 Riffat Mehboob1,2*
Riffat Mehboob1,2* Maher Kurdi3
Maher Kurdi3 Mursleen Ahmad4
Mursleen Ahmad4 Syed Amir Gilani1
Syed Amir Gilani1 Sidra Khalid2Hisham Nasief5Abeer Mirdad6Husam Malibary7Sahar Hakamy8Amber Hassan1Meshari Alaifan9Ahmed Bamaga10,11
Sidra Khalid2Hisham Nasief5Abeer Mirdad6Husam Malibary7Sahar Hakamy8Amber Hassan1Meshari Alaifan9Ahmed Bamaga10,11 Syed Adnan Shahzad12*
Syed Adnan Shahzad12*- 1Research Unit, Faculty of Allied Health Sciences, The University of Lahore, Lahore, Pakistan
- 2Lahore Medical Research Center, LLP, Lahore, Pakistan
- 3Department of Pathology, Faculty of Medicine in Rabigh, King Abdulaziz University, Jeddah, Saudi Arabia
- 4Department of Medicine, Sahiwal Medical College, Sahiwal, Pakistan
- 5Department of Obstetric and Gynecology, Faculty of Medicine, King Abdulaziz University and Hospital, Jeddah, Saudi Arabia
- 6Pediatric Department, East Jeddah Hospital, Jeddah, Saudi Arabia
- 7Department of Internal Medicine, Faculty of Medicine, King Abdul Aziz University, Jeddah, Saudi Arabia
- 8Center of Excellence in Genomic Research, King Abdulaziz University, Jeddah, Saudi Arabia
- 9Department of Paediatrics, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia
- 10Paediatric Department, King Faisal Specialist Hospital and Research Center, Jeddah, Saudi Arabia
- 11Neurology and Pediatric Department, Faculty of Medicine, King Abdulaziz University Hospital, King Abdulaziz University, Jeddah, Saudi Arabia
- 12Faculty of Medicine and University Hospital of Cologne, Institute of Virology, University of Cologne, Cologne, Germany
Background: Sudden infant death syndrome (SIDS) is a tragic incident which remains a mystery even after post-mortem investigation and thorough researches.
Methods: This comprehensive review is based on the genes reported in the molecular autopsy studies conducted on SIDS so far. A total of 20 original studies and 7 case reports were identified and included in this analysis. The genes identified in children or adults were not included. Most of the genes reported in these studies belonged to cardiac channel and cardiomyopathy. Cardiac channel genes in SIDS were scrutinized for further analysis.
Results: After screening and removing the duplicates, 42 unique genes were extracted. When the location of these genes was assessed, it was observed that most of these belonged to Chromosomes 11, 1 and 3 in sequential manner. The pathway analysis shows that these genes are involved in the regulation of heart rate, action potential, cardiac muscle cell contraction and heart contraction. The protein-protein interaction network was also very big and highly interactive. SCN5A, CAV3, ALG10B, AKAP9 and many more were mainly found in these cases and were regulated by many transcription factors such as MYOG C2C1 and CBX3 HCT11. Micro RNA, “hsa-miR-133a-3p” was found to be prevalent in the targeted genes.
Conclusions: Molecular and computational approaches are a step forward toward exploration of these sad demises. It is so far a new arena but seems promising to dig out the genetic cause of SIDS in the years to come.
Introduction
Sudden Infant Death Syndrome (SIDS) is described as a diagnosis of exclusion and is defined as unanticipated passing away of an infant < 1 year of age that remains unsolved even after thorough inquiries that includes a review of the clinical history, performance of complete autopsy, and examination of the death scene (1). Matturri et al. (2) proposed the definitions of SIDS as “the sudden death of an infant under 1 year of age which remains unexplained after a thorough case investigation, including the performance of a complete autopsy, examination of the death scene, and review of the clinical history” should be modified by adding, at the end, the following: “a complete autopsy with an in-depth histopathologic analysis of the cardiorespiratory innervation and specialized myocardium, performed only by an experienced, reliable pathologist.” In 2004, Krous et al. defined SIDS as “The sudden unexpected death of an infant less than 1 year of age, with onset of the fatal episode apparently occurring during sleep, that remains unexplained after a thorough investigation, including performance of a complete autopsy and review of the circumstances of death and the clinical history.” This definition was elaborated by Ottaviani as, “the findings in the cardiac conduction and autonomic nervous systems detected in SIDS can be morphological substrates for the sudden unexpected death (3). In 2016, Goldstein et al. (4) reported that “there is no consensus on the use of the term SIDS, as external factors, such as prone sleep position or bed sharing, may at times explain the cause of death as positional asphyxia or accidental suffocation.”
Therefore, an in-depth histopathological analysis of the cardiac conduction system and autonomic nervous system by expert pathologists is mandatory (3). According to the American Heart Association (5), congenital heart diseases occur in 0.8% of full term live births. Anomalies in the cardiac conduction system and cardiac channelopathies should be considered to be included in congenital heart disease (6). SIDS is diagnosed by eliminating all other known illnesses. Incidence of SIDS spikes between 2 and 4 months of age and greater than 90% happens in the period of first 6 months of the life of infants. Due to the global campaign of “Back To sleep” along with other campaigns, 50-90% drop in the cases of SIDS has been recorded since 1990 (7).
SIDS and Sudden intrauterine death (SIUD) have multifactorial etiology comprising of congenital anomalies of the cardiac conduction system and the autonomic nervous system, respiratory rhythm generation and arousal from sleep (8). Based on these considerations, the new common definition for the SIDS-SIUDS complex is “The sudden death of a fetus after the 25th gestational week or infant under 1 year of age which is unexpected by history and remains unexplained after a thorough case investigation, including examination of the death scene, performance of a general autopsy and examination of the fetal adnexa” (3). Routine autopsy fails to disclose any cause of death in such cases and a more in depth histopathological analysis of the cardiac conduction system and autonomic nervous system is required. Molecular autopsy should also be followed in this regard.
Triple risk model for SIDS as described by Filiano and Kinney, suggests three factors: a vulnerable infant, a critical developmental period in homeostatic control, and an exogenous stressor(s) (9). All three factors, when present together, may lead to SIDS. Neuropathological and Cardiovascular conduction system abnormalities are the main culprits involved in the vulnerability of the infant or fetus for Sudden perinatal death when it combines with the genetic predisposition (3). Sudden cardiac death (SCD) is defined as the unexpected death without an obvious non-cardiac cause that occurs within 1 h of witnessed symptom or within 24 h of unwitnessed symptom onset (10). SCD occurs by at least these three mechanisms: Ischemia induced by a perturbation in a coronary artery with rapid development of ventricular fibrillation, arrhythmia occuring in an early-stage of acute myocardial infarction associated with coronary thrombosis, primary ventricular arrhythmia not associated with new onset ischemia, related to a cardiomyopathy or channelopathy (11).
Genetic predisposition is responsible for vulnerability of an infant toward SIDS. So, genetics does not play its role alone, but it interacts with the epidemiological factors and stressors acting in a critical developmental phase of the infant (12). It is important to understand the genetics associated with SIDS in order to prevent such casualties by taking care of modifiable risk factors in vulnerable infants. So far there is a scarcity of literature available due to limited research on this aspect which led us to write a comprehensive and updated review. Genetics play its role either directly such as in cases with medium-chain acyl-coenzyme A dehydrogenase deficiency and cardiac arrest due to long QT syndrome or indirectly by acting as a predisposing factor for SIDS (13, 14). Inheritance pattern of genes contributing to SIDS is polygenic and acts in combination with environmental risk factors, e.g., male gender, prone sleeping position, carelessness of parents, winter, seasonal variations, night, botulism etc. Immune system, cardio-respiratory function and neurophysiology related genes have been prominent so far in association with SIDS (13).
Inflammation and infections are also contributing factors in SIDS (15). Viral infections, particularly of the respiratory tract, are also among one of the etiological factors for SIDS. Epidemiology and pathology related to SIDS, provides evidence for this concept. But the results are conflicting regarding the isolation of specific viruses due to technical issues, lack of proper controls and many other reasons. However, another very important contributing factor for SIDS is an immunological response (16). Mucosal immune system is activated in SIDS in different studies (17, 18). Higher IgG and IgA immunocyte density in the tonsils of SIDS victims has been observed (18). Elevated expression of CD45+ stromal leucocytes and highly variable expression of human leukocyte antigen-antigen D(HLA-DR) and enhanced expression of HLA class I and II have also been observed in salivary glands in SIDS. It shows that the immune system is stimulated in SIDS with the release of cytokines, which then up-regulate the epithelial expression of HLA-DR (19). Bacteriological findings have also been reported and Staphylococcal aureus and staphylococcal endotoxins have been found to be prevalent in SIDS (20).
Altered immunological homeostasis has been hypothesized as an important factor for SIDS etiology. Cytokines are essential mediators for infant health by monitoring the cell activity during inflammation. The pro-inflammatory cytokines such as IL-1α, IL-1β, IL-6, IL-8, IL-12, IL-18, TNF-α, and IFN-γ potentiates inflammation. The anti-inflammatory cytokines, e.g., interleukin-1 receptor antagonist (IL-1ra), IL-4, IL-10, IL-11, and IL-13 control them. Cytokine genes and their variants may explain the vulnerability of infants to infection. The genes encoding IL-1, IL-6, IL-10, and TNF-α are the most studied in SIDS. Genes involved in the immune system are important in SIDS and specific haplotypes in the IL-10 gene promoter have been associated with SIDS due to infection (21). In toddlers who had SIDS, polymorphism inside the gene IL10 (inflammatory cytokine interleukin 10) at promoter place could impel the reduction in production of antibodies or it can act as a substitute to enhance production of inflammatory cytokine (22). Immunogenetics and cytokine network are very complex to understand and need further exploration (23).
According to certain studies, genetic anomalies that are associated with channelopathies (arrhythmia) may also play a part in SIDS. Main lethal arrhythmias give rise to sudden cardiac death due to genetic variations such as: CPVT (catecholaminergic polymorphic ventricular tachycardia), LQTS (long QT syndrome), SQTS (short QT syndrome) and BrS (Brugada syndrome) (24). So far about 30 genes have been found linked with that of major principal cardiac arrhythmic ailment but mainly 4 genes (RyR2, SCN5A, KCNQ1, and KCNH2) have been taken into account. The chief answerable for each ailment: SQTS (KCNH2 20% and KCNQ1 30%), CPVT (RyR2 50%), BrS (SCN5A 25%), and LQTS (SCN5A 15%, KCNH2 30% and KCNQ1 40%) (25).
Ten to fifteen percent of the SIDS cases are thought to be caused by cardiac arrhythmias (26), particularly CPVT and LQT (27, 28). Furthermore, polymorphism in genes that linked to sleep trouble disorders have been seen to be effective as a potential hazard in terms of genes for SIDS (29). Phenomenon of polymorphism is also acknowledged in 5-HTT (serotonin transporter gene) that will enhance activity of serotonin transporter, on the contrary lessening the concentrations of serotonin at endings of nerves (30, 31). In addition, polymorphism was recognized in genes that affected the enhancement of autonomic apprehensive device (EN1, TLX3, ECE1, RET, PHOX2a) (32).
The progress of SIDS lingers to be solved, but 1 out of 10 cases is linked with mutations of cardiac ion channel gene, it also include the genes that encode the βeta subunits (33). p.S206L in SCN4B and p.V54G and p.V36M mutations in SCN3B have been associated to SIDS (34). Significantly, p.V36M mutation in SCN3B has also been associated to ventricular fibrillation of unknown cause, which is a possible lethal cardiac arrhythmia, and p.S206L in SCN4B also give rise to unusual excitableness in ventricular muscle cells of rat (34, 35). On the other hand there has been one occurrence of SIDS in a baby with a p.R214Q in β1B, which is also linked with Brugada Syndrome (36). β1B regulates function of Nav1.5, significantly assigning a causal method for cardiac malfunction associated with SCN1B (37). Up till now, no variations in SCN2B have been associated to SIDS. A huge figure of genes are thought to be the genetic reason of SIDS. Regardless of innovative progress in the turf of medical science, up to this time a very few facts with regard to genetics of SIDS are present (38). Similar to other heart ailments, variation in ion channel gene is as well a potential cause for SIDS (39).
