 Baiyu Lyu
Baiyu Lyu Yan Dong
Yan Dong Juan Kang
Juan Kang- Department of Pediatrics, Shanghai Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China
Background: The nucleus accumbens associated 1 (NACC1) gene is a transcription factor member of the BTB/POZ family. A de novo heterozygous c.892C>T (p.Arg298Trp) variant in the NACC1 may define a syndrome characterized by intellectual disability, infantile epilepsy, congenital cataract, and feeding difficulties.
Case Presentation: We report a new case with a neurodevelopmental disorder characterized by severe intellectual disability, infantile epilepsy, congenital cataract, and feeding difficulties. Brain MRI reveals brain dysplasia. We observe a de novo heterozygous c.892C>T (p.Arg298Trp) variant in the NACC1 gene in this case. Now, the child regularly goes to the hospital for rehabilitation training (once a month). Sodium Valproate (10 mg/kg/day) and Clobazam (10 mg/kg/day) are used in the treatment of epilepsy. A total of three articles were screened, and two papers were excluded. The search revealed one article related to a syndrome caused by a de novo heterozygous c.892C>T (p.Arg298Trp) variant in the NACC1; they screened the main clinical features of eight cases of a syndrome, which were summarized and analyzed.
Conclusions: The NACC1 gene is a member of the BTB/POZ family of transcription factors. A de novo heterozygous c.892C>T (p.Arg298Trp) variant in the NACC1 may define a syndrome characterized by intellectual disability, infantile epilepsy, congenital cataract, and feeding difficulties. At present, there is no effective cure. In the future, we need more cases to determine the phenotype–genotype correlation of NACC1 variants. Many questions remain to be answered, and many challenges remain to be faced. Future transcriptional studies may further clarify this rare, recurrent variant, and could potentially lead to targeted therapies.
Introduction
NACC1 gene is a member of the BTB/POZ family of transcription factors (1). Many studies have found that NACC1 is highly expressed in various tumors (2). The highly expressed NACC1 is closely related to cell viability, migration, and tumor recurrence (3, 4). It has also been identified as a BTB gene associated with cancer. It has been reported that the NACC1 gene is associated with intellectual disability. Some studies suggest that the NACCI is a genome-wide significant disease-associated gene.
The prevalence of intellectual disability (ID) in newborns is 2–3% (5). In most cases, the cause is unknown (5), while 0.5% suffer from severe intellectual disability (ID), which is mostly genetically related (6). Congenital cataract (CC) refers to the genetic abnormality caused by lens opacity at birth. The prevalence of CC in every 10,000 children is 2.2–13.6 (7). Schoch et al. reported that the NACC1 gene mutations are related to a syndrome characterized by epilepsy, cataracts, feedings difficulties, and neurodevelopmental disorders (1). Although the gene coverage is relatively high, the single loss of function and the lower-than-expected number of missense variants indicates that mutations in NACC1 are affected by selection and are very rare. The predicted pathogenic variants may increase human disease risks (1). We will review the literature and report that a new case on the NACC1 c.892C>T (p.Arg298Trp) variant causes a syndrome. This article aims to expand the knowledge on phenotypic features and introduce the biological role of NACC1. This is the first case report from China and the eighth so far.
Clinical Report
The patient was a 4-year-old Chinese Han female of de novo variant c.892C>T (p.Arg298Trp) in the NACC1 that causes a syndrome characterized by severe intellectual disability, infantile epilepsy, congenital cataract, and feeding difficulties. The patient was born at 37+3 weeks of gestation to non-consanguineous Chinese parents with one healthy twin boy and no family history of epilepsy or disability syndromes. She was born, and the pregnancy was uneventful. She was admitted to the neonatal intensive care unit on the third day of life for neonatal hyperbilirubinemia, with good recovery.
When she was 3 months old, she could not stand her head upright, and a white substance was visible in front of the pupil of the left eye, which moved with the eyeball. Ophthalmology was diagnosed as a congenital cataract.
