating suture patency, with a single disrupted allele causing synostosis (36). Goos et al. further investigated BCL11B variants in relation to craniosynostosis (29). A mouse model confirmed that the de novo BCL11B missense mutation (p.Arg3Ser) in their patient could have a causative effect on the development of craniosynostosis. Furthermore, they found several rare variants in a British cohort of craniosynostosis patients. However, these variants were dismissed as polymorphisms because of low impact or because they were inherited from a healthy parent. The authors suggest that only a specific subset of BCL11B mutations may cause craniosynostosis.
Our patients inherited the BCL11B variants from phenotypically normal parents. This could suggest incomplete penetrance, which may be similar to other craniosynostosis syndromes such as TCF12 and SMAD 6-related craniosynostosis and could potentially complicate genetic counseling (32, 50–52). Notably, patients reported in previous BCL11B studies mainly were carriers of de novo mutations. Although Combined Annotation Dependent Depletion (CADD) scores in our patients vary, patient A, C and D are in the same range as CADD scores of variants previously reported (27–29, 33, 34). Although a low CADD score could potentially indicate a benign variant, it is remarkable that four patients have now presented with craniosynostosis bearing a missense variant in exon 4 of BCL11B. Expression of this missense variant in blood excluded non-sense mediated RNA decay in patients A and B. Hence a dominant negative effect or an altered function of the mutated protein could be an explanation of the more severe phenotype.
To our knowledge, only one previous study reported on a patient with a CDH with a BCL11B missense mutation [p.(Arg350Cys); CADD score: 31] (30). We now report on a second patient with a BCL11B missense variant in exon 4 with CDH. Although further studies remain necessary to assess if BCL11B is a causative factor in this CDH phenotype the fact that BCL11B is involved in pathways that overlap with pathways that have been previously linked to CDH support this observation. BCL11B likely interacts with a CDH causative gene (NR2F2) as well as with other CDH candidate genes (CREBBP, EP300, CHD4) (1, 53). The genetic etiologies of both CDH and craniosynostosis are highly complex and definitive genetic pathways remain to be further elucidated. Although the combination of CDH and craniosynostosis is rare, the fact that multiple syndromes are associated with both craniosynostosis and CDH suggests a possible overlap between craniosynostosis and CDH pathways. For instance, NR2F2, a causative CDH gene with which BCL11B has been shown to interact (54, 55), appears to have a function in mesenchymal cell differentiation in embryogenesis including a regulatory function in myogenesis, chondrogenesis, and osteogenesis (56). These processes are disturbed in CDH and craniosynostosis. NR2F2 also regulates Runx2, which has been reported to be overexpressed in some types of craniosynostosis, and has a function in the retinoic acid signaling pathway regulation, which is key in the development of CDH (1, 56–61). Further studies are needed to establish definitive CDH and craniosynostosis pathways and to assess if these pathways are interlinked.
The phenotype of our patients is similar to previously reported clinical phenotypes of patients with BCL11B missense mutations, which included craniosynostosis and CDH (6, 27, 28). Therefore, it is implied that both CDH and craniosynostosis may be features associated with BCL11B missense mutations. Further functional studies are required to assess if these variants are coincidental findings or if they indeed have a causative effect on craniosynostosis and CDH.
Patients with missense mutations in BCL11B appear to be affected more severely than patients with loss of function or other types of BCL11B mutations (28). In a previous study, the most severely affected patient carried a missense mutation in a zinc-finger domain in exon 4 (patient EII-I, Table 1) (28). BCL11B encodes for a zing finger protein transcription factor and function both as a transcriptional activator as well as a repressor (35, 62). Lessel et al. hypothesized that these missense mutations may not only lead to a loss of DNA binding but also to novel DNA binding sites in other genes (28). Similar observations were made in a previous study, which demonstrated that a mutation p.(Asn441Lys) led to both decreased binding of BCL11B to original target DNA sites as well as to the promotion of novel target DNA binding sites (27). We hypothesize that also these BCL11B missense variants may either have a dominant negative effect or induce new target genes, thereby causing a more severe phenotype in patients with these missense variants /mutations as compared to eg loss of function type of mutations. Future studies should further examine the in vivo effect of these specific BCL11B mutations in animal models to establish if the described variants are indeed disease causing mutants or coincidental findings, as shown by Goos et al. for the mutation in their patient (29).
