 Jenna Diaz1†
Jenna Diaz1† Ashley G. Fonseca1*†Richard Arboleda2‡Alejandro Frade1Maria Pilar Gennaro3‡Parul Jayakar3‡Paula Schleifer3‡Erick Hernandez2‡
Ashley G. Fonseca1*†Richard Arboleda2‡Alejandro Frade1Maria Pilar Gennaro3‡Parul Jayakar3‡Paula Schleifer3‡Erick Hernandez2‡- 1Department of Medical Education, Nicklaus Children's Hospital, Miami, FL, United States
- 2Department of Pediatric Gastroenterology, Hepatology, and Nutrition, Nicklaus Children's Hospital, Miami, FL, United States
- 3Department of Neurogenetics, Nicklaus Children's Hospital, Miami, FL, United States
Background: Wilson disease (WD) and glucose transporter type 1 (GLUT1) deficiency syndrome are two syndromes with different modes of inheritance but share certain similarities on neurological presentation. To date we have not found previous reports of an association between these two disorders.
Case Presentation: Here we describe a 9-year-old male with global developmental delay that presented with intermittent and sudden onset weakness that first occurred at age 3. He was diagnosed with a mutation in the SLC2A1 (Solute Carrier Family 2 Member 1) gene, which results in GLUT1 deficiency. A ketogenic diet could not be started because of unexplained elevated liver enzymes. Due to his liver enzymes' persistent elevation, further investigations demonstrated mildly decreased ceruloplasmin levels, high basal 24-h urinary copper excretion, and an elevated hepatic parenchymal copper concentration on liver biopsy, consistent with WD. Genetic testing revealed two separate mutations in the ATP7B (ATPase Copper Transporting Beta) gene, consistent with WD. The patient was treated with a low copper diet, zinc acetate, and trientine hydrochloride. When liver enzymes normalized, he was subsequently started on a ketogenic diet with improvement in neurological symptoms. His neurological symptoms were most likely secondary to GLUT1 deficiency syndrome, as WD's neurological symptoms are primarily observed in the second decade of life.
Conclusion: Recent studies have demonstrated the importance of genetic testing upon unexplained persistent elevation of liver enzymes. This case highlights the importance of carefully evaluating a patient with an unexplained liver disorder, even in the presence of primary neurological disease, as it can have significant therapeutic implications.
Introduction
Glucose transporter type 1 (GLUT1) deficiency is caused by impaired glucose transport regulation into the brain due to a defect in the SLC2A1 gene on chromosome 1p34.2 and can be inherited in an autosomal dominant manner. However, most acquire the disease via a spontaneous mutation in utero (1). Patients often present in infancy with developmental delay, acquired microcephaly, intractable seizures, and dystonia. Later in life, they may have ataxia, paroxysmal neurologic events, and paroxysmal exertion-induced dyskinesia with or without epilepsy (1). The hallmark of GLUT1 deficiency is low CSF glucose concentration with normoglycemia (2).
Wilson disease (WD) is an autosomal recessive disorder of copper metabolism caused by a mutation in the ATP7B (ATPase Copper Transporting Beta) gene on chromosome 13q14.3 (3, 4). Mutations in the ATP7B gene can lead to detrimental copper accumulation. The prevalence of specific mutations in Wilson disease varies by geographic location. The p.H1069Q mutation is one of the most common mutations and has a population allelic frequency of 10–40%. Other common mutations that exist in the ATP7B gene include p. E1064A, p.R778L, p.G943S, and p.M769V (5). Patients with WD typically present with liver disorder (40–60%) in children >2 years of age, neurological manifestations (40–50%) associated to Kayser-Fleischer (KF) rings (90–100%) in children >10 years of age, and psychiatric symptoms (10–25%) in children >15 years of age (6, 7). Early diagnosis and treatment of both GLUT1 deficiency and WD is crucial and drastically improves long-term outcomes. Here we describe a patient who presented with characteristics of both disorders.
Case Description
We present a case with global developmental delay and ataxia due to GLUT1 that subsequently was diagnosed with WD due to persistently elevated liver enzymes. This patient was born from a non-consanguineous family. The mother was a 35-year-old healthy woman of Salvadoran descent, and the father, 32 years old, was of Cuban descent. While the father was diagnosed with type 1 diabetes mellitus at age 4, there was no family history of seizures, intellectual disability, hearing loss, pregnancy losses, neurocutaneous syndromes, or metabolic diseases.