Furthermore, variations in a number of heart channel genes have also been taken into consideration as a reason of SIDS and variations in genes associated with ion channels of heart (KCNH2, LQTS,SCN5 and KCNQ) in 9.5 % of SIDS cases (40, 41). In a study, 38 out of 47 victims of SIDS were examined for alternatives in KCNH2, SCN5A, RYR2, and KCNQ1, exhibited that 8 (17%) of the victims of SIDS had variations in gene disturbing role of ion channels. Mutations in these genes will give rise to disorders of malfunctioning of ion channels which can lead to arrhythmias and sudden death due to hereditary heart ailments (42).
This study was planned to review the previous literature regarding the genetic predisposition in SIDS and investigate a link between genetic and epidemiological factors associated with SIDS.
Methods
This is an in silico study based on the existing data reported in literature related to the genes associated with SIDS. Very few studies have been found and the genes identified in these studies were gathered and analyzed for various properties through bioinformatics tools. These studies included 20 original articles and 7 case reports. After deleting the repetitive genes, a total of 42 unique genes were identified which were explored further. We have analyzed these genes for computational properties, pathway analysis, gene enrichment analysis, histone methylation and other properties.
We employed ShinyGo (https://academic.oup.com/bioinformatics/article/36/8/2628/5688742), for enrichment, pathway and gene characterization. All analysis were performed on ShinyGo V0.61. For histone mark enrichment, mircoRNAs and transcription factors targets we used an integrative web enrichment (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3637064/). Protein-protein association network analysis were performed using String database.
Results
Genomic Architecture of Genes Involved in SIDS
The chromosome 11 harbored the maximum number of genes selected for the study, followed by chromosome 1 and 3 (Table 1; Figure 1A). The genes on chromosome 11 included the genes encoding for: potassium voltage-gated family channel (subfamilies Q and E), sodium channel beta subunits and potassium inwardly rectifying channel (subfamily J). The characteristics of SIDS genes are compared with the rest in the genome. Chi-square and Student's t-tests are run to see if the particular genes have special characteristics when compared with all the other genes in the human genome (Figures 1A–D). Figure 1B indicates that SIDS genes have more transcripts or isoforms, showing the complexity of the network. SIDS genes have been observed to contain more exons as compared to the random genes as shown in Figure 1C. Another interesting aspect observed through the computational analysis is that most of the SIDS genes are coding, which means that they code for proteins and involved in the regulatory mechanisms (Figure 1D).
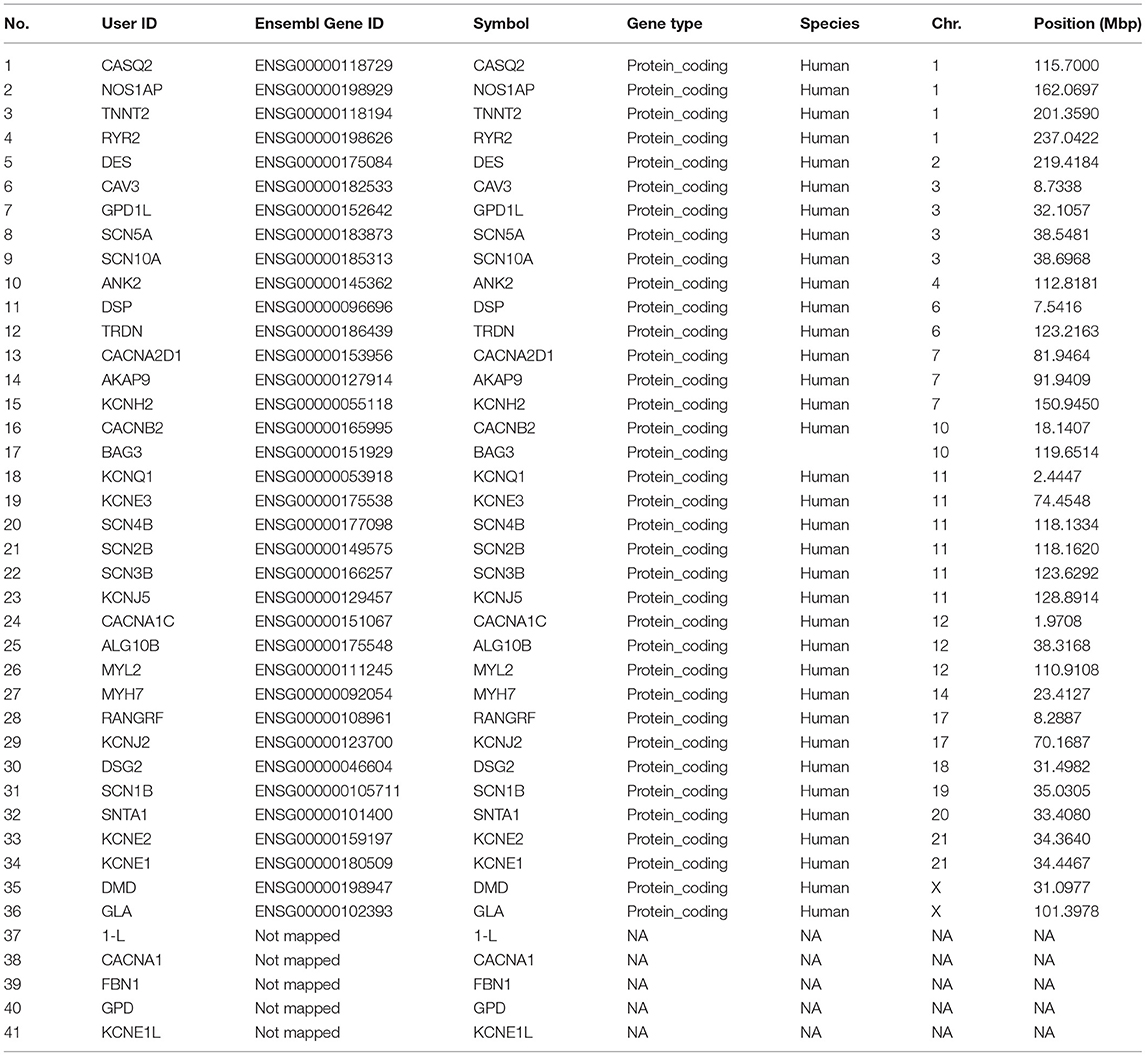
Table 1. Genes extracted and investigated for association.
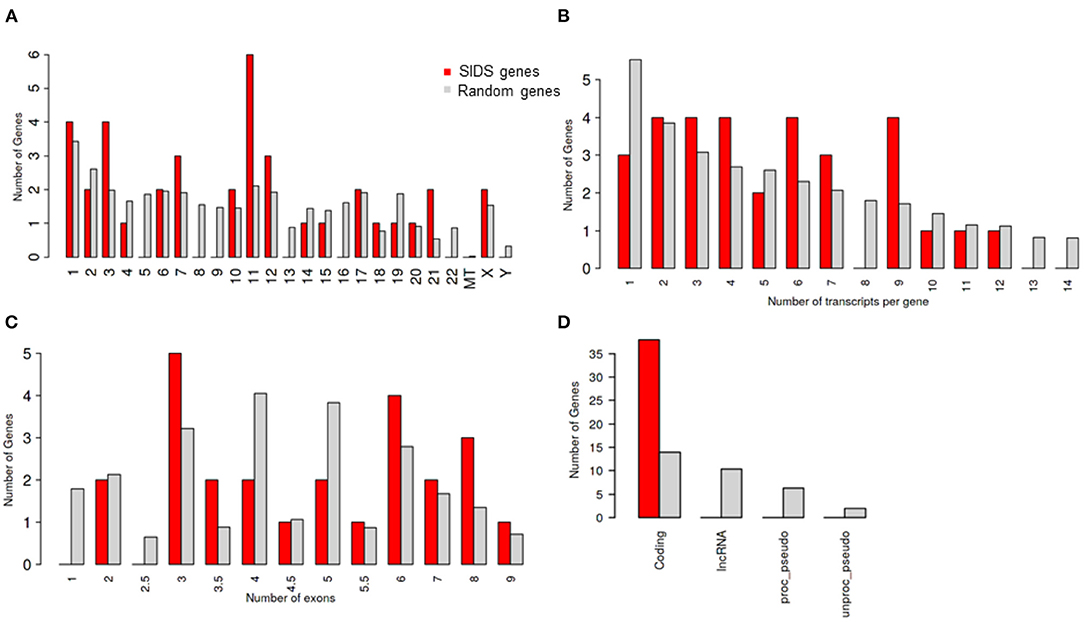
Figure 1. Genomic architecture of genes involves in SIDS (A) Distribution of SIDS genes on chromosomes Chi-squared test P = 0.43 (B) Number of transcripts isoforms per coding genes Chi-squared test P = 0.65 (C) Number of exons SIDS Chi-squared test P = 0.66 (D) Distribution of Gene type Chi-squared test P = 1.2 E-10.
The potassium voltage-gated (Kv) channels are the prototype members of membrane signaling proteins and they comprise the most complex, largest and evolutionary conserved family of ion channels (43). They are ubiquitous to all eukaryotic cells and are versatile in retaining membrane potential, balancing the cell volume and harmonizing the neural electric excitability (44). These channels allow millions of ions to selectively pass the membrane in a second and upon change in voltage or ligand concentration, their gates can open and close in milliseconds. The movement of solutes across the cell membrane produces a voltage difference that results in the generation of electric current. Homeostasis makes the fast-electrical signaling possible by maintaining the environment and content of the cell (increased concentration of sodium ions in extracellular fluid and blood and increased concentration of potassium ions inside the cytoplasm) (43). This homeostasis is maintained by the movement of ions across the membrane through the voltage gated channels. Humans contain 12 subfamilies of the Kv channels that comprise 40 genes encoding for the pore forming subunit of the channels. In the neurons innervating the muscle and cardiac muscle cells, they regulate the waveform of action potential (45).
The potassium voltage-gated channel subfamily Q member 1 (KCNQ1) encodes a voltage gated potassium channel. Two major functions of the KCNQ1 includes the repolarization of the cardiac tissue ensuing action potential and transport of water and salt in epithelial tissues (46). It contributes to diverse physiological processes by complexing with the subfamily E regulatory subunit 1 (KCNE1). There are five genes in the KCNE family that encode the regulatory subunit of transmembrane spanning ion channel. Their primary function is to complex with the Kv channels (47). Mutations in KCNQ1 and KCNE1 leads to malfunctioning of the channel and thus cause Romano-Ward syndrome and Jervell and Lange-Nielsen syndrome (48, 49).
In the present investigation, three sodium channel beta subunits genes were linked with SIDS. The Voltage gated sodium channels consist of a pore forming α subunit joined by two β subunits, thus forming a heterotrimeric complex that was first identified for their involvement in the upstroke of action potential. In mammals, the SCN (1-10) A genes encode the α subunit and SCN (1-4) B genes encode the β subunits. The sodium voltage-gated channel beta subunits (encoded by SCN4B, SCN2B and SCN3B genes) are not only the regulatory subunits of the transmembrane glycoprotein complexes but they also perform several functions independent of the α subunit and are involved in regulating cell excitability, affecting brain development and conferring distinct channel pharmacology (50). Defects in SCN4B, 2B and 3B genes (besides other problems) could result in different syndromes including SIDS (1, 51, 52).
A member of the potassium inwardly rectifying channel subfamily J was also found to be associated with SIDS—KCNJ5. This gene encodes the integral membrane protein of the inwardly rectifying potassium channels. Conditional to their type and location, these channels perform a variety of physiological functions. They permit the flow of potassium ions in rather out of cell and are regulated by G-proteins (53).
There were four genes on each of the chromosome 1 and 3 linked to SIDS. The genes on chromosome 1 included the Calsequestrin-2 (CASQ2), Nitric Oxide Synthase 1 Adaptor Protein (NOS1AP), Troponin T type 2 (TNNT2) and Ryanodine Receptor 2 (RYR2) genes.