At 7 months of age, the girl still could not stand her head upright and turn over. She could chase sounds and laughs when people tease her, and she has difficulty feeding (only bottle feeding, and easy to choke). Also, at 7 months of age, she suffered from her first seizure characterized by eye blinking, cyanosis around the mouth, and ankylosis of limbs. EEG findings show infantile epilepsy.
At 2 years of age, on clinical observation, she was behind her peers in intellectual development. She was diagnosed with intellectual disability by Wechsler Intelligence Scale for Children Fourth Edition-Chinese. Brain MRI showed widening frontal and temporal sulci and widening the subarachnoid space in the frontal and temporal regions. The volume of the white matter in the center of the semioval was small, and the corpus callosum was thin. At the same time, as rehabilitation training, levetiracetam (10 mg/kg/day) therapy was used for epilepsy.
When she was 4 years old, She could turn over, but she could not sit alone and walk and talk. Hip x-ray suggests congenital dislocation of the hip. At the same time, epilepsy was not well-controlled. Now, Sodium Valproate (10 mg/kg/day) and Clobazam (10 mg/kg/day) are used in the treatment of epilepsy. Congenital cataract lens removal surgery was carried out when the child was 4 years old. A timeline with relevant data from the patient episode of care is shown in Figure 1.
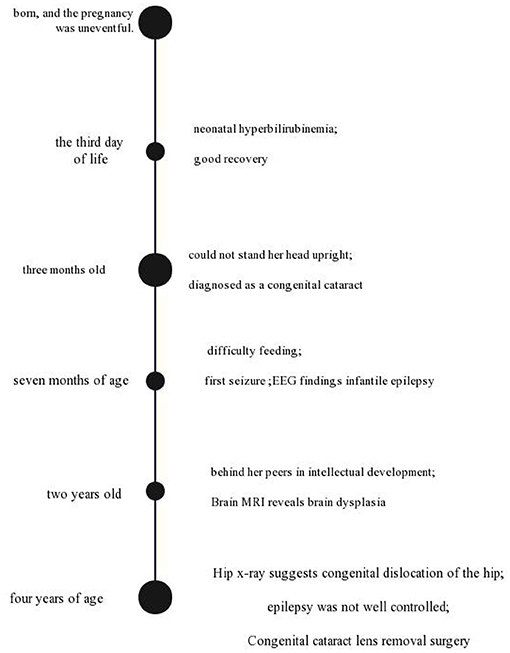
Figure 1. A timeline with relevant data from the patient episode of care.
Diagnostic Assessment
To confirm the diagnosis, sequencing was performed with the consent of the family members. Whole exon sequencing of the girl and parents and twin brother showed that the child was de novo heterozygous c.892C>T (p.Arg298Trp) variant in the NACC1 (GenBank NM_052876_3), as shown in Figure 2. Now, the child regularly goes to the hospital for rehabilitation training (once a month). Sodium Valproate (10 mg/kg/day) and Clobazam (10 mg/kg/day) are used in the treatment of epilepsy.
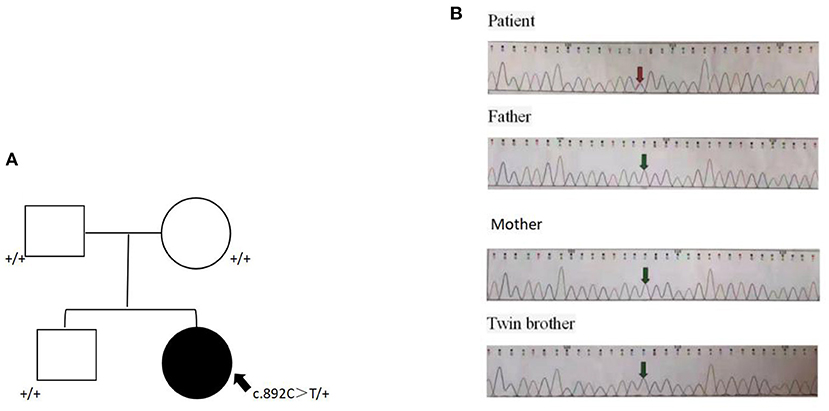
Figure 2. De novo c.892C>T (p.Arg298Trp) missense change in NACC1. (A) Pedigree of the family. A heterozygous missense was detected in the proband but not the parents. (B) Sanger validation of the missense variant in the proband and parents. Arrow indicates the site of the variant.