The patient's developmental delay became evident at 18 months. His vocabulary was limited, and he did not walk until 20 months. Initial workup performed by the neurology service, including complete blood count (CBC), comprehensive metabolic panel (CMP), creatine phosphokinase test (CPK), and Fragile X, were all found to be normal. Magnetic resonance imaging (MRI) of the brain was unremarkable. At age 3, he started having ataxic episodes. Videographic assessment demonstrated episodes of falling to the floor due to a collapse of the right leg. He would try to stand but immediately fall back to the ground without loss of consciousness. Such ataxic episodes subsided but recurred at 5 years of age.
Video electroencephalography (vEEG) showed paroxysmal events of sudden abnormal movements with bilateral hand dyskinesia that were non-rhythmic and non-synchronous. However, the videos showed no electroencephalographic seizures or postictal state. Hence, the patient was diagnosed with paroxysmal exercise-induced dyskinesia.
Genetic testing revealed a pathogenic heterozygous mutation c.988 C>T (p.Arg330) in the exon 8 of the SLC2A1 gene by ACMG criteria PVS1, PM2, and PP5. This variant creates a premature translational stop signal resulting in an absent or disrupted protein product. This loss of function in the SLC2A1 gene has been previously reported in individuals with GLUT1 deficiency (2). Parental testing was negative for the SLC2A1 gene mutation.
During this workup, increasing levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were discovered. A ketogenic diet for his GLUT1 deficiency could not be started because of unexplained elevated liver enzymes. Thus, he was placed on a low carbohydrate diet. An ultrasound of the abdomen showed a heterogeneous liver appearance and a contracted gallbladder. Laboratory results, including prothrombin time (PT), partial thromboplastin time (PTT), antithrombin III activity, protein C, protein S, lupus anticoagulation, cardiolipin antibody, homocysteine level, hepatitis panel, Epstein Barr virus serology, thyroid-stimulating hormone (TSH), lead level, tissue transglutaminase, and alpha 1 antitrypsin phenotype were all unremarkable. The patient was not on any hepatotoxic medication. Ceruloplasmin level was low at 15 mg/dL (NL 28.6–56.1 mg/dL for age and sex) (8) and a basal 24-h urinary copper excretion was elevated at 127 mcg/24 h (NL 15–60 mcg/24 h).
Ophthalmologic exam was unremarkable. Repeat MRI of the brain was normal. The liver biopsy showed moderate (50–60%) macrovesicular steatosis, mild portal inflammation with lymphocytes and eosinophils, and grade II to III periportal fibrosis with slender portal septa (Figures 1A,B). The hepatic parenchymal copper concentration was 1,243 mcg/g dry weight (NL 10-35 mcg/g), suggesting WD. The diagnosis was confirmed by genetic testing, which revealed two pathogenic heterozygous variants of the ATP7B gene. One ATP7B allele showed a c.3207C>A (p.H1069Q) transversion on exon 14, which causes a substitution of histidine (CAC) to glutamine (CAA), and classified as pathogenic by ACMG criteria PM1, PP3, and PP5 (4). The second ATP7B allele showed c.3263 T>A (p.Leu1088Ter) on exon 15, which creates a pre-mature stop codon causing premature protein truncation, and classified as pathogenic by ACMG criteria PVS1, PP5, PM2, and PP3. Both variants have been reported to be associated with WD (9). Additionally, both parents demonstrated to be carriers for WD upon parental testing.
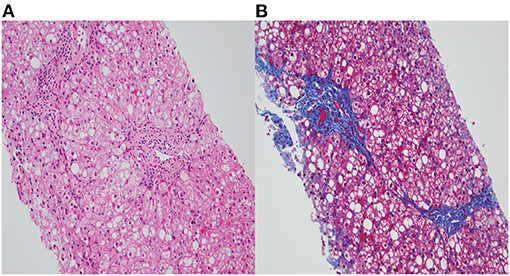
Figure 1. (A) Fatty change and ballooned hepatocytes with mild portal chronic inflammation (H&E, X200). (B) Portal fibrosis with slender septa extending from portal tracts and surrounding steatosis (Trichrome stain, X200).
Management of WD was initiated at the time of diagnosis. The patient was started on a copper-restricted diet, trientine hydrochloride 250 mg BID, and zinc acetate 25 mg BID, eventually leading to decreased urine copper excretion and normalization of liver enzymes.