The protein encoded by CASQ2 is a calcium binding protein that serves as a calcium depot for muscle function in the cardiac muscles. The precise control of calcium ions regulates the normal contraction and relaxation in the heart muscles which is essential for maintaining regular heart rhythm (54). If there occurs an alteration in calcium ions concentration then the gene TNNT2 encoded protein regulates the muscle contraction. Mutations in TNNT2 lead to dilated cardiomyopathy (enlarged heart effecting pumping) or familial hypertrophic cardiomyopathy (abnormal thickening of heart muscles leading to difficulty in pumping blood) (55, 56). The RYR2 gene encoded protein makes channels for the transport of calcium ions in the heart myocytes. These channels are embedded in the sarcoplasmic reticulum (outer membrane of cell structure) thus acting as storage depot for calcium ions. The RYR2 channels regulated the release of calcium ions (from the sarcoplasmic reticulum to the surrounding cells) in myocytes in response to the signals. Increase in calcium ion concentration leads to heart muscle contraction. The cardiac myocytes relax when the calcium ions are taken back to the sarcoplasmic reticulum (57).
The genes on chromosome 3 included the Caveolin-3 (CAV3), Glycerol-3-Phosphate Dehydrogenase 1 Like (GPD1L) and sodium voltage gated channels alpha subunits 5 and 10 (SCN5A and SCN10A) genes. CAV3 gene encodes the protein caveolin-3 that plays a role in muscle development along with the other two members of the family; CAV-1 and−2. CAV-3 is also involved in the signaling pathways and energy metabolism (58, 59). The GPD1L gene encodes a cytoplasmic protein associated with the cell membrane where it binds to the SCN5A. Mutations in the GPD1L has been reported to cause Brugada syndrome type 2 (BRS2) as well as SIDS (60). Mutations in CAV3 are responsible for the malfunctioning of SCN5A that also becomes a potential cause of SIDS (61).
Gene Enrichment in Biological Processes
Gene enrichment analysis was performed to recognize the over represented genes in our data set that may have been associated with SIDS. The biological pathways that were enriched in certain genes were analyzed. The 40 genes undertaken in this study were found to be regulating different biological pathways (Table 2; Figure 2). Most of the genes were associated with cardiac related functions. Nearly 70% of the genes studied were involved in regulation of the heart rate by cardiac conduction and cardiac muscle cell action potential. About 80% of the genes were linked to cardiac contraction.
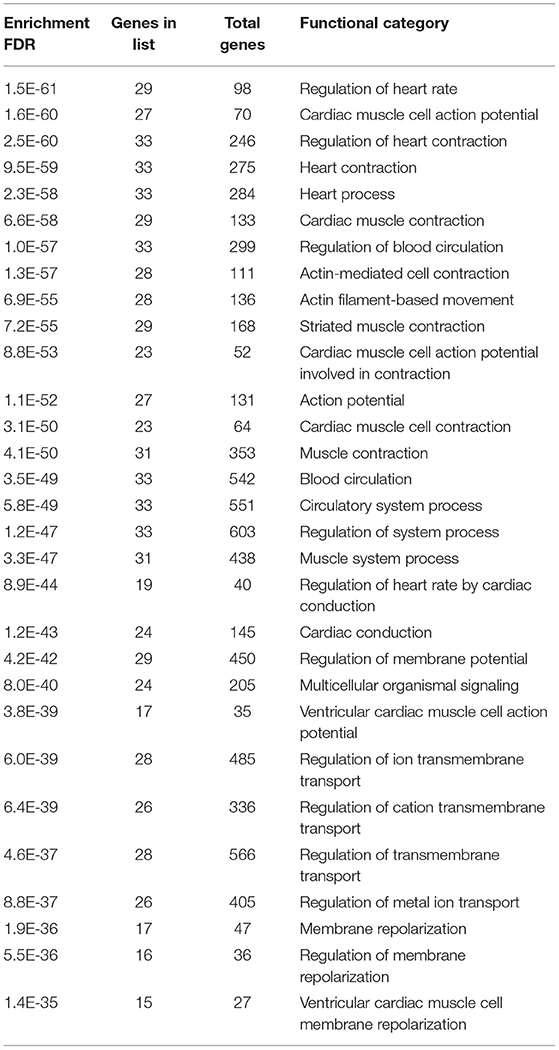
Table 2. Gene enrichment showing the number of genes associated to a particular biological process.

Figure 2. Pathway analysis showing the over represented SIDS genes in different biological pathways.
A normal heart contracts at regular intervals in a continuous fashion to supply oxygenated blood to the whole body and bring back the deoxygenated blood to the lungs (62). To maintain normal heart rhythm, a specialized tract of myocardial cell constitute the cardiac conduction system (63). The mechanical functioning of the heart depends on the production and propagation of action potential—electrical functioning. Between the two impulses, the heart contracts and relaxes followed by a period of refractoriness. This electrical functioning is manifested in the successive activation of cells in specialized regions of the heart—the pacemaker, and the propagation of action through the ventricles. The electrical activity of the myocardial cells is associated to the production of action potential in heart cells and its propagation along with the electromagnetic functioning of ventricles depends on the electrical coupling between cells mediated by gap junctions (64, 65). Activation and subsequent inactivation of ion channels that conduct depolarization (influx of Na+ and Ca2+) and repolarization (efflux of K+) generates myocardial action potential (66). The unidirectional propagation of excitation through the myocardium and normal cardiac rhythm generates due to the distinct waveforms of action potential in different regions of the heart that occurs due to the differences in the expression and/or properties of underlying ion channels (66–69). Inherited mutations in the genes encoding the ion channels or myocardial disease can lead to changes in the functional expression and properties of ion channels resulting in alterations in the action potential waveforms, and/or propagation thus predisposing the heart to life threatening arrhythmias (70). Structural anomalies in the conduction system can lead to lethal cardiac arrhythmias or heart block that might be linked to SIDS (71). Nearly 85 % of the sudden deaths are of cardiac origin—sudden cardiac death (72).
Gene Regulation by Histone Modification
A ubiquitous and integral component of eukaryotic gene regulation is histone. The DNA is wrapped around histone octamers forming a structure called nucleosomes thus changing the accessibility and structure of DNA (73). Post-translational modifications in histones may occur by covalent addition of chemical groups (acetyl, methyl, phosphoryl, and small proteins) at specific residues (74), and occur frequently in the extruding tails of DNA that can change the nucleosome structure and also modulate gene expression by recruiting protein involved in gene regulation (73). The set of genes undertaken in the present study were found to be involved in four different epigenetic modifications: H3K27me3, H3K9me3, H3K4me3, and H3K36me3 (Figure 3; Table 3).
Figure 3. Pathway network illustrating the genes interaction in biological processes.
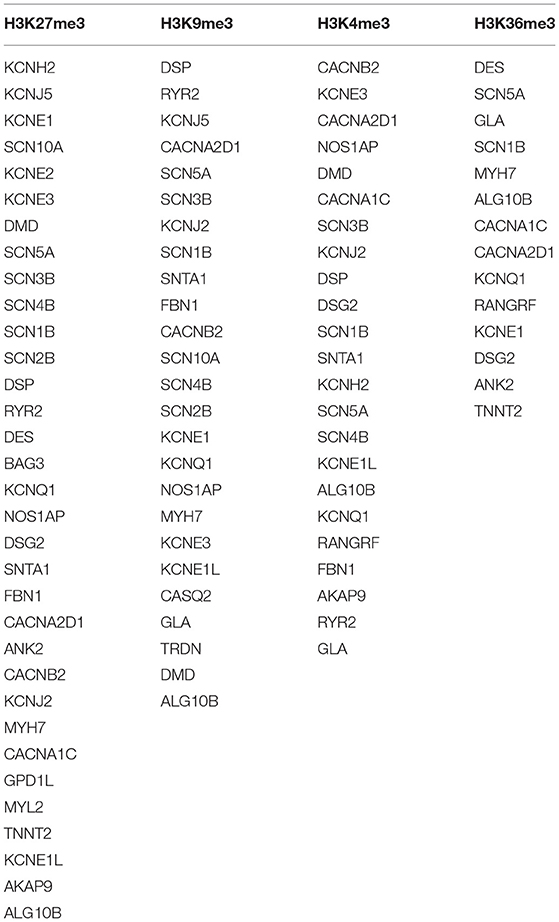
Table 3. Four different histone modifications found in present study.
Histone methylation is a type of epigenetic modification that allows alteration on chromatin structure without changing the underlying genomic sequence. They usually occur on lysine or arginine residue (75). H3K27me3 is a tri-methylation at the 27th lysine residue of the DNA packaging protein, Histone H3. This epigenetic modification forms heterochromatic regions thus downregulating the nearby genes (76).
The tri-methylated H3K27 signifies a highly conserved histone modification that is strongly linked to inactive gene promoters (77). This transcriptional silencing is produced and maintained by evolutionary-conserved histone-modifying enzymes—Polycomb Repressive Complexes (PRCs) (78). Along with H3K27me3, tri-methylation of histone 3 lysine 9 (H3K9me3) are the best-known histone modifications related to gene expression and heterochromatin. These modifications are carried out by enzymatic “writers” and are rectified by enzymes—“eraser,” to remove the modifications (79). H3K27me3 controls the expression of developmentally regulated genes (80, 81). H3K9me3 is abundantly found in human active gene promoters in several cell types and it represses the repetitive DNA elements and silencing factors encoding genes (79, 82).
Tri-methylation of histone 3 lysine 4 (H3K4me3) upregulates transcription by stabilizing the formation of pre-initiation complex (83). It binds to different protein folds that are present in chromatin remodeling and histone modifying factors, thus changes in levels of H3K4me3 can attribute to change in transcription (84). The tri-methylation of histone 3 at lysine 36 (H3K36me3) is firmly associated with actively transcribed genome regions and plays role in transcriptional activation. This modification is known to mark active genes. There is evidence to indicate a role of H3K36me3 in the DNA damage response, thus a reduced level of H3K36me3 could be linked with a decline in DNA repair efficiency (75).
Aberrant histone methylation plays a significant role in causing cancer and differentiation of stem cells. The transcriptional inhibition by H3K27me3 regulates the gene expression, affects chromatin structure, transcriptional activity of neighboring genes, availability of transcription factors, thus also normal cell development and disease progression (85, 86). In colorectal cancer patients, decreased levels of H3K27me3 lead to poor prognosis and resistance to chemotherapy (87). Also, loss of polycomb repression reduces H3K27me3 level, that leads to troublesome prognosis of several cancers. Mutations in the histone 3 drive tumorigenesis in pediatric high-grade gliomas (88). H3K27me3 and H3K9me3 may also play a role in colorectal cancer due to alteration in expression level (89).
As a result of excess and abnormal methylation reactions, cancer cells require a higher amount of methionine and thus become methionine addicted (90–92). This addiction prevents cancer cells from proliferation under methionine restriction. Histone methylation status of H3K9me3 and H3K4me3 were volatile in cancer cells and their levels were drastically dropped be methionine restriction in cancer cells (93).
Role of Transcription Factors in Genes Involved in SIDS
Transcription factors are the proteins that regulate the transcription rate by interacting with the regulatory DNA sequences lying 5' upstream to the target genes. There are different families of transcription factors including the helix-turn-helix, helix-loop-helix, zinc finger, beta sheet motifs and basic protein-leucine zipper (94). Different transcription factors are correlated with the genes involved in SIDS (Figure 4). The maximum number of genes (12 genes) were found to be linked with the Myogenic differentiation 1 (MyoD1) transcription factor that promotes the expression of muscle-specific genes and has role in muscle differentiation along with myogenic factor 5 (MYF5) and Myogenin (MYOG) (95). The MYOD1 plays a crucial role in accelerating the transcription of p21 (a cyclin dependent kinase that acts as a tumor suppressor) and myogenin to remove the cells from cell cycle and terminate multiplication of differentiated myocytes (96). Functional dysregulation and abnormal expression of MyoD1 has been reported in retinoblastoma (97), head and neck cancer (98), breast cancer (99) and lung adenocarcinoma (100). The MyoD1 was also significantly under expressed in gastric cancer (101).