Literature Review
Using “NACC1” and “intellectual disability” as keywords, the Chinese and English databases (PubMed, CNKI, Wanfang, and VIP databases) were searched for papers published up to July 15, 2021. A total of three articles were screened. Two papers (8, 9) were excluded because of cases without infantile epilepsy, congenital cataract, and feeding difficulties. The critical phenotypic features of all patients with NACC1 c.892C>T (p.Arg298Trp) variant published to date are summarized in Table 1.
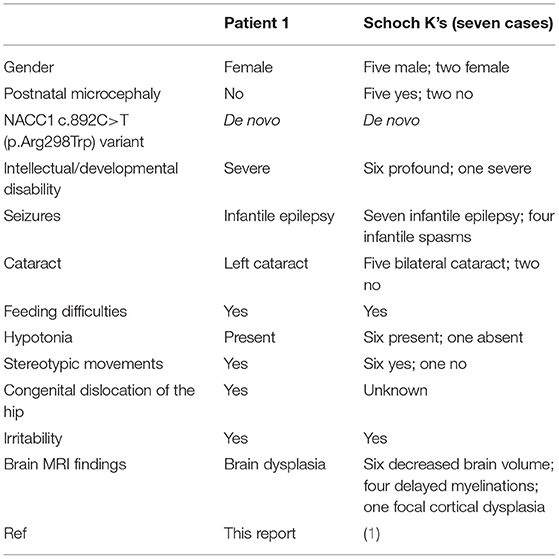
Table 1. Phenotypic features of all patients with NACC1 c.892C>T (p.Arg298Trp) variant published to date.
Discussion
In this case, a de novo heterozygous c.892C>T (p.Arg298Trp) variant in the NACC1 causes a severe intellectual disability, infantile epilepsy, congenital cataract, profound developmental delays, feeding difficulties, and congenital dislocation of the hip. Previously reported microduplication/microdeletion syndrome involving chromosome 19p13.13 includes most NACC1 genes (10). Previous research has shown that mosaic mutations frequently occur in individuals diagnosed with autism spectrum disorders and intellectual disability (11). One individual with intellectual disability (IQ of ~45), schizo-affective disorder, and autism has been noted with a de novo missense allele c.1402C>T (p.Arg468Cys) in NACC1 (6). Another patient with autism spectrum disorder was distinguished to have a de novo splicing variant (probably LoF), c.946þ2T>C, in NACC1 (12).
Many studies have found that the NACC1 is highly expressed in a variety of tumors, such as ovarian cancer (3, 13), cervical cancer (3, 4), endometrial cancer (3, 14), breast cancer, renal cell cancer, pancreatic cancer (15, 16), and melanoma (17). Highly expressed NACC1 goes hand in hand with cell proliferation, migration and invasion, chemotherapy resistance, tumor recurrence, and poor prognosis (4). NACC1 knockout mouse embryos and newborns have lower survival rates (18).
Nucleus accumbens-associated protein 1 (NAC1) is encoded by the NACC1 gene (15) and is a member of the BTB/POZ protein family (19). BTB/POZ proteins can recruit histone deacetylases (20), ubiquitin-ligases (21), and corepressors (22) to construct complexes that modulate gene expression and typically modify chromatin conformation (19).
NAC1 was initially identified and cloned as a cocaine-inducible transcript from the nucleus accumbens, a unique forebrain structure involved in addictive behavior and reward motivation (15). Subsequently, NAC1 has been shown to play critical roles in a variety of biological processes, including pathogenesis of human cancer (23), the proliferation of embryonic stem cells, maintenance of stemness (24), participation in the psychomotor response of rats after taking cocaine (25), and targeting substrates to cullin-based E3 ligases for ubiquitin-dependent proteasome degradation (26). NAC1 plays an essential role in the pluripotency of embryonic stem cells (27) through direct transcriptional regulation of c-Myc (28). Furthermore, It has recently been demonstrated that NAC1 represses neuroectodermal fate selection and promotes mesendodermal in embryonic stem cells, in concert with the pluripotency transcription factors, Tcf3, Oct4, and Sox2 (15, 29). NAC1 is essential to generate iPSC (15). NAC1 is also a necessary part of RIG-I-like receptor-mediated innate immune responses against viral infection (19).