Management of the GLUT1 deficiency with a ketogenic diet was initiated after normalization of his liver enzymes, which resulted in improvement of ataxia and seizures (Figure 2).
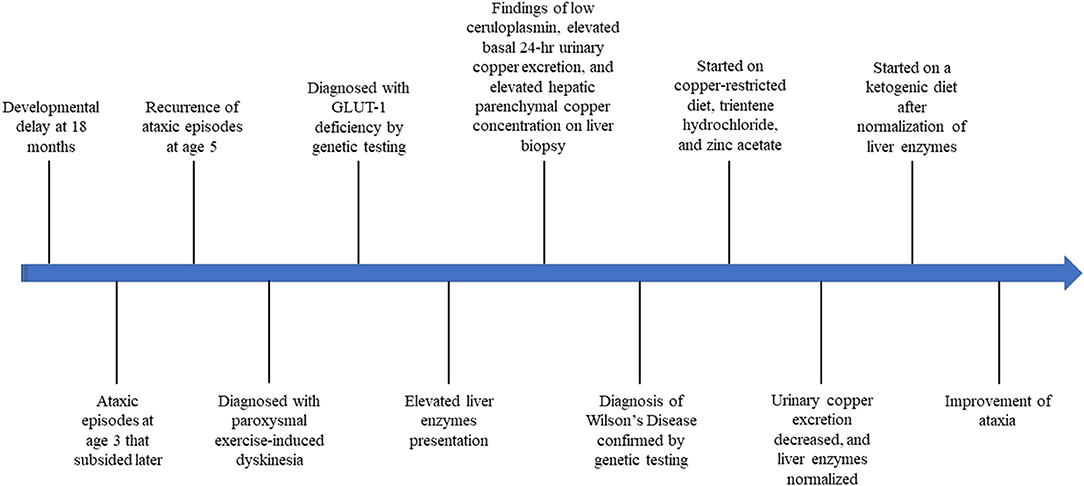
Figure 2. Timeline of our patient's presentation throughout diagnosis and treatment of Wilson disease and GLUT-1 deficiency.
Discussion
After an extensive literature review, we were unable to identify any other reported cases with an association with both WD and GLUT1 deficiency. These two disorders have different genetic modes of inheritance and two different loci on two different chromosomes. Simultaneous occurrence of an autosomal dominant disorder with an autosomal recessive disorder is extremely rare, especially with no history of consanguinity in the family. The presence of GLUT1 deficiency in our patient is most likely due to a de novo mutation, as a great number of de novo mutations occur in dominant genetic disorders (10). Moreover, the presence of WD in our patient could be related to parents coming from different countries of origin but Hispanic in nature.
Based on this patient's age of presentation and hepatic and neurological involvement, metabolic liver disorders including mitochondrial depletion syndrome (MDS) and Niemann-Pick disease type C (NPC) must be included in the differential diagnosis. MDS is associated to a group of autosomal recessive disorders characterized by a reduction in mitochondrial DNA, thus limiting energy production in several organs including the musculoskeletal, liver, and brain. Presentation includes seizures, hypotonia, developmental delay, feeding problems, and liver dysfunction (11). Moreover, NPC is a lysosomal disorder whose presentation is age-dependent, progressing from early perinatal and infantile liver involvement to hypotonia, developmental delay, ataxia, seizures, and psychiatric manifestations (12). Both disorders are diagnosed by molecular genetic testing.
This patient's early developmental delay presentation led to an extensive neurological workup and diagnosis of paroxysmal exertional dyskinesia with a seizure component secondary to GLUT1 deficiency. Cerebellar ataxia might present later in life in almost 30% of WD patients. These patients can present with an ataxic gait (wide-based gait with tandem walking), intentional tremor, dysdiadochokinesis, impaired coordination of fine hand movements, and ataxic speech (13–15). Moreover, KF rings, a copper deposition at the cornea's Descemet membrane, appear in 90–100% of patients with WD and neurological and psychiatric symptoms in >10 years of age (6). However, such type of ataxia and ocular findings were not seen in our patient.