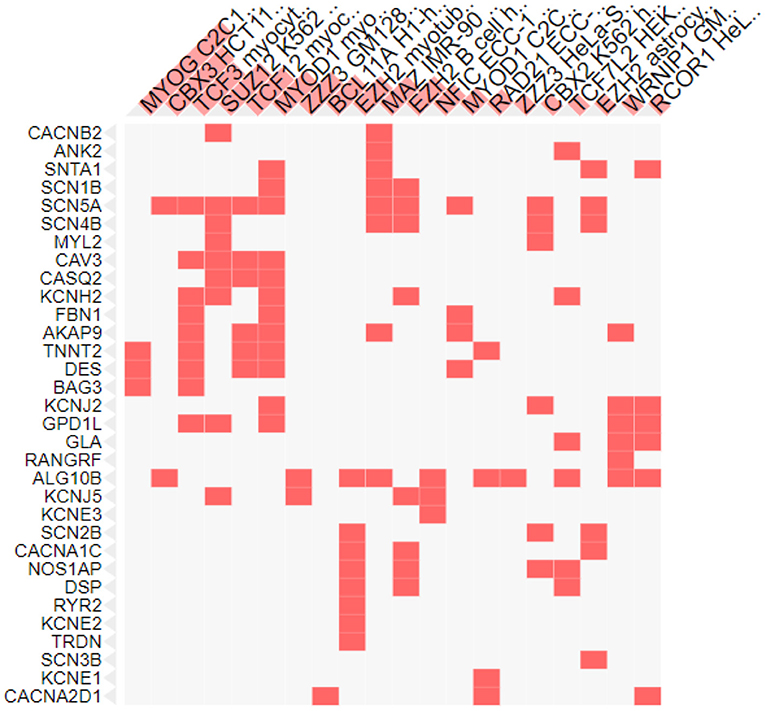
Figure 4. Transcription factors associated with SIDS genes. X-axis shows the transcription factors and y axis shows the genes under study.
Nine genes were also associated with the Transcription factor 3 (TCF3), encoded by the TCF3 gene and belongs to the helix-loop-helix family [E protein (class 1)] of transcription factors. These proteins are essential for the B and T lymphocyte development, thus play a crucial role in lymphopoiesis. Deletion or under expression of this gene is linked with lymphoid malignancies including pre-B-cell acute lymphoblastic leukemia and childhood leukemia (102). TCFs was recognized as the key regulator in the wilms tumor (pediatric kidney tumor) (103) and was found to act as a promoter in cervical cancer and associated with the development of cervical squamous cell carcinoma (104).
The lowest number of genes were found to be linked to the MYOG and chromobox protein. Three of the genes were associated with the muscle-specific transcription factor MYOG (Figure 4) essential for skeletal muscle development. It is a member of the Myogenic Regulatory Factor family of transcription factors that are main controllers of vertebrate muscle gene expression at early and adult myogenesis (105, 106). Severe muscle deficiencies were found in MYOG gene knockout mouse (107). Any abnormality in Myog transcription factor can lead to defects in regulation of muscle homeostasis, muscle growth and development, and reduced myofiber growth (108).
Two genes under study were associated with a transcription factor from the heterochromatin protein 1 family member, Chromobox protein homolog 3 (CBX3), that in involved in a variety of cellular processes like: DNA repair, gene regulation (by transcriptional activation or repression), cell differentiation and growth and epigenetic modification (109–114). CBX3 is upregulated in many cancers and plays an important role in the progression of tumors. The expression of CBX3 was elevated in human colorectal cancer (115), prostate cancer (116), breast cancer (117) and pancreatic cancer (118). In breast and lung cancer over expression of CBX3 leads to poor prognosis (117, 119).
The Key Regulator of Putative SIDS Genes—“mir-133a-3p”
MicroRNA (miRNA) are a set of highly conserved, endogenous, small non-coding RNAs molecules that range in size from 18 to 23 nucleotides and supress the gene expression by interacting with 3' untranslated regions (UTR) of target mRNAs (120). The miRNAs are significant in carcinogenesis and a number of evidences support that by adapting to the pathophysiological progressions, they may act as oncogenes or tumor suppressor genes (121–124). miRNAs are predicted as a tool for early diagnosis and treatment of cancer patients to extend their survival time (125–128).
In our candidate set of SIDS genes, miR-133a-3p is the highest reported (Figure 5). MiR-133-3p is a member of the miR-133 family that has two copies, miR-133a-1 and miR-133a-2, in humans. miR-133a-3p has multifaceted functions such as controlling proliferation and differentiation of myoblast (129), constraining proliferation of embryonic cardiomyocytes (130) and eluding genetic cardiac hypertrophy (131). In several human malignancies, miR-133a-3p is one of the recurrently downregulated miRNA which proposes that it may have a crucial role in tumor development in numerous malignancies, including non-small cell lung cancer (132), colorectal cancer (133), breast cancer (134), ovarian cancer (135), bladder cancer (136) and prostate cancer (137). The miR-208b-3p was the second in line in our set of genes but it was not regulating to the extent as miR-133-3p does. The miR-208b is among those miRNA species that are outstanding candidate as biomarkers for the diagnosis and prognosis of different diseases owing to their remarkable stability and detectability in peripheral blood or plasma, since the levels of these circulating miRNAs are symptomatically altered in individuals in several diseases (138–141). For the detection of cardiac diseases, cardiac specific miR-208b-3p have been utilized as promising biomarkers (142).
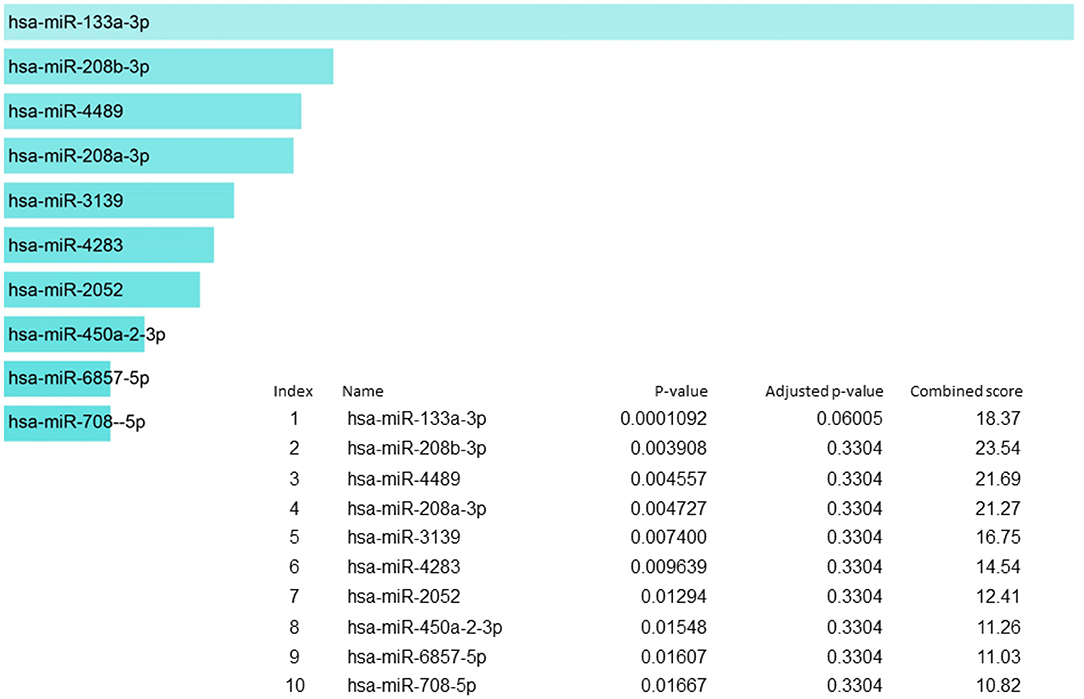
Figure 5. miRNA regulation of SIDS genes showing the key regulator miR-133a-3p at the highest position.
Discussion
SIDS is multifactorial and its cause remains undiagnosed so far. Genetic factors are least known and least explored. SIDS may be associated with variants in various genes. This study highlights the fact that genetic studies related to SIDS are scarce and there is insufficient literature reported so far. As there is genetic diversity due to genetic polymorphisms in different populations and also within same populations, so there is a dire need to explore SIDS on these lines.
A recent study from Germany (143) evaluated 31 SIDS cases via Next Generation Sequencing and reported the facts similar to our findings that cardiac channelopathy and cardiac myopathy genes should be investigated to find a link with SIDS. They used the pulmonary tissue samples after autopsy. We have also found the genes involved in cardiac channelopathy and cardiac myopathy to be associated with SIDS in various studies. In approximately 30% cases of SIDS, cardiac channelopathy linked genes were observed to be a monogenic cause of deaths (144). In 3.5% of the SIDS cases, a genetic polymorphism in a cardiac myopathy gene was reported (145). Although it is very important to detect and evaluate the genetic variants in order to counsel the family in a SIDS case, but it should be evaluated and labeled carefully as “pathogenic” or “informative” as 4.3% was found to be pathogenic and 13% as informative in the recent study mentioned above (143) which is much lower that assumed previously.
Various case reports have identified genetic predisposition in previously healthy infants who became victims of SIDS later on. In one report, SIDS with dysgenesis of the Testes syndrome (SIDDT), which is a rare condition caused by TSPYL1 gene, was reported for the first time in 2004. It is characterized by sudden cardiac or respiratory arrest, disordered testicular development, neurologic dysfunction. exome sequencing revealed homozygosity for a frameshift variant in TSPYL1 (c.725_726delTG, p.Val242GlufsTer52) consistent with a diagnosis of SIDDT, explaining many of the clinical features (146). Cerebellum has also been reported in a recent case series of four infant deaths and upon investigation, cerebellar heterotopia of infancy was found which is a distinct malformation of the cerebellum. It should be further explored (147). Exome sequencing was done in 10 SIDS cases and SCN1A was identified in two victims with hippocampal abnormalities. One had SCN1A G682V and other had 2 SCN1A variants in cis: L1296M and E1308D variants which were previously known to be associated with epilepsy (148).
In a rare case report of 15 day old infant who died unexpectedly and suddenly, whole exome sequencing was performed and two novel mutations in the CLCKB gene were found and lead to a molecular diagnosis of Bartter's syndrome Type III. This case highlights the importance of whole genome sequencing in such cases for proper diagnosis (149). In another case report, a 3 month old infant was found dead while sleeping and postmortem CT scan showed fatty attenuation in the liver parenchyma. Matrix assisted laser desorption/ionization imaging mass spectrometry (MALDI–IMS) analysis was conducted to further investigate the underlying mechanism of lipid accumulation. This revealed a significant accumulation of C14:1 acylcarnitine in the liver, proposing a very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency. Genetic analysis showed two novel mutations in ACADVL gene (150).
In a recent study, brain, heart, liver and kidney weights of SIDS victims were compared to controls. There was a large samples size of 291 SIDS and 294 controls obtained from Australia and Russia. Brain, thymus, liver and body weights were significantly higher among SIDS as compared to controls. Heart weight was significantly less in SIDS cases. Major difference was observed among thymus and brain weights and sizes. Heavier thymus points out to a possible infectious link to SIDS demises and larger brain sizes and weights shows a possible neuropathological roles (151). Larger thymus highlights the possible involvement of immunological genes which may be explored in this context. Genes related to neurotransmitters, their receptors and cardiopulmonary system should also be investigated further. Larger livers show a possible role of biochemical parameters and fatty acids (152) and need to be studied in detail. In another study, the frequency of extramedullary haematopoiesis (EMH) was observed to be significantly higher in SIDS victims as compared to controls which may be due to anemia associated with intrauterine hypoxia or infections (153).
Heterozygous gene mutation in PHOX2B, was reported in another case report of 28 day old female infant who died while lying on her bed in prone position. There were no pathological findings in postmortem investigation. PHOX2B gene is associated nervous system development and responsible for breathing control. Any mutation in this gene may lead to congenital central hypoventilation syndrome (154). Neonatal hypertrophic cardiomyopathy is a genetic disorder caused by mutations in MYBPC3 gene (p.Glu258Lys and IVS25-1G>A). It is associated with cardiac myopathy in neonates and leading cause of mortality and morbidity (155). In another case report, a sudden death of a 1 month old male infant while asleep was reported who was apparently healthy before the demise. This case was attributed to a combination of unavoidable events of congenital cardiac alterations, neuropathological alterations in brainstem and mutation in transporter gene (156). A case report on SIDS victim two female infants who were sisters as well, revealed a homozygous L2HGDH gene mutation. This gene is known to be associated with inherited metabolic disorders if mutated but it was first time reported in SIDS victims in this report published in 2009 (157).