Although we are the first to report on the co-occurrence of CDH and craniosynostosis in a patient with a BCL11B missense mutation, the co-occurrence of CDH and craniosynostosis has been described previously for several syndromes, such as Apert's syndrome and craniofrontonasal syndrome (7–22). Craniosynostosis syndromes comprise ~30% of all craniosynostosis cases (63). Syndromic craniosynostosis is highly heterogeneous and is often associated with extracranial anomalies, including neurologic, limb, ophthalmologic and cardiac anomalies. Most craniosynostosis syndromes have an autosomal dominant inheritance pattern, although many cases arise from de novo mutations (32, 64). We conducted a literature search to identify which craniosynostosis syndromes are associated with CDH. We found nine syndromes to include both CDH as well as craniosynostosis as an associated feature, based on searches in OMIM, Medline, Science Direct and a gray literature search in Google Scholar. Table 2A summarizes the genetic anomalies as well as the main features of each syndrome reported to include both craniosynostosis as well as CDH. In addition, we found seven isolated case reports that describe patients presenting with both craniosynostosis as well as CDH, which are shown in Table 2B. The supplemental information includes Table 2 with a full reference list. Although CDH appears to be a rare feature in craniosynostosis syndromes, craniosynostosis is often treatable with surgical intervention and has high survival rates. In contrast, CDH is a potentially lethal disorder and is associated strongly with poor long-term clinical outcome (66–68). Awareness of this rare feature in craniosynostosis syndromes therefore is key.
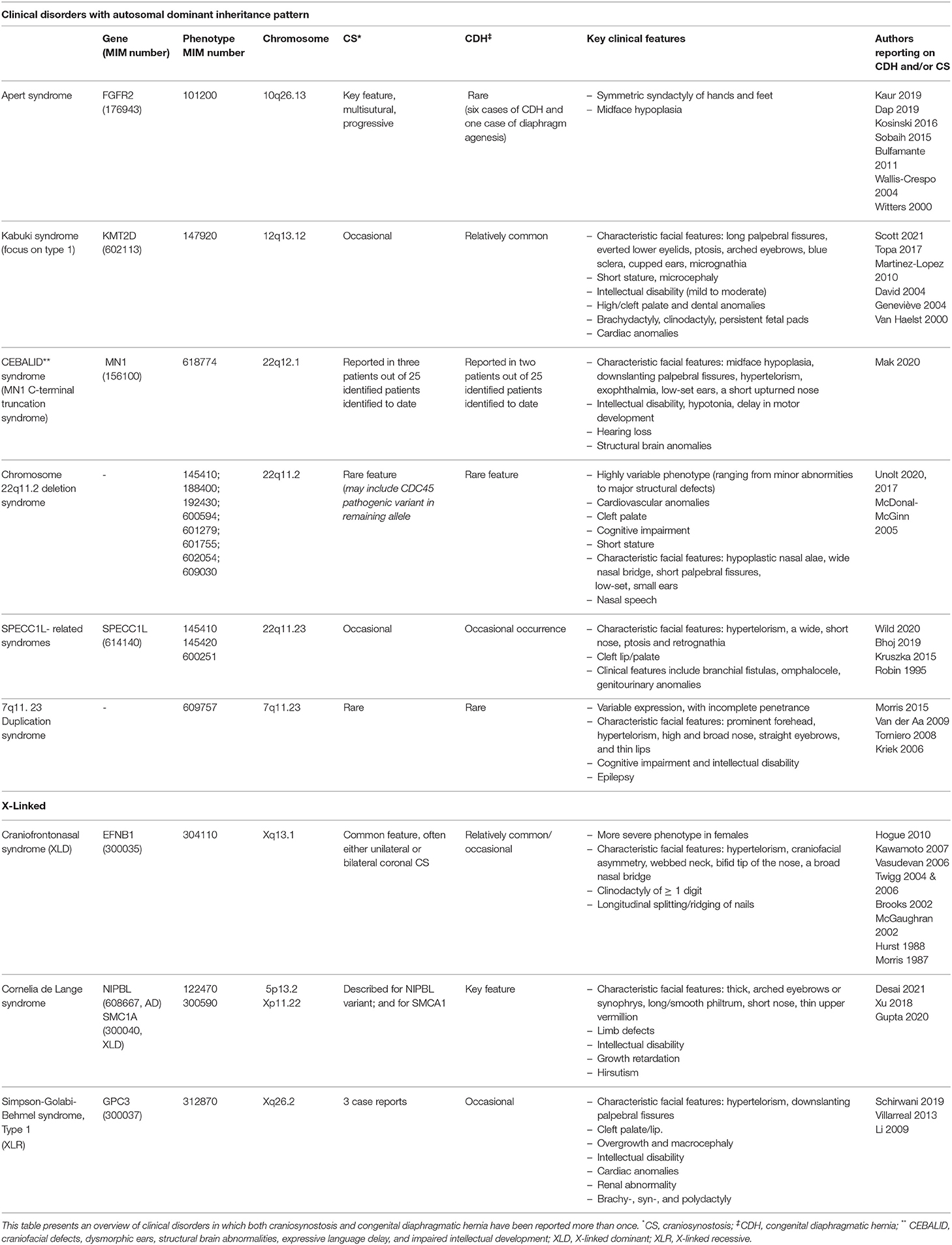
Table 2A. Overview of disorders characterized by co-occurrence of craniosynostosis and congenital diaphragmatic hernia.
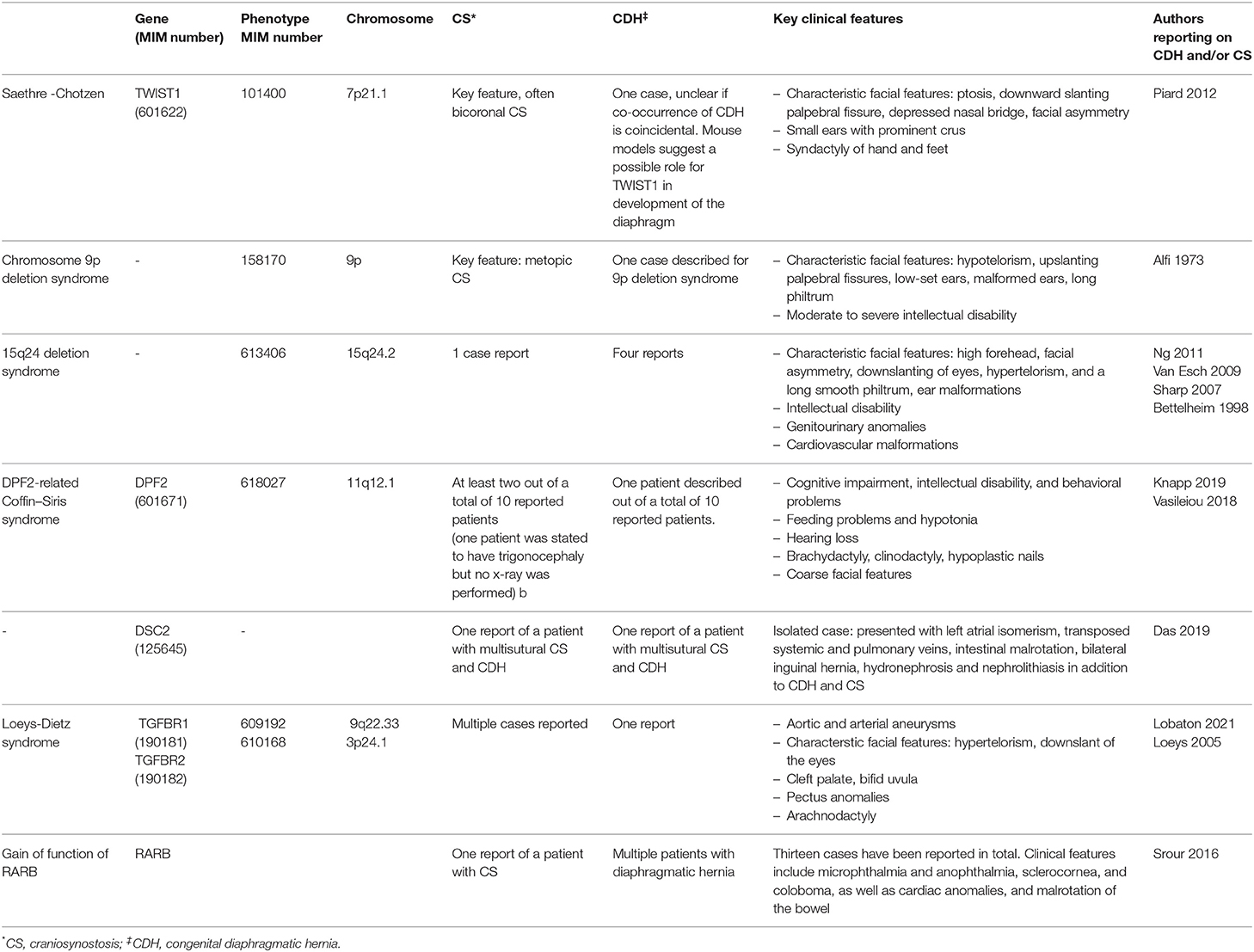
Table 2B. Isolated case reports on the co-occurrence of craniosynostosis and congenital diaphragmatic hernia.