Although pediatric WD commonly presents in the first decade of life, WD's neurological symptoms are mostly observed in the second decade (16, 17). Therefore, his neurological symptoms were most likely secondary to GLUT1 deficiency. GLUT 1 serves as a transporter in the central nervous system (CNS), and, in its absence, the patient will present with a normal blood glucose concentration and a CSF glucose concentration of <60 mg/dL (1). The prognosis of GLUT1 deficiency varies, but most respond well to a ketogenic diet. A ketogenic diet comprises a high-fat and low-carbohydrate diet, which maintains ketosis and allows the brain to use the ketone bodies as an alternative energy source. Ketogenic diets are currently considered the sole treatment for those with GLUT1 deficiency (2, 18). Liver enzymes alongside abdominal ultrasonography need to be closely monitored as studies have shown that a long-term ketogenic diet can induce parenchymal liver injury, hepatic steatosis, and formation of gallstones (19).
Moreover, our patient had elevated liver enzymes prior to initiation of ketogenic diet, likely related to WD and not usually consistent with GLUT1 deficiency. WD is caused by a mutation in the ATP7B gene, which encodes a copper transporting P-type ATPase necessary for copper excretion through the plasma and the bile. ATP7B is highly expressed in the liver but is also found in other organs such as the kidney, placenta, mammary glands, brain, and lung (5). If such a defect exists in the ATP7B gene, it can lead to a progressive toxic copper accumulation in these organs. Most children with WD present with liver disease manifesting with incidental and asymptomatic elevated liver enzymes, hepatomegaly, acute hepatitis, or cirrhosis. If WD is not appropriately diagnosed and treated, such copper accumulation can lead to liver failure and/or irreversible brain damage (7). Therefore, WD must be considered in the differential diagnosis of a patient with unexplained elevated liver enzymes, in the presence of another confirmed genetic condition since delay in diagnosis will delay early treatment. Recent studies have demonstrated the importance of performing genetic sequencing of the ATP7B gene in the initial investigation (20).
Conclusion
This case highlights the importance of carefully evaluating a patient with an unexplained liver disorder, even in the presence of primary neurological disease, as it can have significant therapeutic implications.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Written informed consent was obtained from the relevant individual(s), and/or minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
JD, AGF, and AF contributed with the elaboration, drafting, and final approval of the work. RA, MG, PJ, PS, and EH contributed with the revision and final approval of the work, and offered professional guidance. All authors agree to be accountable for all aspects of this work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the patient's family for their support of this work. The authors also thank Dr. Liset Pelaez for providing us optimal biopsy imaging for this patient.
Abbreviations
ALT, Alanine aminotransferase; ATOX1, Antioxidant protein 1; AST, Aspartate aminotransferase; ATP7B, ATPase Copper Transporting Beta; CTR1, Cell through copper transporter 1; CNS, Central Nervous System; CBC, Complete blood count; CMP, Comprehensive metabolic panel; CPK, Creatine phosphokinase test; GLUT1, Glucose transporter type 1; KF, Kayser-Fleischer; MRI, Magnetic resonance imaging; MDS, Mitochondrial depletion syndrome; NPC, Niemann-Pick disease type C; PTT, Partial thromboplastin time; PT, Prothrombin time; SLC2A1, Solute Carrier Family 2 Member 1; TSH, Thyroid-stimulating hormone; TGN, Trans-Golgi network; vEEG, Video electroencephalography; WD, Wilson disease.
References
1. NIH. Glucose Transporter Type 1 Deficiency Syndrome. NIH.gov. Available online at: https://rarediseases.info.nih.gov/diseases/9265/glucose-transporter-type-1-deficiency-syndrome (accessed June 13, 2021).
2. Pearson TS, Pons R, Engelstad K, Kane SA, Goldberg ME, De Vivo DC. Paroxysmal eye-head movements in Glut1 deficiency syndrome. Neurology. (2017) 88:1666–73. doi: 10.1212/WNL.0000000000003867
3. Bhatnagar N, Lingaiah P, Lodhi JS, Karkhur Y. Pathological fracture of femoral neck leading to a diagnosis of Wilson's disease: a case report and review of literature. J Bone Metab. (2017) 24:135–9. doi: 10.11005/jbm.2017.24.2.135
4. Weiss KH. Wilson disease. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al., editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (1999).