Twin pregnancy and birth is also a risk factor for increased complications and SIDS. Koehler et al., published a review in 2001 on SIDS and Simultaneous Sudden Infant Death Syndrome (SSIDS) cases based on worldwide review of literature published from 1991 to 1998. SSIDS cases included the death of a pair of twins occurring suddenly without known reason. 41 SSIDS cases were found in this given time duration and they proposed a three point criteria for the cases to be considered as SSIDS and these included the location of death, a summary of the circumstances surrounding the deaths and the evaluation of these cases in terms of a proposed definition of SSIDS. According to this criteria, only 12 pairs of twins met all the three criteria and no conclusions were drawn (158). In a case report published in 2013, both the 10 weeks old twin infants died while sleeping on their backs. They were healthy and well before the sad event. Post-mortem investigations revealed petechial hemorrhages on the visceral pleura, epicardial surface and thymus gland. Histological findings showed pulmonary edema, intra-alveolar hemorrhage and minor lymphocytic infiltration in both infants, indicative of infection (159).
SIDS was significantly more common in monozygotic twins and of same gender during a retrospective study in England and Wales from 1993 to 2003 conducted by the office of National Statistics (160). In another study with conflicting results from United States, 23,464 singleton SIDS and 1,056 twin deaths were reported during 1987-1991. It was concluded that twins do not appear to be an increased risk factor for SIDS, twins dying of SIDS is uncommon while twins dying on same day is extremely rare incident (161). In another case report, a sudden infant death occurred in a 22 month old male twin infant while the other remained healthy. They contracted gastroenteritis and were given an antibiotic containing pivalic acid which is sometimes causing hypocarnitinemia. After post-mortem investigations and later on the genetic investigations, it was revealed that the fatal incident happened due to the deficiency of fatty acid oxidation accelerated by an antibiotic containing pivalic acid and the virus infection in this SIDS victim was containing the the thermolabile variant of carnitine palmitoyl transferase 2 (CPT2) gene. All factors alone are not fatal but cause SIDS when come in combination (162).
A case of sudden death of 1 month old male infant was reported by Donatella Mecchia et al. (156), attributing the death to cardiac, nervous system and genetic involvement. Polymorphism in serotonin transporter gene 5-HTT was observed (156). In another case report, a genetic analysis of 104 sudden cardiac death related genes was performed through Next Generation Sequencing (NGS). NGS identified 7 variants, 2 variations in genes AKAP9, EN1 were known previously as pathogenic, four variations in genes KCNE3, PKP2, VCL, and TTN had no clinical significance known, 7th variation in gene TTN was novel (163). In a molecular autopsy study of 50 cases of sudden young death <45 years old, during a period of 2 years was performed by exome sequencing. For exome, variants in TTN gene were the most abundant, having 56 variants, followed by MUC16 having 53 variants and SSPO having 49 variants. TTN is the gene encoding titin which is the largest protein of the human genome having 305 kbs and is involved in cardiomyopathies. MUC16 or mucin 16 is a 132 kb gene coding for mucin protein which is a glycoprotein involved in cancers and SSPO (subcommissural organ spondin) is a 58 kb gene that codes for a protein involved in neuronal aggregation. All the above cases had negative autopsy findings but molecular autopsy revealed these genetic variations (164). Although these cases were not SIDS but it must be explored in SIDS as well and similar strategy may by used for molecular autopsy of SIDS.
In a whole exome sequencing study of 161 SIDS cases from Europe, 192 genes associated with cardiovascular and metabolic diseases were investigated. Potentially causative genes were identified in 20% of the SIDS cases, 9% of the genes were associated with channelopathies, 7% with cardiomyopathies, 1% with metabolic disorders (144). A molecular study conducted on 41 cases of sudden unexpected death in infancy, analyzed 86 sudden cardiac death related genes. 63 variants in 35 cases were validated. Most of these variants were located on cardiomyopathy genes or channelopathy genes (165). NGS was performed as a molecular autopsy tool for 16 cases of sudden unexpected deaths in youngs, <35 years old. Twenty three genes associated with inherited cardiac channelopathies were screened. An average 200 variants per case were identified and after correlating them with the clinical data and molecular findings, 4 likely pathogenic variants were identified in three cases in the genes KCNH2, ANK2, SCN5A, and RYR2 (166). In a molecular autopsy on 141 SIDS cases, <6 months of age and 133 non/infants (19-58 years old) six major channelpathy genes, KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, and RyR2 were characterize in 274 cases which were autopsy negative. 22 previously known variants linked to cardiac channelopathy and 24 novel variants were identified among infants (13.5%) and non-infants (19.5%). Most of these variants involved SCN5A gene (68.4% in infants and 50% in non-infants) which is a sodium channel alpha subunit. This study highlighted the role of cardiac channel genes in sudden deaths of infants and non-infants (167).
In our previous studies, including post-mortem studies, in silico and in vitro studies, we have proposed the involvement of Substance P/Neurokinin 1 Receptor and the gene encoding SP, Tachykinin 1 (Tac 1) to be involved in SIDS (168–172). We emphasize further studies to be conducted on this pathway as it is involved in the breathing control mechanism and a genetic mutation in its gene can be deleterious. We have also proposed the involvement of neuropathology in COVID-19 as well and proposed the same SP/ NK-1R mechanism which contributes in the neuropathology of SIDS. The same phenomenon may also be involved for lesser implications in children and infants as compared to the adults (173). Current study also presents the involvement of these candidate genes in other biological processes. We have also highlighted the importance of gene expression and its association with SIDS by discovering the histone methylations, gene enrichment pathways, transcription factors and micro RNAs reported so far. Gene regulation has utmost importance in these cases and must be investigated further. This study provides a comprehensive analysis and presents the genetic determinants which need further investigation. It will pave way for other researchers who are trying to find out the genetic causes of SIDS. Next Generation sequencing may be an important tool in this regard.
Conclusions
A number of genes mainly potassium and sodium channel genes may be associated with the devastating phenomenon of SIDS. Future studies should be conducted to explore the properties of these genes in relation to SIDS so that vulnerable infants and neonates may be diagnosed timely before the sad demise. Preventive measures can be adopted to avoid such incidences. Next generation sequencing and careful interpretation of the gene variants is required to reveal these facts.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author Contributions
RM and SAS planned, conceived, and designed and supervised the study. MK, MA, SAG, and SK did data extraction and write-up. HN, AM, HM, SH, and AH worked on revisions, write-up, and analysis. MA and AB did analysis, write-up, and critical review. All the authors have contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Winkel BG, Yuan L, Olesen MS, Sadjadieh G, Wang Y, Risgaard B, et al. The role of the sodium current complex in a nonreferred nationwide cohort of sudden infant death syndrome. Heart Rhythm. (2015) 12:1241–9. doi: 10.1016/j.hrthm.2015.03.013
2. Matturri L, Lavezzi AM, Rossi L. Proposal to modify the defi nition of SIDS, with regard to the post-mortem exam. In: Proceedings of the 7th International Conference on SIDS. Florence. (2002) p. 103.
3. Ottaviani G. Defining sudden infant death and sudden intrauterine unexpected death syndromes with regard to anatomo-pathological examination. Front Pediatr. (2016) 4:103. doi: 10.3389/fped.2016.00103
4. Goldstein RD, Kinney HC, Willinger M. Sudden unexpected death in fetal life through early childhood. Pediatrics. (2016) 137:e20154661. doi: 10.1542/peds.2015-4661
5. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation. (2016) 133:e38–360. doi: 10.1161/CIR.0000000000000350
6. Ottaviani G, Buja LM. Congenital heart disease: pathology, natural history, and interventions. In: Buja LM, Butany J, editors. Cardiovascular Pathology. 4th ed. Waltham, MA: Elsevier. (2016) p. 611–47.
7. Andreasen C, Refsgaard L, Nielsen JB, Sajadieh A, Winkel BG, Tfelt-Hansen J, et al. Mutations in genes encoding cardiac ion channels previously associated with sudden infant death syndrome (SIDS) are present with high frequency in new exome data. Can J Cardiol. (2013) 29:1104–9. doi: 10.1016/j.cjca.2012.12.002
8. Ottaviani G. Crib Death – Sudden Infant Death Syndrome (SIDS). Sudden Infant and Perinatal Unexplained Death: The Pathologist's Viewpoint. 2nd ed. Heidelberg: Springer International Publishing (2014).
9. Filiano JJ, Kinney HC. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Biol Neonate. (1994) 65:194–7. doi: 10.1159/000244052
10. Ottaviani G, Buja LM. Anatomopathological changes of the cardiac conduction system in sudden cardiac death, particularly in infants: advances over the last 25 years. Cardiovasc Pathol. (2016) 25:489–99. doi: 10.1016/j.carpath.2016.08.005
11. Buja LM, Vander Heide RS. Pathobiology of ischemic heart disease: past, present and future. Cardiovasc Pathol. (2016) 25:214–20. doi: 10.1016/j.carpath.2016.01.007
12. Kaler J, Hussain A, Lee S. Manifestation and pathogenesis of sudden infant death syndrome: a review. Crit Rev Eukaryot Gene Expr. (2020) 30:111–20. doi: 10.1615/CritRevEukaryotGeneExpr.2020033009
13. Opdal SH, Rognum TO. Gene variants predisposing to SIDS: current knowledge. Forensic Sci Med Pathol. (2011) 7:26–36. doi: 10.1007/s12024-010-9182-9
14. Courts C, Madea B. Genetics of the sudden infant death syndrome. Forensic Sci Int. (2010) 203:25–33. doi: 10.1016/j.forsciint.2010.07.008
15. Opdal SH. Cytokines, Infection, and Immunity, in SIDS Sudden Infant and Early Childhood Death: The Past, the Present and the Future. Duncan JR and Byard RW, editors. Adelaide (2018).