NAC1 plays a vital role in normal neurologic function by maintaining synaptic plasticity and reducing protein turnover in dendritic cells (1, 30). As a transcriptional repressor, NAC1 regulates cell growth, senescence, epithelial–mesenchymal transition, and autophagy. NAC1 prevents cytotoxicity by stabilizing hypoxia-inducible factor-1α and regulating glucose levels, thereby playing a role in the hypoxic tumor environment (31). NAC1 forms a homodimer or heterodimers with other binding partners through the BTB/POZ domain and exerts transcriptional inhibition through the recruitment of histone deacetylase (32). It has also been predicted that the BEN domain participates in chromatin organization and transcriptional regulation by mediating protein–DNA and protein–protein interactions (33). Although most of the functions of NAC1 are related to transcriptional regulation, it is also associated with cytokinesis through the NACC-1/actin/profilin-1 complex and the post-translational role of the protein in cancer cells (28, 31).
The c. 892C> T transition occurs at a CpG dinucleotide within an arginine codon, a highly variable CpG pattern associated with de novo events at many loci in setting advanced paternal age. However, advanced paternal age was not a common feature of this group (1). The p.Arg298Trp variant is located outside the BTB/POZ domain that is important for cancer progression. However, it is unknown whether individuals with the p.Arg298Trp variant are at risk for cancer (1). It has been reported that a missense variant located in the BEN domain is associated with intellectual disability (6). Because of the characteristic phenotype related to the p.Arg298Trp variant, future transcriptional studies may further clarify this rare, recurrent variant, potentially leading to targeted therapies.
The number of cases is still limited, many questions remain to be answered, and many challenges remain to be faced. In this case, after the diagnosis, the patient's families were very depressed when they learned that it is a rare disease for which there is no effective cure at present. However, they did not give up hope. They believe that future medical advancement will find a way to cure the disease.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Shanghai Ninth People's Hospital, Shanghai Jiaotong University School of Medicine. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the legal guardian for publication of this case report and any accompanying images.
Author Contributions
BL and YD conceived and supervised the study. BL analyzed data. BL and JK wrote the manuscript. BL, YD, and JK made manuscript revisions. All authors approved the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are very grateful for the cooperation and support of the family.
References
1. Schoch K, Meng L, Szelinger S, Bearden DR, Stray-Pedersen A, Busk OL, et al. A recurrent de novo variant in NACC1 causes a syndrome characterized by infantile epilepsy, cataracts, and profound developmental delay. Am J Hum Genet. (2017) 100:343–51. doi: 10.1016/j.ajhg.2016.12.013
2. Zhang H, Zhang R, Zhang G, Liu W, Ma Z, Yue C, et al. Clinical significance of miR-1298 in cervical cancer and its biological function in vitro. Oncol Lett. (2021) 21:401. doi: 10.3892/ol.2021.12662
3. Li L, Yu H, Ren Q. MiR-218-5p Suppresses the progression of retinoblastoma through targeting NACC1 and inhibiting the AKT/mTOR signaling pathway. Cancer Manag Res. (2020) 12:6959–67. doi: 10.2147/CMAR.S246142
4. Yeasmin S, Nakayama K, Rahman MT, Rahman M, Ishikawa M, Katagiri A, et al. Biological and clinical significance of NAC1 expression in cervical carcinomas: a comparative study between squamous cell carcinomas and adenocarcinomas/adenosquamous carcinomas. Hum Pathol. (2012) 43:506–19. doi: 10.1016/j.humpath.2011.05.021
5. Markovic-Jovanovic SR, Milovanovic JD, Jovanovic AN, Zivkovic JB, Balovic AD, Nickovic V, et al. Comorbidities in children with intellectual disabilities. Birth Defects Res. (2020) 112:54–61. doi: 10.1002/bdr2.1587
6. Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. (2014) 511:344–7. doi: 10.1038/nature13394
7. Wang X, Qin Y, Abudoukeremuahong A, Dongye M, Zhang X, Wang D, et al. Elongated axial length and myopia-related fundus changes associated with the Arg130Cys mutation in the LIM2 gene in four Chinese families with congenital cataracts. Ann Transl Med. (2021) 9:235. doi: 10.21037/atm-20-4275
8. Pavinato L, Villamor-Payà M, Sanchiz-Calvo M, Andreoli C, Gay M, Vilaseca M, et al. Functional analysis of TLK2 variants and their proximal interactomes implicates impaired kinase activity and chromatin maintenance defects in their pathogenesis. J Med Genet. (2020). doi: 10.1136/jmedgenet-2020-107281. [Epub ahead of print].