5. Chang IJ, Hahn SH. The genetics of Wilson disease. Handb Clin Neurol. (2017) 142:19–34. doi: 10.1016/B978-0-444-63625-6.00003-3
6. Litwin T, Dusek P, Szafrański T, Dziezyc K, Czonkowska A, Rybakowski JK. Psychiatric manifestations in Wilson's disease: possibilities and difficulties for treatment. Ther Adv Psychopharmacol. (2018) 8:199–211. doi: 10.1177/2045125318759461
7. Socha P, Janczyk W, Dhawan A, Baumann U, D'Antiga L, Tanner S, et al. Wilson's disease in children: a position paper by the hepatology committee of the European society for paediatric gastroenterology, hepatology and nutrition: a position paper by the hepatology committee of the European society for paediatric gastroenterology, hepatology and nutrition. J Pediatr Gastroenterol Nutr. (2018) 66:334–44. doi: 10.1097/MPG.0000000000001787
8. Soldin SJ, Hicks JM, Bailey J, Beatey J, Watson P. Pediatric reference ranges for β2-microglobulin and ceruloplasmin. Pharmacia Beta Microglobul RIA. (1997) 43:S199.
9. Deguti MM, Genschel J, Cancado EL, Barbosa ER, Bochow B, Mucenic M, et al. Wilson disease: novel mutations in the ATP7B gene and clinical correlation in Brazilian patients. Hum Mutat. (2004) 23:398. doi: 10.1002/humu.9227
10. Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. (2016) 17:241. doi: 10.1186/s13059-016-1110-1
11. El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics. (2013) 10:186–98. doi: 10.1007/s13311-013-0177-6
12. Patterson M. Niemann-Pick disease type C. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (1993–2021).
13. Lorincz MT. Neurologic Wilson's disease: Wilson's disease. Ann N Y Acad Sci. (2010) 1184:173–87. doi: 10.1111/j.1749-6632.2009.05109.x
14. Litwin T, Dusek P, Czlonkowska A. Neurological manifestations in Wilson's disease –possible treatment options for symptoms. Expert Opin Orphan Drugs. (2016) 4:719–28. doi: 10.1080/21678707.2016.1188003
15. Litwin T, Dušek P, Czonkowska A. Symptomatic treatment of neurologic symptoms in Wilson disease. Handb Clin Neurol. (2017) 142:211–23. doi: 10.1016/B978-0-444-63625-6.00018-5
16. Rukunuzzaman M. Wilson's disease in Bangladeshi children: analysis of 100 cases. Pediatr Gastroenterol Hepatol Nutr. (2015) 18:121–7. doi: 10.5223/pghn.2015.18.2.121
17. Kim TJ, Kim IO, Kim WS, Cheon JE, Moon SG, Kwon JW, et al. MR imaging of the brain in Wilson disease of childhood: findings before and after treatment with clinical correlation. AJNR Am J Neuroradiol. (2006) 27:1373–8.
18. Lee M. The use of ketogenic diet in special situations: expanding use in intractable epilepsy and other neurologic disorders. Korean J Pediatr. (2012) 55:316–21. doi: 10.3345/kjp.2012.55.9.316
19. Arslan N, Guzel O, Kose E, Yilmaz U, Kuyum P, Aksoy B, et al. Is ketogenic diet treatment hepatotoxic for children with intractable epilepsy? Seizure. (2016) 43:32–8. doi: 10.1016/j.seizure.2016.10.024
Keywords: glucose transporter 1 (GLUT1) deficiency, ataxia, elevated liver enzymes, ATP7B, SLC2A1, case report, Wilson disease
Citation: Diaz J, Fonseca AG, Arboleda R, Frade A, Gennaro MP, Jayakar P, Schleifer P and Hernandez E (2021) Case Report: The Association of Wilson Disease in a Patient With Ataxia and GLUT-1 Deficiency. Front. Pediatr. 9:750593. doi: 10.3389/fped.2021.750593
Received: 30 July 2021; Accepted: 07 September 2021;
Published: 05 October 2021.
Edited by:
Consolato M. Sergi, Children's Hospital of Eastern Ontario (CHEO), CanadaReviewed by:
Mervin Burnett, University of Alberta, CanadaVikrant Sood, The Institute of Liver and Biliary Sciences (ILBS), India
Copyright © 2021 Diaz, Fonseca, Arboleda, Frade, Gennaro, Jayakar, Schleifer and Hernandez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashley G. Fonseca, ashley.fonseca@nicklaushealth.org
†These authors have contributed equally to this work and share first authorship
‡These authors share senior authorship