17. Stoltenberg L, Saugstad OD, Rognum TO. Sudden infant death syndrome victims show local immunoglobulin M response in tracheal wall and immunoglobulin A response in duodenal mucosa. Pediatr Res. (1992) 31:372–5. doi: 10.1203/00006450-199204000-00013
18. Stoltenberg L, Vege A, Saugstad OD, Rognum TO. Changes in the concentration and distribution of immunoglobulin-producing cells in SIDS palatine tonsils. Pediatr Allergy Immunol. (1995) 6:48–55. doi: 10.1111/j.1399-3038.1995.tb00258.x
19. Thrane PS, Rognum TO, Brandtzaeg P. Up-regulated epithelial expression of HLA-DR and secretory component in salivary glands: reflection of mucosal immunostimulation in sudden infant death syndrome. Pediatr Res. (1994) 35:625–8. doi: 10.1203/00006450-199405000-00017
20. Highet AR, Goldwater PN. Staphylococcal enterotoxin genes are common in Staphylococcus aureus intestinal flora in Sudden Infant Death Syndrome (SIDS) and live comparison infants. FEMS Immunol Med Microbiol. (2009) 57:151–5. doi: 10.1111/j.1574-695X.2009.00592.x
21. Opdal SH. IL-10 gene polymorphisms in infectious disease and SIDS. FEMS Immunol Med Microbiol. (2004) 42:48–52. doi: 10.1016/j.femsim.2004.06.006
22. Korachi M, Pravica V, Barson AJ, Hutchinson IV, Drucker DB. Interleukin 10 genotype as a risk factor for sudden infant death syndrome: determination of IL-10 genotype from wax-embedded postmortem samples. FEMS Immunol Med Microbiol. (2004) 42:125–9. doi: 10.1016/j.femsim.2004.06.008
23. Ferrante L, Opdal SH. Sudden infant death syndrome and the genetics of inflammation. Front Immunol. (2015) 6:63. doi: 10.3389/fimmu.2015.00063
24. Sweeting J, Semsarian C. Cardiac abnormalities and sudden infant death syndrome. Paediatr Respir Rev. (2014) 15:301–6. doi: 10.1016/j.prrv.2014.09.006
25. Campuzano O, Sanchez-Molero O, Allegue C, Coll M, Mademont-Soler I, Selga E, et al. Post-mortem genetic analysis in juvenile cases of sudden cardiac death. Forensic Sci Int. (2014) 245:30–7. doi: 10.1016/j.forsciint.2014.10.004
26. Skinner JR, Crawford J, Smith W, Aitken A, Heaven D, Evans CA, et al. Prospective, population-based long QT molecular autopsy study of postmortem negative sudden death in 1 to 40 year olds. Heart Rhythm. (2011) 8:412–9. doi: 10.1016/j.hrthm.2010.11.016
27. Tester DJ, Dura M, Carturan E, Reiken S, Wronska A, Marks AR, et al. A mechanism for sudden infant death syndrome (SIDS): stress-induced leak via ryanodine receptors. Heart Rhythm. (2007) 4:733–9. doi: 10.1016/j.hrthm.2007.02.026
28. Ackerman MJ. Cardiac causes of sudden unexpected death in children and their relationship to seizures and syncope: genetic testing for cardiac electropathies. Semin Pediatr Neurol. (2005) 12:52–8. doi: 10.1016/j.spen.2005.02.002
29. Poetsch M, Todt R, Vennemann M, Bajanowski T. That's not it, either-neither polymorphisms in PHOX2B nor in MIF are involved in sudden infant death syndrome (SIDS). Int J Legal Med. (2015) 129:985–9. doi: 10.1007/s00414-015-1213-3
30. Weese-Mayer DE, Zhou L, Berry-Kravis EM, Maher BS, Silvestri JM, Marazita ML. Association of the serotonin transporter gene with sudden infant death syndrome: a haplotype analysis. Am J Med Genet A. (2003) 122:238–45. doi: 10.1002/ajmg.a.20427
31. Narita N, Narita M, Takashima S, Nakayama M, Nagai T, Okado N. Serotonin transporter gene variation is a risk factor for sudden infant death syndrome in the Japanese population. Pediatrics. (2001) 107:690–2. doi: 10.1542/peds.107.4.690
32. Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Curran ME, Silvestri JM, et al. Sudden infant death syndrome: case-control frequency differences at genes pertinent to early autonomic nervous system embryologic development. Pediatr Res. (2004) 56:391–5. doi: 10.1203/01.PDR.0000136285.91048.4A
33. Van Norstrand DW, Ackerman MJ. Sudden infant death syndrome: do ion channels play a role? Heart Rhythm. (2009) 6:272–8. doi: 10.1016/j.hrthm.2008.07.028
34. Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, et al. Sudden infant death syndrome-associated mutations in the sodium channel beta subunits. Heart Rhythm. (2010) 7:771–8. doi: 10.1016/j.hrthm.2010.01.032
35. Valdivia CR, Medeiros-Domingo A, Ye B, Shen WK, Algiers TJ, Ackerman MJ, et al. Loss-of-function mutation of the SCN3B-encoded sodium channel {beta}3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc Res. (2010) 86:392–400. doi: 10.1093/cvr/cvp417
36. Hu D, Barajas-Martínez H, Medeiros-Domingo A, Crotti L, Veltmann C, Schimpf R, et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na(v)1.5 and K(v)4.3 channel currents. Heart Rhythm. (2012) 9:760–9. doi: 10.1016/j.hrthm.2011.12.006
37. Patino GA, Brackenbury WJ, Bao Y, Lopez-Santiago LF, O'Malley HA, Chen C, et al. Voltage-gated Na+ channel beta1B: a secreted cell adhesion molecule involved in human epilepsy. J Neurosci. (2011) 31:14577–91. doi: 10.1523/JNEUROSCI.0361-11.2011
38. Chockalingam P, Wilde AA. Inherited arrhythmia syndromes leading to sudden cardiac death in the young: a global update and an Indian perspective. Indian Heart J. (2014) 66:S49–57. doi: 10.1016/j.ihj.2013.11.008
39. Pei H, Yu Q, Su X, Wang Z, Zhao H, Yang D, et al. New features of electrocardiogram in a case report of arrhythmogenic right ventricular cardiomyopathy: a care-compliant article. Medicine. (2016) 95:e3442. doi: 10.1097/MD.0000000000003442
40. Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. (2007) 115:361–7. doi: 10.1161/CIRCULATIONAHA.106.658021
41. Otagiri T., Kijima K, Osawa M, Ishii K, Makita N, Matoba R, et al. Cardiac ion channel gene mutations in sudden infant death syndrome. Pediatr Res. (2008) 64:482–7. doi: 10.1203/PDR.0b013e3181841eca
42. Hertz CL, Christiansen SL, Larsen MK, Dahl M, Ferrero-Miliani L, Weeke PE, et al. Genetic investigations of sudden unexpected deaths in infancy using next-generation sequencing of 100 genes associated with cardiac diseases. Eur J Hum Genet. (2016) 24:817–22. doi: 10.1038/ejhg.2015.198
43. Yellen G. The voltage-gated potassium channels and their relatives. Nature. (2002) 419:35–42. doi: 10.1038/nature00978
44. Chandy KG, Williams CB, Spencer RH, Aguilar BA, Ghanshani S, Tempel BL, et al. A family of three mouse potassium channel genes with intronless coding regions. Science. (1990) 247:973. doi: 10.1126/science.2305265
45. Hille B. Ionic channels in excitable membranes. Current problems and biophysical approaches. Biophysical J. (1978) 22:283–94. doi: 10.1016/S0006-3495(78)85489-7
46. Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: from gene to physiological function. Physiology. (2005) 20:408–16. doi: 10.1152/physiol.00031.2005
47. Abbott GW. KCNE1 and KCNE3: The yin and yang of voltage-gated K(+) channel regulation. Gene. (2016) 576:1–13. doi: 10.1016/j.gene.2015.09.059
48. Dixit G, Dabney-Smith C, Lorigan GA. The membrane protein KCNQ1 potassium ion channel: Functional diversity and current structural insights. Biochimica et Biophysica Acta (BBA) – Biomembranes. (2020) 1862:183148. doi: 10.1016/j.bbamem.2019.183148
49. Faridi R, Tona R, Brofferio A, Hoa M, Olszewski R, Schrauwen I, et al. Mutational and phenotypic spectra of KCNE1 deficiency in Jervell and Lange-Nielsen Syndrome and Romano-Ward Syndrome. Hum Mutat. (2019) 40:162–76. doi: 10.1002/humu.23689
50. Hull JM, Isom LL. Voltage-gated sodium channel β subunits: the power outside the pore in brain development and disease. Neuropharmacology. (2018) 132:43–57. doi: 10.1016/j.neuropharm.2017.09.018
51. Xiong H, Yang Q, Zhang X, Wang P, Chen F, Liu Y, et al. Significant association of rare variant p.Gly8Ser in cardiac sodium channel β4-subunit SCN4B with atrial fibrillation. Ann Hum Genet. (2019) 83:239–48. doi: 10.1111/ahg.12305
52. Yereddi NR, Cusdin FS, Namadurai S, Packman LC, Monie TP, Slavny P, et al. The immunoglobulin domain of the sodium channel β3 subunit contains a surface-localized disulfide bond that is required for homophilic binding. FASEB J. (2013) 27:568–80. doi: 10.1096/fj.12-209445
53. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. (2010) 90:291–366. doi: 10.1152/physrev.00021.2009
54. Cerrone M, Napolitano C, Priori SG. Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated to impaired Ca(2+) regulation. Heart Rhythm. (2009) 6:1652–9. doi: 10.1016/j.hrthm.2009.06.033
55. Durand JB, Bachinski LL, Bieling LC, Czernuszewicz GZ, Abchee AB, Yu QT, et al. Localization of a gene responsible for familial dilated cardiomyopathy to chromosome 1q32. Circulation. (1995) 92:3387–9. doi: 10.1161/01.CIR.92.12.3387
56. Bosè F, Renna LV, Fossati B, Arpa G, Labate V, Milani V, et al. TNNT2 missplicing in skeletal muscle as a cardiac biomarker in myotonic dystrophy type 1 but not in myotonic dystrophy type 2. Front Neurol. (2019) 10:992. doi: 10.3389/fneur.2019.00992
57. Bhuiyan ZA, van den Berg MP, van Tintelen JP, Bink-Boelkens MT, Wiesfeld AC, Alders M, et al. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation. (2007) 116:1569–76. doi: 10.1161/CIRCULATIONAHA.107.711606
58. Minetti C, Sotgia F, Bruno C, Scartezzini P, Broda P, Bado M, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. (1998) 18:365–8. doi: 10.1038/ng0498-365
59. Gazzerro E, Sotgia F, Bruno C, Lisanti MP, Minetti C. Caveolinopathies: from the biology of caveolin-3 to human diseases. Eur J Hum Genet. (2010) 18:137–45. doi: 10.1038/ejhg.2009.103
60. Valdivia CR, Ueda K, Ackerman MJ, Makielski JC. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol. (2009) 297:H1446–52. doi: 10.1152/ajpheart.00513.2009
61. Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, Ackerman MJ. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. (2007) 4:161–6. doi: 10.1016/j.hrthm.2006.11.030
62. Gordan R, Gwathmey JK, Xie LH. Autonomic and endocrine control of cardiovascular function. World J Cardiol. (2015) 7:204–14. doi: 10.4330/wjc.v7.i4.204
63. Munshi NV. Gene regulatory networks in cardiac conduction system development. Circ Res. (2012) 110:1525–37. doi: 10.1161/CIRCRESAHA.111.260026
64. Kanno S, Saffitz JE. The role of myocardial gap junctions in electrical conduction and arrhythmogenesis. Cardiovasc Pathol. (2001) 10:169–77. doi: 10.1016/S1054-8807(01)00078-3
65. Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. (2003) 83:1359–400. doi: 10.1152/physrev.00007.2003
66. Antzelevitch C, Dumaine R. Electrical heterogeneity in the heart: physiological, pharmacological and clinical implications, in comprehensive physiology. In: Page E, Fozzard HA, Solaro RJ, editors. The Handbook of Physiology. New York, NY: Oxford University Press (2002) p. 654–92.