Conclusion
This report implies that both CDH as well as craniosynostosis are features of BCL11B missense mutations. However, further studies are required to establish if BCL11B missense mutations are indeed a causative factor or if our finding was coincidental.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
LG, AK, IM, and MD contributed to the study's conception, wrote the manuscript, and provided critical revisions. LG, AG, and MD assessed the clinical and phenotypical features of the patients. FM and FJ assisted with data collection and provided genetic information. AG, QB, DV, and EB provided critical revision. All authors approved the article for submission.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the patients for their participation in this study. In addition, the authors would like to thank J. A. C. Goos and B. Versluijs for their contribution in collecting the data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.772800/full#supplementary-material
References
1. Kammoun M, Souche E, Brady P, Ding J, Cosemans N, Gratacos E, et al. Genetic profile of isolated congenital diaphragmatic hernia revealed by targeted next-generation sequencing. Prenat Diagn. (2018) 38:654–63. doi: 10.1002/pd.5327
2. McGivern MR, Best KE, Rankin J, Wellesley D, Greenlees R, Addor MC, et al. Epidemiology of congenital diaphragmatic hernia in Europe: a register-based study. Arch Dis Child Fetal Neonatal Ed. (2015) 100:F137–44. doi: 10.1136/archdischild-2014-306174
3. Gallot D, Boda C, Ughetto S, Perthus I, Robert-Gnansia E, Francannet C, et al. Prenatal detection and outcome of congenital diaphragmatic hernia: a French registry-based study. Ultrasound Obstet Gynecol. (2007) 29:276–83. doi: 10.1002/uog.3863
4. Paoletti M, Raffler G, Gaffi MS, Antounians L, Lauriti G, Zani A. Prevalence and risk factors for congenital diaphragmatic hernia: a global view. J Pediatr Surg. (2020) 55:2297–307. doi: 10.1016/j.jpedsurg.2020.06.022
5. Zaiss I, Kehl S, Link K, Neff W, Schaible T, Sutterlin M, et al. Associated malformations in congenital diaphragmatic hernia. Am J Perinatol. (2011) 28:211–8. doi: 10.1055/s-0030-1268235
6. Robert E, Kallen B, Harris J. The epidemiology of diaphragmatic hernia. Eur J Epidemiol. (1997) 13:665–73. doi: 10.1023/A:1007395727819
7. Kaur R, Mishra P, Kumar S, Sankar MJ, Kabra M, Gupta N. Apert syndrome with congenital diaphragmatic hernia: another case report and review of the literature. Clin Dysmorphol. (2019) 28:78–80. doi: 10.1097/MCD.0000000000000261
8. Bulfamante G, Gana S, Avagliano L, Fabietti I, Gentilin B, Lalatta F. Congenital diaphragmatic hernia as prenatal presentation of Apert syndrome. Prenat Diagn. (2011) 31:910–1. doi: 10.1002/pd.2788
9. Dap M, Bach-Segura P, Bertholdt C, Menzies D, Masutti JP, Klein O, et al. Variable phenotypic expression of Apert syndrome in monozygotic twins. Clin Case Rep. (2019) 7:54–7. doi: 10.1002/ccr3.1915
10. Kosinski P, Luterek K, Wielgos M. Diaphragmatic hernia as an early ultrasound manifestation of Apert syndrome. Ginekol Pol. (2016) 87:830. doi: 10.5603/GP.2016.0097
11. Sobaih BH, AlAli AA. A third report of Apert syndrome in association with diaphragmatic hernia. Clin Dysmorphol. (2015) 24:106–8. doi: 10.1097/MCD.0000000000000083