9. Bellucco FT, de Mello CB, Meloni VA, Melaragno MI. Malan syndrome in a patient with 19p13.2p13.12 deletion encompassing NFIX and CACNA1A genes: case report and review of the literature. Mol Genet Genom Med. (2019) 7:e997. doi: 10.1002/mgg3.997
10. Jorge R, Silva C, Agueda S, Doria S, Leao M. Intellectual disability and overgrowth-A new case of 19p13.13 microdeletion syndrome with digital abnormalities. Am J Med Genet A. (2015) 167A:2839–43. doi: 10.1002/ajmg.a.37280
11. Freed D, Pevsner J. The contribution of mosaic variants to autism spectrum disorder. PLoS Genet. (2016) 12:e1006245. doi: 10.1371/journal.pgen.1006245
12. Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. (2014) 515:216–21. doi: 10.1038/nature13908
13. Nakayama K, Nakayama N, Davidson B, Sheu JJ, Jinawath N, Santillan A, et al. A BTB/POZ protein, NAC-1, is related to tumor recurrence and is essential for tumor growth and survival. Proc Natl Acad Sci USA. (2006) 103:18739–44. doi: 10.1073/pnas.0604083103
14. Ishikawa M, Nakayama K, Yeasmin S, Katagiri A, Iida K, Nakayama N, et al. NAC1, a potential stem cell pluripotency factor expression in normal endometrium, endometrial hyperplasia and endometrial carcinoma. Int J Oncol. (2010) 36:1097–103. doi: 10.3892/ijo_00000591
15. Nakayama N, Sakashita G, Nagata T, Kobayashi N, Yoshida H, Park SY, et al. Nucleus accumbens-associated protein 1 binds DNA directly through the BEN domain in a sequence-specific manner. Biomedicines. (2020) 8:608. doi: 10.3390/biomedicines8120608
16. Nakayama N, Sakashita G, Nariai Y, Kato H, Sinmyozu K, Nakayama JI, et al. Cancer-related transcription regulator protein NAC1 forms a protein complex with CARM1 for ovarian cancer progression. Oncotarget. (2018) 9:28408–20. doi: 10.18632/oncotarget.25400
17. Jiao H, Jiang S, Wang H, Li Y, Zhang W. Upregulation of LINC00963 facilitates melanoma progression through miR-608/NACC1 pathway and predicts poor prognosis. Biochem Biophys Res Commun. (2018) 504:34–9. doi: 10.1016/j.bbrc.2018.08.115
18. Yap KL, Sysa-Shah P, Bolon B, Wu RC, Gao M, Herlinger AL, et al. Loss of NAC1 expression is associated with defective bony patterning in the murine vertebral axis. PLoS ONE. (2013) 8:e69099. doi: 10.1371/journal.pone.0069099
19. Xia Z, Xu G, Nie L, Liu L, Peng N, He Q, et al. NAC1 potentiates cellular antiviral signaling by bridging MAVS and TBK1. J Immunol. (2019) 203:1001–11. doi: 10.4049/jimmunol.1801110
20. Rui J, Liu H, Zhu X, Cui Y, Liu X. Epigenetic silencing of CD8 genes by ThPOK-mediated deacetylation during CD4 T cell differentiation. J Immunol. (2012) 189:1380–90. doi: 10.4049/jimmunol.1201077
21. Mathew R, Seiler MP, Scanlon ST, Mao AP, Constantinides MG, Bertozzi-Villa C, et al. BTB-ZF factors recruit the E3 ligase cullin 3 to regulate lymphoid effector programs. Nature. (2012) 491:618–21. doi: 10.1038/nature11548