67. Antzelevitch C. Molecular genetics of arrhythmias and cardiovascular conditions associated with arrhythmias. J Cardiovasc Electrophysiol. (2003) 14:1259–72. doi: 10.1046/j.1540-8167.2003.03316.x
68. Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. (2004) 84:431–88. doi: 10.1152/physrev.00025.2003
69. Nerbonne JM, Guo W. Heterogeneous expression of voltage-gated potassium channels in the heart: roles in normal excitation and arrhythmias. J Cardiovasc Electrophysiol. (2002) 13:406–9. doi: 10.1046/j.1540-8167.2002.00406.x
70. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. (2005) 85:1205–53. doi: 10.1152/physrev.00002.2005
71. Matturri L Ottaviani G, Ramos SG, Rossi L. Sudden infant death syndrome (SIDS):a study of cardiac conduction system. Cardiovasc Pathol. (2000) 9:137–45. doi: 10.1016/S1054-8807(00)00035-1
72. Magi S, Lariccia V, Maiolino M, Amoroso S, Gratteri S. Sudden cardiac death: focus on the genetics of channelopathies and cardiomyopathies. J Biomed Sci. (2017) 24:56. doi: 10.1186/s12929-017-0364-6
73. Murray SC, Lorenz P, FS Howe, Wouters M, Brown T, Xi S, et al. H3K4me3 is neither instructive for, nor informed by, transcription. bioRxiv. (2019) 2019:709014. doi: 10.1101/709014
74. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. (2011) 21:381–95. doi: 10.1038/cr.2011.22
75. Sun Z, Zhang Y, Jia J, Fang Y, Tang Y, Wu H, et al. H3K36me3, message from chromatin to DNA damage repair. Cell Biosci. (2020) 10:9. doi: 10.1186/s13578-020-0374-z
76. Ferrari Karin J, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stützer A, et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell. (2014) 53:49–62. doi: 10.1016/j.molcel.2013.10.030
77. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. (2007) 129:823–37. doi: 10.1016/j.cell.2007.05.009
78. Ma Z, Wang H, Cai Y, Wang H, Niu K, Wu X, et al. Epigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in Drosophila. Elife. (2018) 7:e35368. doi: 10.7554/eLife.35368
79. Ninova M, Fejes Tóth K, Aravin AA. The control of gene expression and cell identity by H3K9 trimethylation. Development. (2019) 146:dev181180. doi: 10.1242/dev.181180
80. Aloia L, Di Stefano B, Di Croce L. Polycomb complexes in stem cells and embryonic development. Development. (2013) 140:2525. doi: 10.1242/dev.091553
81. Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. Genome regulation by polycomb and trithorax: 70 years and counting. Cell. (2017) 171:34–57. doi: 10.1016/j.cell.2017.08.002
82. Cano-Rodriguez D, Gjaltema RA, Jilderda LJ, Jellema P, Dokter-Fokkens J, Ruiters MH, et al. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat Commun. (2016) 7:12284. doi: 10.1038/ncomms12284
83. Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. (2013) 152:1021–36. doi: 10.1016/j.cell.2013.01.052
84. Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. (2007) 25:15–30. doi: 10.1016/j.molcel.2006.12.014
85. Jiang W, Wang J, Zhang Y. Histone H3K27me3 demethylases KDM6A and KDM6B modulate definitive endoderm differentiation from human ESCs by regulating WNT signaling pathway. Cell Res. (2013) 23:122–30. doi: 10.1038/cr.2012.119
86. Shpargel KB, Starmer J, Yee D, Pohlers M, Magnuson T. KDM6 Demethylase independent loss of histone H3 Lysine 27 trimethylation during early embryonic development. PLOS Genet. (2014) 10:e1004507. doi: 10.1371/journal.pgen.1004507
87. Wang Q, Chen X, Jiang Y, Liu S, Liu H, Sun X. Elevating H3K27me3 level sensitizes colorectal cancer to oxaliplatin. J Mol Cell Biol. (2020) 12:125–37. doi: 10.1093/jmcb/mjz032
88. Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. (2013) 24:660–72. doi: 10.1016/j.ccr.2013.10.006
89. Carvalho S, Freitas M, Antunes L, Monteiro-Reis S, Vieira-Coimbra M, Tavares A, et al. Prognostic value of histone marks H3K27me3 and H3K9me3 and modifying enzymes EZH2, SETDB1 and LSD-1 in colorectal cancer. J Cancer Res Clin Oncol. (2018) 144:2127–37. doi: 10.1007/s00432-018-2733-2
90. Hoffman RM, Erbe RW. High in vivo rates of methionine biosynthesis in transformed human and malignant rat cells auxotrophic for methionine. Proc Natl Acad Sci. (1976) 73:1523. doi: 10.1073/pnas.73.5.1523
91. Hoffman RM. Development of recombinant methioninase to target the general cancer-specific metabolic defect of methionine dependence: a 40-year odyssey. Exp Opin Biol Ther. (2015) 15:21–31. doi: 10.1517/14712598.2015.963050
92. Kawaguchi K, Han Q, Li S, Tan Y, Igarashi K, Murakami T, et al. Efficacy of recombinant methioninase (rMETase) on recalcitrant cancer patient-derived orthotopic xenograft (PDOX) mouse models: a review. Cells. (2019) 8:410. doi: 10.3390/cells8050410
93. Yamamoto J, Han Q, Inubushi S, Sugisawa N, Hamada K, Nishino H, et al. Histone methylation status of H3K4me3 and H3K9me3 under methionine restriction is unstable in methionine-addicted cancer cells, but stable in normal cells. Biochem Biophys Res Commun. (2020) 533:1034–8. doi: 10.1016/j.bbrc.2020.09.108
94. Adcock IM, Caramori G. Chapter 31 - Transcription factors. In: Barnes PJ, et al., editors. Asthma and COPD (Second Edition). Oxford: Academic Press (2009). p. 373–80.
95. Akizawa Y, Kanno H, Kawamichi Y, Matsuda Y, Ohta H, Fujii H, et al. Enhanced expression of myogenic differentiation factors and skeletal muscle proteins in human amnion-derived cells via the forced expression of MYOD1. Brain Dev. (2013) 35:349–55. doi: 10.1016/j.braindev.2012.05.012
96. Milewska M, Grabiec K, Grzelkowska-Kowalczyk K. [Interactions of proliferation and differentiation signaling pathways in myogenesis]. Postepy Hig Med Dosw. (2014) 68:516–26. doi: 10.5604/17322693.1101617
97. Rajabi HN, Takahashi C, Ewen ME. Retinoblastoma protein and MyoD function together to effect the repression of Fra-1 and in turn cyclin D1 during terminal cell cycle arrest associated with myogenesis*. J Biol Chem. (2014) 289:23417–27. doi: 10.1074/jbc.M113.532572
98. Peng G, Masood K, Gantz O, Sinha U. Neuromuscular electrical stimulation improves radiation-induced fibrosis through Tgf-B1/MyoD homeostasis in head and neck cancer. J Surg Oncol. (2016) 114:27–31. doi: 10.1002/jso.24265
99. Zhang Q, Liu XY, Li S, Zhao Z, Li J, Cui MK, et al. Repression of ESR1 transcription by MYOD potentiates letrozole-resistance in ERα-positive breast cancer cells. Biochem Biophys Res Commun. (2017) 492:425–33. doi: 10.1016/j.bbrc.2017.08.082
100. Jiang H, Wang H, Wang S, Pei Z, Fu Z, Fang C, et al. Expression of ERCC1, TYMS, RRM1, TUBB3, non-muscle myosin II, myoglobin and MyoD1 in lung adenocarcinoma pleural effusions predicts survival in patients receiving platinum-based chemotherapy. Mol Med Rep. (2015) 11:3523–32. doi: 10.3892/mmr.2014.3141
101. Wu F, Qin Y, Jiang Q, Zhang J, Li F, Li Q, et al. MyoD1 suppresses cell migration and invasion by inhibiting FUT4 transcription in human gastric cancer cells. Cancer Gene Ther. (2020) 27:773–84. doi: 10.1038/s41417-019-0153-3
102. Ben-Ali M, Yang J, Chan KW, Ben-Mustapha I, Mekki N, Benabdesselem C, et al. Homozygous transcription factor 3 gene (TCF3) mutation is associated with severe hypogammaglobulinemia and B-cell acute lymphoblastic leukemia. J Allergy Clin Immunol. (2017) 140:1191–4.e4. doi: 10.1016/j.jaci.2017.04.037
103. Kehl T, Schneider L, Kattler K, Stöckel D, Wegert J, Gerstner N, et al. The role of TCF3 as potential master regulator in blastemal Wilms tumors. Int J Cancer. (2019) 144:1432–43. doi: 10.1002/ijc.31834
104. Luo L, Zhang H, Nian S, Lv C, Ni B, Wang D, et al. Up-regulation of transcription factor 3 is correlated with poor prognosis in cervical carcinoma. Int J Gynecol Cancer. (2017) 27:1422–30. doi: 10.1097/IGC.0000000000001032
105. Hernández-Hernández JM, García-González EG, Brun CE, Rudnicki MA. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin Cell Dev Biol. (2017) 72:10–8. doi: 10.1016/j.semcdb.2017.11.010
106. Zammit PS. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin Cell Dev Biol. (2017) 72:19–32. doi: 10.1016/j.semcdb.2017.11.011
107. Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, et al. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. (1993) 364:501–6. doi: 10.1038/364501a0
108. Ganassi M, Badodi S, Wanders K, Zammit PS, Hughes SM. Myogenin is an essential regulator of adult myofibre growth and muscle stem cell homeostasis. Elife. (2020) 9:e60445. doi: 10.7554/eLife.60445
109. Maison C, Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol. (2004) 5:296–304. doi: 10.1038/nrm1355
110. Hediger F, Gasser SM. Heterochromatin protein 1: don't judge the book by its cover! Curr Opin Genet Dev. (2006) 16:143–50. doi: 10.1016/j.gde.2006.02.013
111. Lomberk G, Wallrath L, Urrutia R. The heterochromatin protein 1 family. Genome Biol. (2006) 7:228. doi: 10.1186/gb-2006-7-7-228
112. Kwon SH, Workman JL. The heterochromatin protein 1 (HP1) family: put away a bias toward HP1. Mol Cells. (2008) 26:217–27.
113. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. (2007) 8:286–98. doi: 10.1038/nrg2005
114. Chen LY, Cheng CS, Qu C, Wang P, Chen H, Meng ZQ, et al. CBX3 promotes proliferation and regulates glycolysis via suppressing FBP1 in pancreatic cancer. Biochem Biophys Res Commun. (2018) 500:691–7. doi: 10.1016/j.bbrc.2018.04.137
115. Liu M, Huang F, Zhang D, Ju J, Wu XB, Wang Y, et al. Heterochromatin protein HP1γ promotes colorectal cancer progression and is regulated by miR-30a. Cancer Res. (2015) 75:4593–604. doi: 10.1158/0008-5472.CAN-14-3735
116. Slezak J, Truong M, Huang W, Jarrard D. HP1γ expression is elevated in prostate cancer and is superior to Gleason score as a predictor of biochemical recurrence after radical prostatectomy. BMC Cancer. (2013) 13:148. doi: 10.1186/1471-2407-13-148
117. Liang YK, Lin HY, Chen CF, Zeng D. Prognostic values of distinct CBX family members in breast cancer. Oncotarget. (2017) 8:92375–87. doi: 10.18632/oncotarget.21325
118. Chen L-Y, Cheng CS, Qu C, Wang P, Chen H, Meng ZQ, et al. Overexpression of CBX3 in pancreatic adenocarcinoma promotes cell cycle transition-associated tumor progression. Int J Mol Sci. (2018) 19:1768. doi: 10.3390/ijms19061768
119. Chang SC, Lai YC, Chen YC, Wang NK, Wang WS, Lai JI. CBX3/heterochromatin protein 1 gamma is significantly upregulated in patients with non-small cell lung cancer. Asia Pac J Clin Oncol. (2018) 14:e283–8. doi: 10.1111/ajco.12820
120. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
121. Liu H, Li W, Chen C, Pei Y, Long X. MiR-335 acts as a potential tumor suppressor miRNA via downregulating ROCK1 expression in hepatocellular carcinoma. Tumour Biol. (2015) 36:6313–9. doi: 10.1007/s13277-015-3317-2
122. Zhang X, Tang W, Li R, He R, Gan T, Luo Y, et al. Downregulation of microRNA-132 indicates progression in hepatocellular carcinoma. Exp Ther Med. (2016) 12:2095–101. doi: 10.3892/etm.2016.3613
123. Motawi TK, Shaker OG, El-Maraghy SA, Senousy MA. Serum MicroRNAs as potential biomarkers for early diagnosis of hepatitis C virus-related hepatocellular carcinoma in Egyptian patients. PLoS ONE. (2015) 10:e0137706. doi: 10.1371/journal.pone.0137706
124. Zhang JG, Shi Y, Hong DF, Song M, Huang D, Wang CY, et al. MiR-148b suppresses cell proliferation and invasion in hepatocellular carcinoma by targeting WNT1/β-catenin pathway. Sci Rep. (2015) 5:8087. doi: 10.1038/srep08087
125. Liu Y, Ren F, Rong M, Luo Y, Dang Y, Chen G. Association between underexpression of microrna-203 and clinicopathological significance in hepatocellular carcinoma tissues. Cancer Cell Int. (2015) 15:62. doi: 10.1186/s12935-015-0214-0
126. Yao H, Liu X, Chen S, Xia W, Chen X. Decreased expression of serum miR-424 correlates with poor prognosis of patients with hepatocellular carcinoma. Int J Clin Exp Pathol. (2015) 8:14830–5. Available online at: www.ijcep.com/ISSN:1936-2625/IJCEP0016755
127. Huang CS, Yu W, Cui H, Wang YJ, Zhang L, Han F, et al. Increased expression of miR-21 predicts poor prognosis in patients with hepatocellular carcinoma. Int J Clin Exp Pathol. (2015) 8:7234–8.