12. Wallis-Crespo MC, Gilbert-Barness E. Pathology teach and tell: acrocephalosyndactyly type I (Apert syndrome). Fetal Pediatr Pathol. (2004) 23:71–8. doi: 10.1080/15227950490423160
13. Witters I, Devriendt K, Moerman P, van Hole C, Fryns JP. Diaphragmatic hernia as the first echographic sign in Apert syndrome. Prenat Diagn. (2000) 20:404–6. doi: 10.1002/(SICI)1097-0223(200005)20:5<404::AID-PD813>3.0.CO;2-0
14. Brooks AS, van Dooren M, Hoogeboom J, Gischler S, Willems PJ, Tibboel D. Congenital diaphragmatic hernia in a female patient with craniofrontonasal syndrome. Clin Dysmorphol. (2002) 11:151–3. doi: 10.1097/00019605-200204000-00019
15. Hogue J, Shankar S, Perry H, Patel R, Vargervik K, Slavotinek A. A novel EFNB1 mutation (c.712delG) in a family with craniofrontonasal syndrome and diaphragmatic hernia. Am J Med Genet Part A. (2010) 152A:2574–7. doi: 10.1002/ajmg.a.33596
16. Hurst J, Baraitser M. Craniofrontonasal dysplasia. J Med Genet. (1988) 25:133–4. doi: 10.1136/jmg.25.2.133
17. Kawamoto HK, Heller JB, Heller MM, Urrego A, Gabbay JS, Wasson KL, et al. Craniofrontonasal dysplasia: a surgical treatment algorithm. Plast Reconstr Surg. (2007) 120:1943–56. doi: 10.1097/01.prs.0000287286.12944.9f
18. McGaughran J, Rees M, Battin M. Craniofrontonasal syndrome and diaphragmatic hernia. Am J Med Genet. (2002) 110:391–2. doi: 10.1002/ajmg.10176
19. Morris CA, Palumbos JC, Carey JC. Delineation of the male phenotype in carniofrontonasal syndrome. Am J Med Genet. (1987) 27:623–31. doi: 10.1002/ajmg.1320270315
20. Twigg SR, Kan R, Babbs C, Bochukova EG, Robertson SP, Wall SA, et al. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci USA. (2004) 101:8652–7. doi: 10.1073/pnas.0402819101
21. Twigg SR, Matsumoto K, Kidd AM, Goriely A, Taylor IB, Fisher RB, et al. The origin of EFNB1 mutations in craniofrontonasal syndrome: frequent somatic mosaicism and explanation of the paucity of carrier males. Am J Hum Genet. (2006) 78:999–1010. doi: 10.1086/504440
22. Vasudevan PC, Twigg SR, Mulliken JB, Cook JA, Quarrell OW, Wilkie AO. Expanding the phenotype of craniofrontonasal syndrome: two unrelated boys with EFNB1 mutations and congenital diaphragmatic hernia. Eur J Hum Genet. (2006) 14:884–7. doi: 10.1038/sj.ejhg.5201633
23. Cornelissen M, Ottelander B, Rizopoulos D, van der Hulst R, Mink van der Molen A, van der Horst C, et al. Increase of prevalence of craniosynostosis. J Craniomaxillofac Surg. (2016) 44:1273–9. doi: 10.1016/j.jcms.2016.07.007
24. de Jong T, Bannink N, Bredero-Boelhouwer HH, van Veelen ML, Bartels MC, Hoeve LJ, et al. Long-term functional outcome in 167 patients with syndromic craniosynostosis; defining a syndrome-specific risk profile. J Plast Reconstr Aesthet Surg. (2010) 63:1635–41. doi: 10.1016/j.bjps.2009.10.029
25. Maliepaard M, Mathijssen IM, Oosterlaan J, Okkerse JM. Intellectual, behavioral, and emotional functioning in children with syndromic craniosynostosis. Pediatrics. (2014) 133:e1608–15. doi: 10.1542/peds.2013-3077
26. Mathijssen IMJ, Craniosynostosis Working Group. Guideline. Updated guideline on treatment and management of craniosynostosis. J Craniofac Surg. (2021) 32:371–450. doi: 10.1097/SCS.0000000000007035