22. Melnick A, Ahmad KF, Arai S, Polinger A, Ball H, Borden KL, et al. In-depth mutational analysis of the promyelocytic leukemia zinc finger BTB/POZ domain reveals motifs and residues required for biological and transcriptional functions. Mol Cell Biol. (2000) 20:6550–67. doi: 10.1128/MCB.20.17.6550-6567.2000
23. Shih Ie M, Nakayama K, Wu G, Nakayama N, Zhang J, Wang TL. Amplification of the ch19p13.2 NACC1 locus in ovarian high-grade serous carcinoma. Mod Pathol. (2011) 24:638–45. doi: 10.1038/modpathol.2010.230
24. Ma T, Wang Z, Guo Y, Pei D. The C-terminal pentapeptide of Nanog tryptophan repeat domain interacts with Nac1 and regulates stem cell proliferation but not pluripotency. J Biol Chem. (2009) 284:16071–81. doi: 10.1074/jbc.M109.005041
25. Acuna-Hidalgo R, Bo T, Kwint MP, van de Vorst M, Pinelli M, Veltman JA, et al. Post-zygotic point mutations are an underrecognized source of de novo genomic variation. Am J Hum Genet. (2015) 97:67–74. doi: 10.1016/j.ajhg.2015.05.008
26. Shen H, Korutla L, Champtiaux N, Toda S, LaLumiere R, Vallone J, et al. NAC1 regulates the recruitment of the proteasome complex into dendritic spines. J Neurosci. (2007) 27:8903–13. doi: 10.1523/JNEUROSCI.1571-07.2007
27. Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. (2008) 132:1049–61. doi: 10.1016/j.cell.2008.06.004
28. Ruan Y, He J, Wu W, He P, Tian Y, Xiao L, et al. Nac1 promotes self-renewal of embryonic stem cells through direct transcriptional regulation of c-Myc. Oncotarget. (2017) 8:47607–18. doi: 10.18632/oncotarget.17744
29. Choi H, Park HJ, Kim H, Kim J, Lee YK, Kim J. Nac1 facilitates pluripotency gene activation for establishing somatic cell reprogramming. Biochem Biophys Res Commun. (2019) 518:253–8. doi: 10.1016/j.bbrc.2019.08.043
30. Shashi V, Pena LD, Kim K, Burton B, Hempel M, Schoch K, et al. De novo truncating variants in ASXL2 are associated with a unique and recognizable clinical phenotype. Am J Hum Genet. (2016) 99:991–9. doi: 10.1016/j.ajhg.2016.08.017
31. Morita K, Fujii T, Itami H, Uchiyama T, Nakai T, Hatakeyama K, et al. NACC1, as a target of MicroRNA-331-3p, regulates cell proliferation in urothelial carcinoma cells. Cancers. (2018) 10:347. doi: 10.3390/cancers10100347
32. Korutla L, Wang P, Jackson TG, Mackler SA. NAC1, a POZ/BTB protein that functions as a corepressor. Neurochem Int. (2009) 54:245–52. doi: 10.1016/j.neuint.2008.12.008
Keywords: NACC1, intellectual disability, infantile epilepsy, congenital cataract, feeding difficulties
Citation: Lyu B, Dong Y and Kang J (2021) A New Case of de novo Variant c.892C>T (p.Arg298Trp) in NACC1: A First Case Report From China. Front. Pediatr. 9:754261. doi: 10.3389/fped.2021.754261
Received: 06 August 2021; Accepted: 04 October 2021;
Published: 15 November 2021.
Edited by:
Hong Ni, Children's Hospital of Soochow University, ChinaReviewed by:
Kuokuo Li, Anhui Medical University, ChinaFrancesca Felicia Operto, University of Salerno, Italy
Copyright © 2021 Lyu, Dong and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Kang, a2FuZ2p1YW45OUBxcS5jb20=
†These authors have contributed equally to this work