128. Chen Z, Huang Z, Ye Q, Ming Y, Zhang S, Zhao Y, et al. Prognostic significance and anti-proliferation effect of microRNA-365 in hepatocellular carcinoma. Int J Clin Exp Pathol. (2015) 8:1705–11. Available online at: www.ijcep.com/ISSN:1936-2625/IJCEP0004398
129. Koutsoulidou A, Mastroyiannopoulos NP, Furling D, Uney JB, Phylactou LA. Expression of miR-1, miR-133a, miR-133b and miR-206 increases during development of human skeletal muscle. BMC Dev Biol. (2011) 11:34. doi: 10.1186/1471-213X-11-34
130. Babiarz JE, Ravon M, Sridhar S, Ravindran P, Swanson B, Bitter H, et al. Determination of the human cardiomyocyte mRNA and miRNA differentiation network by fine-scale profiling. Stem Cells Dev. (2012) 21:1956–65. doi: 10.1089/scd.2011.0357
131. Li Y, Cai X, Guan Y, Wang L, Wang S, Li Y, et al. Adiponectin upregulates MiR-133a in cardiac hypertrophy through AMPK activation and reduced ERK1/2 phosphorylation. PLoS ONE. (2016) 11:e0148482. doi: 10.1371/journal.pone.0148482
132. Wang LK, Hsiao TH, Hong TM, Chen HY, Kao SH, Wang WL, et al. MicroRNA-133a suppresses multiple oncogenic membrane receptors and cell invasion in non-small cell lung carcinoma. PLoS ONE. (2014) 9:e96765. doi: 10.1371/journal.pone.0096765
133. Zhang W, Liu K, Liu S, Ji B, Wang Y, Liu Y. MicroRNA-133a functions as a tumor suppressor by targeting IGF-1R in hepatocellular carcinoma. Tumour Biol. (2015) 36:9779–88. doi: 10.1007/s13277-015-3749-8
134. Cui W, Zhang S, Shan C, Zhou L, Zhou Z. microRNA-133a regulates the cell cycle and proliferation of breast cancer cells by targeting epidermal growth factor receptor through the EGFR/Akt signaling pathway. FEBS J. (2013) 280:3962–74. doi: 10.1111/febs.12398
135. Guo J, Xia B, Meng F, Lou G. miR-133a suppresses ovarian cancer cell proliferation by directly targeting insulin-like growth factor 1 receptor. Tumour Biol. (2014) 35:1557–64. doi: 10.1007/s13277-013-1215-z
136. Yoshino H, Chiyomaru T, Enokida H, Kawakami K, Tatarano S, Nishiyama K, et al. The tumour-suppressive function of miR-1 and miR-133a targeting TAGLN2 in bladder cancer. Br J Cancer. (2011) 104:808–18. doi: 10.1038/bjc.2011.23
137. Kojima S, Chiyomaru T, Kawakami K, Yoshino H, Enokida H, Nohata N, et al. Tumour suppressors miR-1 and miR-133a target the oncogenic function of purine nucleoside phosphorylase (PNP) in prostate cancer. Br J Cancer. (2012) 106:405–13. doi: 10.1038/bjc.2011.462
138. Gilad S, Meiri E, Yogev Y, Benjamin S, Lebanony D, Yerushalmi N, et al. Serum microRNAs are promising novel biomarkers. PLoS ONE. (2008) 3:e3148. doi: 10.1371/journal.pone.0003148
139. Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. (2008) 18:997–1006. doi: 10.1038/cr.2008.282
140. Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. (2008) 105:10513–8. doi: 10.1073/pnas.0804549105
141. Wang K, Zhang S, Weber J, Baxter D, Galas DJ. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. (2010) 38:7248–59. doi: 10.1093/nar/gkq601
142. Devaux Y, Vausort M, Goretti E, Nazarov PV, Azuaje F, Gilson G, et al. Use of circulating microRNAs to diagnose acute myocardial infarction. Clin Chem. (2012) 58:559–67. doi: 10.1373/clinchem.2011.173823
143. Koffer J, Scheiper-Welling S, Verhoff MA, Bajanowski T, Kauferstein S. Post-mortem genetic investigation of cardiac disease-associated genes in sudden infant death syndrome (SIDS) cases. Int J Legal Med. (2021) 135:207–12. doi: 10.1007/s00414-020-02394-x
144. Neubauer J, Lecca MR, Russo G, Bartsch C, Medeiros-Domingo A, Berger W, et al. Post-mortem whole-exome analysis in a large sudden infant death syndrome cohort with a focus on cardiovascular and metabolic genetic diseases. Eur J Hum Genet. (2017) 25:404–9. doi: 10.1038/ejhg.2016.199
145. Brion M, Allegue C, Santori M, Gil R, Blanco-Verea A, Haas C, et al. Sarcomeric gene mutations in sudden infant death syndrome (SIDS). Forensic Sci Int. (2012) 219:278–81. doi: 10.1016/j.forsciint.2012.01.018
146. Slater B, Glinton K, Dai H, Lay E, Karaviti L, Mizerik E, et al. Sudden infant death with dysgenesis of the testes syndrome in a non-Amish infant: a case report. Am J Med Genet A. (2020) 182:2751–4. doi: 10.1002/ajmg.a.61842
147. Matschke J, Sperhake JP, Wilke N, Püschel K, Glatzel M. Cerebellar heterotopia of infancy in sudden infant death syndrome: an observational neuropathological study of four cases. Int J Legal Med. (2020). 134:2143–7. doi: 10.1007/s00414-020-02316-x
148. Brownstein CA, Goldstein RD, Thompson CH, Haynes RL, Giles E, Sheidley B, et al. SCN1A variants associated with sudden infant death syndrome. Epilepsia. (2018) 59:e56–62. doi: 10.1111/epi.14055
149. Lopez HU, Haverfield E, Chung WK. Whole-exome sequencing reveals CLCNKB mutations in a case of sudden unexpected infant death. Pediatr Dev Pathol. (2015) 18:324–6. doi: 10.2350/14-08-1543-CR.1
150. Takahashi Y, Sano R, Nakajima T, Kominato Y, Kubo R, Takahashi K, et al. Combination of postmortem mass spectrometry imaging and genetic analysis reveals very long-chain acyl-CoA dehydrogenase deficiency in a case of infant death with liver steatosis. Forensic Sci Int. (2014) 244:e34–7. doi: 10.1016/j.forsciint.2014.08.031
151. Goldwater PN, Kelmanson IA, Little BB. Increased thymus weight in sudden infant death syndrome compared to controls: The role of sub-clinical infections. Am J Hum Biol. (2020) 2020:e23528. doi: 10.1002/ajhb.23528
152. Fogerty AC, Ford GL, Willcox ME, Clancy SL. Liver fatty acids and the sudden infant death syndrome. Am J Clin Nutr. (1984) 39:201–8. doi: 10.1093/ajcn/39.2.201
153. Toro K, Hubay M, Keller E. Extramedullary haematopoiesis in liver of sudden infant death cases. Forensic Sci Int. (2017) 170:15–9. doi: 10.1016/j.forsciint.2006.08.027
154. Ventura F, Barranco R, Bachetti T, Nozza P, Fulcheri E, Palmieri A, et al. Medico-legal investigation in an explicable case of congenital central hypoventilation syndrome due to a rare variant of the PHOX2B gene. J Forensic Leg Med. (2018) 58:1–5. doi: 10.1016/j.jflm.2018.04.009
155. Marziliano N, Merlini PA, Vignati G, Orsini F, Motta V, Bandiera L, et al. A case of compound mutations in the MYBPC3 gene associated with biventricular hypertrophy and neonatal death. Neonatology. (2012) 102:254–8. doi: 10.1159/000339847
156. Mecchia D, Casale V, Oneda R, Matturri L, Lavezzi AM. Sudden death of an infant with cardiac, nervous system and genetic involvement–a case report. Diagn Pathol. (2013) 8:159. doi: 10.1186/1746-1596-8-159
157. Jequier Gygax M, Roulet-Perez E, Meagher-Villemure K, Jakobs C, Salomons GS, Boulat O, et al. Sudden unexpected death in an infant with L-2-hydroxyglutaric aciduria. Eur J Pediatr. (2009) 168:957–62. doi: 10.1007/s00431-008-0869-9
158. Koehler SA, Ladham S, Shakir A, Wecht CH. Simultaneous sudden infant death syndrome: a proposed definition and worldwide review of cases. Am J Forensic Med Pathol. (2001) 22:23–32. doi: 10.1097/00000433-200103000-00004
159. Huang P. Sudden twin infant death on the same day: a case report and review of the literature. Forensic Sci Med Pathol. (2013) 9:225–30. doi: 10.1007/s12024-013-9429-3
160. Pharoah PO, Platt MJ. Sudden infant death syndrome in twins and singletons. Twin Res Hum Genet. (2007) 10:644–8. doi: 10.1375/twin.10.4.644
161. Malloy MH, Freeman DH. Sudden infant death syndrome among twins. Arch Pediatr Adolesc Med. (1999) 153:736–40. doi: 10.1001/archpedi.153.7.736
162. Takahashi Y, Sano R, Kominato Y, Kubo R, Takahashi K, Nakajima T, et al. A case of sudden unexpected infant death involving a homozygotic twin with the thermolabile CPT2 variant, accompanied by rotavirus infection and treatment with an antibiotic containing pivalic acid. Leg Med. (2016) 22:13–7. doi: 10.1016/j.legalmed.2016.07.005
163. Campuzano O, Allegue C, Sarquella-Brugada G, Coll M, Mates J, Alcalde M, et al. The role of clinical, genetic and segregation evaluation in sudden infant death. Forensic Sci Int. (2014) 242:9–15. doi: 10.1016/j.forsciint.2014.06.007
164. Rueda M, Wagner JL, Phillips TC, Topol SE, Muse ED, Lucas JR, et al. Molecular Autopsy for Sudden Death in the Young: Is Data Aggregation the Key? Front Cardiovasc Med. (2017) 4:72. doi: 10.3389/fcvm.2017.00072
165. Santori M, Blanco-Verea A, Gil R, Cortis J, Becker K, Schneider PM, et al. Broad-based molecular autopsy: a potential tool to investigate the involvement of subtle cardiac conditions in sudden unexpected death in infancy and early childhood. Arch Dis Child. (2015) 100:952–6. doi: 10.1136/archdischild-2015-308200
166. Farrugia A, Keyser C, Hollard C, Raul JS, Muller J, Ludes B. Targeted next generation sequencing application in cardiac channelopathies: Analysis of a cohort of autopsy-negative sudden unexplained deaths. Forensic Sci Int. (2015) 254:5–11. doi: 10.1016/j.forsciint.2015.06.023
167. Wang D, Shah KR, Um SY, Eng LS, Zhou B, Lin Y, et al. Cardiac channelopathy testing in 274 ethnically diverse sudden unexplained deaths. Forensic Sci Int. (2014) 237: 90–9. doi: 10.1016/j.forsciint.2014.01.014
168. Mehboob R. Substance P/Neurokinin 1 and trigeminal system: a possible link to the pathogenesis in sudden perinatal deaths. Front Neurol. (2017) 8:82. doi: 10.3389/fneur.2017.00082
169. Lavezzi AM, Mehboob R, Matturri L. Developmental alterations of the spinal trigeminal nucleus disclosed by substance P immunohistochemistry in fetal and infant sudden unexplained deaths. Neuropathology. (2011) 31:405–13. doi: 10.1111/j.1440-1789.2010.01190.x
170. Mehboob R, Shahzad AS, Hashmi AM, Ahmad FJ. Vertebrate Specific oncogenic TAC1 has unconventional networking properties. HealthMed. (2014) 8:843–9.
171. Alwazzan A, Mehboob R, Hassan A, Perveen S, Sadaf, Gilani SA, et al. Elevated Neurokinin-1 receptor expression in uterine products of conception is associated with first trimester miscarriages. Front Physiol. (2020) 11:554766. doi: 10.3389/fphys.2020.554766
172. Mehboob R. Neurokinin-1 receptor as a potential drug target for COVID-19 treatment. Biomed Pharmacother. (2021) 143:112159. doi: 10.1016/j.biopha.2021.112159
Keywords: sudden infant death syndrome, channelopathies, sodium channels, cardiac channel genes, breathing control, neuropathology
Citation: Mehboob R, Kurdi M, Ahmad M, Gilani SA, Khalid S, Nasief H, Mirdad A, Malibary H, Hakamy S, Hassan A, Alaifan M, Bamaga A and Shahzad SA (2021) Comprehensive Analysis of Genes Associated With Sudden Infant Death Syndrome. Front. Pediatr. 9:742225. doi: 10.3389/fped.2021.742225
Received: 15 July 2021; Accepted: 13 September 2021;
Published: 15 October 2021.
Edited by:
Giulia Ottaviani, University of Milan, ItalyReviewed by:
Paul Nathan Goldwater, University of Adelaide, AustraliaVittorio Fineschi, Sapienza University of Rome, Italy
Copyright © 2021 Mehboob, Kurdi, Ahmad, Gilani, Khalid, Nasief, Mirdad, Malibary, Hakamy, Hassan, Alaifan, Bamaga and Shahzad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Riffat Mehboob, bWVoYm9vYi5yaWZmYXRAZ21haWwuY29t; cHJvZi5yaWZmYXRAbG1yYy5jb20ucGs=; Syed Adnan Shahzad, aW1hZG5hbnN5ZWRAaG90bWFpbC5jb20=