27. Punwani D, Zhang Y, Yu J, Cowan MJ, Rana S, Kwan A, et al. Multisystem anomalies in severe combined immunodeficiency with mutant BCL11B. N Engl J Med. (2016) 375:2165–76. doi: 10.1056/NEJMoa1509164
28. Lessel D, Gehbauer C, Bramswig NC, Schluth-Bolard C, Venkataramanappa S, van Gassen KLI, et al. BCL11B mutations in patients affected by a neurodevelopmental disorder with reduced type 2 innate lymphoid cells. Brain. (2018) 141:2299–311. doi: 10.1093/brain/awy173
29. Goos JAC, Vogel WK, Mlcochova H, Millard CJ, Esfandiari E, Selman WH, et al. A de novo substitution in BCL11B leads to loss of interaction with transcriptional complexes and craniosynostosis. Hum Mol Genet. (2019) 28:2501–13. doi: 10.1093/hmg/ddz072
30. Longoni M, High FA, Qi H, Joy MP, Hila R, Coletti CM, et al. Genome-wide enrichment of damaging de novo variants in patients with isolated and complex congenital diaphragmatic hernia. Hum Genet. (2017) 136:679–91. doi: 10.1007/s00439-017-1774-y
31. Tweedie S, Braschi B, Gray K, Jones TEM, Seal RL, Yates B, et al. Genenames.org: the HGNC and VGNC resources in 2021. Nucleic Acids Res. (2021) 49:D939–46. doi: 10.1093/nar/gkaa980
32. Goos JAC, Mathijssen IMJ. Genetic causes of craniosynostosis: an update. Mol Syndromol. (2019) 10:6–23. doi: 10.1159/000492266
33. CADD. University of Washington Hudson-Alpha Institute for Biotechnology and Berlin Institute of Health. Combined Annotation Dependent Depletion; Single Nucleotide Variant (SNV) Lookup. (2021). Available online at: https://cadd.gs.washington.edu/snv
34. Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. (2021) 13:31. doi: 10.1186/s13073-021-00835-9
35. Lennon MJ, Jones SP, Lovelace MD, Guillemin GJ, Brew BJ. Bcl11b-A critical neurodevelopmental transcription factor-roles in health and disease. Front Cell Neurosci. (2017) 11:89. doi: 10.3389/fncel.2017.00089
36. Kyrylkova K, Iwaniec UT, Philbrick KA, Leid M. BCL11B regulates sutural patency in the mouse craniofacial skeleton. Dev Biol. (2016) 415:251–60. doi: 10.1016/j.ydbio.2015.10.010
37. Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, Mishima Y, et al. Bcl11b is required for differentiation and survival of alphabeta T lymphocytes. Nat Immunol. (2003) 4:533–9. doi: 10.1038/ni927
38. Li L, Leid M, Rothenberg EV. An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science. (2010) 329:89–93. doi: 10.1126/science.1188989
39. Albu DI, VanValkenburgh J, Morin N, Califano D, Jenkins NA, Copeland NG, et al. Transcription factor Bcl11b controls selection of invariant natural killer T-cells by regulating glycolipid presentation in double-positive thymocytes. Proc Natl Acad Sci USA. (2011) 108:6211–6. doi: 10.1073/pnas.1014304108
40. Arlotta P, Molyneaux BJ, Jabaudon D, Yoshida Y, Macklis JD. Ctip2 controls the differentiation of medium spiny neurons and the establishment of the cellular architecture of the striatum. J Neurosci. (2008) 28:622–32. doi: 10.1523/JNEUROSCI.2986-07.2008
41. Golonzhka O, Liang X, Messaddeq N, Bornert JM, Campbell AL, Metzger D, et al. Dual role of COUP-TF-interacting protein 2 in epidermal homeostasis and permeability barrier formation. J Invest Dermatol. (2009) 129:1459–70. doi: 10.1038/jid.2008.392
42. Golonzhka O, Metzger D, Bornert JM, Bay BK, Gross MK, Kioussi C, et al. Ctip2/Bcl11b controls ameloblast formation during mammalian odontogenesis. Proc Natl Acad Sci USA. (2009) 106:4278–83. doi: 10.1073/pnas.0900568106
43. Kastner P, Chan S, Vogel WK, Zhang LJ, Topark-Ngarm A, Golonzhka O, et al. Bcl11b represses a mature T-cell gene expression program in immature CD4(+)CD8(+) thymocytes. Eur J Immunol. (2010) 40:2143–54. doi: 10.1002/eji.200940258
44. Simon R, Baumann L, Fischer J, Seigfried FA, De Bruyckere E, Liu P, et al. Structure-function integrity of the adult hippocampus depends on the transcription factor Bcl11b/Ctip2. Genes Brain Behav. (2016) 15:405–19. doi: 10.1111/gbb.12287
45. Simon R, Brylka H, Schwegler H, Venkataramanappa S, Andratschke J, Wiegreffe C, et al. A dual function of Bcl11b/Ctip2 in hippocampal neurogenesis. EMBO J. (2012) 31:2922–36. doi: 10.1038/emboj.2012.142
46. Uddin MN, Zhang Y, Harton JA, MacNamara KC, Avram D. TNF-alpha-dependent hematopoiesis following Bcl11b deletion in T cells restricts metastatic melanoma. J Immunol. (2014) 192:1946–53. doi: 10.4049/jimmunol.1301976
47. Vanvalkenburgh J, Albu DI, Bapanpally C, Casanova S, Califano D, Jones DM, et al. Critical role of Bcl11b in suppressor function of T regulatory cells and prevention of inflammatory bowel disease. J Exp Med. (2011) 208:2069–81. doi: 10.1084/jem.20102683
48. Kyrylkova K, Kyryachenko S, Biehs B, Klein O, Kioussi C, Leid M. BCL11B regulates epithelial proliferation and asymmetric development of the mouse mandibular incisor. PLoS ONE. (2012) 7:e37670. doi: 10.1371/journal.pone.0037670
49. Albu DI, Feng D, Bhattacharya D, Jenkins NA, Copeland NG, Liu P, et al. BCL11B is required for positive selection and survival of double-positive thymocytes. J Exp Med. (2007) 204:3003–15. doi: 10.1084/jem.20070863
50. Timberlake AT, Choi J, Zaidi S, Lu Q, Nelson-Williams C, Brooks ED, et al. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife. (2016) 5:e20125. doi: 10.7554/eLife.20125
51. Sharma VP, Fenwick AL, Brockop MS, McGowan SJ, Goos JA, Hoogeboom AJ, et al. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat Genet. (2013) 45:304–7. doi: 10.1016/S0140-6736(13)60554-1
52. di Rocco F, Baujat G, Arnaud E, Renier D, Laplanche JL, Daire VC, et al. Clinical spectrum and outcomes in families with coronal synostosis and TCF12 mutations. Eur J Hum Genet. (2014) 22:1413–6. doi: 10.1038/ejhg.2014.57
53. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. (2019) 47:D607–13. doi: 10.1093/nar/gky1131
54. Sidwell T, Rothenberg EV. Epigenetic dynamics in the function of t-lineage regulatory factor Bcl11b. Front Immunol. (2021) 12:669498. doi: 10.3389/fimmu.2021.669498
55. Avram D, Fields A, Pretty On Top K, Nevrivy DJ, Ishmael JE, Leid M. Isolation of a novel family of C(2)H(2) zinc finger proteins implicated in transcriptional repression mediated by chicken ovalbumin upstream promoter transcription factor (COUP-TF) orphan nuclear receptors. J Biol Chem. (2000) 275:10315–22. doi: 10.1074/jbc.275.14.10315
56. Xie X, Qin J, Lin SH, Tsai SY, Tsai MJ. Nuclear receptor chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) modulates mesenchymal cell commitment and differentiation. Proc Natl Acad Sci USA. (2011) 108:14843–8. doi: 10.1073/pnas.1110236108
57. Cuellar A, Bala K, Di Pietro L, Barba M, Yagnik G, Liu JL, et al. Gain-of-function variants and overexpression of RUNX2 in patients with nonsyndromic midline craniosynostosis. Bone. (2020) 137:115395. doi: 10.1016/j.bone.2020.115395
58. Lee KN, Jang WG, Kim EJ, Oh SH, Son HJ, Kim SH, et al. Orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) protein negatively regulates bone morphogenetic protein 2-induced osteoblast differentiation through suppressing runt-related gene 2 (Runx2) activity. J Biol Chem. (2012) 287:18888–99. doi: 10.1074/jbc.M111.311878
59. Kruse SW, Suino-Powell K, Zhou XE, Kretschman JE, Reynolds R, Vonrhein C, et al. Identification of COUP-TFII orphan nuclear receptor as a retinoic acid-activated receptor. PLoS Biol. (2008) 6:e227. doi: 10.1371/journal.pbio.0060227
60. Kardon G, Ackerman KG, McCulley DJ, Shen Y, Wynn J, Shang L, et al. Congenital diaphragmatic hernias: from genes to mechanisms to therapies. Dis Model Mech. (2017) 10:955–70. doi: 10.1242/dmm.028365
61. Clugston RD, Zhang W, Alvarez S, de Lera AR, Greer JJ. Understanding abnormal retinoid signaling as a causative mechanism in congenital diaphragmatic hernia. Am J Respir Cell Mol Biol. (2010) 42:276–85. doi: 10.1165/rcmb.2009-0076OC
62. Kominami R. Role of the transcription factor Bcl11b in development and lymphomagenesis. Proc Jpn Acad Ser B Phys Biol Sci. (2012) 88:72–87. doi: 10.2183/pjab.88.72
63. Wilkie AOM, Johnson D, Wall SA. Clinical genetics of craniosynostosis. Curr Opin Pediatr. (2017) 29:622–8. doi: 10.1097/MOP.0000000000000542
64. Twigg SR, Wilkie AO. A genetic-pathophysiological framework for craniosynostosis. Am J Hum Genet. (2015) 97:359–77. doi: 10.1016/j.ajhg.2015.07.006
65. Srour M, Caron V, Pearson T, Nielsen SB, Levesque S, Delrue MA, et al. Gain-of-function mutations in RARB cause intellectual disability with progressive motor impairment. Hum Mutat. (2016) 37:786–93. doi: 10.1002/humu.23004
66. Congenital Diaphragmatic Hernia Study G, Lally KP, Lally PA, Lasky RE, Tibboel D, Jaksic T, et al. Defect size determines survival in infants with congenital diaphragmatic hernia. Pediatrics. (2007) 120:e651–7. doi: 10.1542/peds.2006-3040
67. Coughlin MA, Werner NL, Gajarski R, Gadepalli S, Hirschl R, Barks J, et al. Prenatally diagnosed severe CDH: mortality and morbidity remain high. J Pediatr Surg. (2016) 51:1091–5. doi: 10.1016/j.jpedsurg.2015.10.082
Keywords: case report, craniosynostosis, congenital diaphragmatic hernia (CDH), BCL11B, craniosynostosis syndromes
Citation: Gaillard L, Goverde A, van den Bosch QCC, Jehee FS, Brosens E, Veenma D, Magielsen F, de Klein A, Mathijssen IMJ and van Dooren MF (2021) Case Report and Review of the Literature: Congenital Diaphragmatic Hernia and Craniosynostosis, a Coincidence or Common Cause? Front. Pediatr. 9:772800. doi: 10.3389/fped.2021.772800
Received: 08 September 2021; Accepted: 01 October 2021;
Published: 26 November 2021.
Edited by:
Thomas Schaible, University of Heidelberg, GermanyReviewed by:
Theo Dassios, King's College Hospital NHS Foundation Trust, United KingdomAugusto Schmidt, University of Miami, United States
Copyright © 2021 Gaillard, Goverde, van den Bosch, Jehee, Brosens, Veenma, Magielsen, de Klein, Mathijssen and van Dooren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda Gaillard, l.gaillard@erasmusmc.nl
†These authors share last authorship