 Nupur Chaphekar
Nupur Chaphekar Prerna Dodeja1
Prerna Dodeja1 Raman Venkataramanan
Raman Venkataramanan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 03 November 2021
Sec. Obstetric and Pediatric Pharmacology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.733823
This article is part of the Research TopicExploring Maternal-Fetal Pharmacology through PBPK Modeling ApproachesView all 11 articles
Pregnancy and the postpartum period are associated with several physiological changes that can alter the pharmacokinetics (PK) and pharmacodynamics (PD) of drugs. For certain drugs, dosing changes may be required during pregnancy and postpartum to achieve drug exposures comparable to what is observed in non-pregnant subjects. There is very limited data on fetal exposure of drugs during pregnancy, and neonatal exposure through transfer of drugs via human milk during breastfeeding. Very few systematic clinical pharmacology studies have been conducted in pregnant and postpartum women due to ethical issues, concern for the fetus safety as well as potential legal ramifications. Over the past several years, there has been an increase in the application of modeling and simulation approaches such as population PK (PopPK) and physiologically based PK (PBPK) modeling to provide guidance on drug dosing in those special patient populations. Population PK models rely on measured PK data, whereas physiologically based PK models incorporate physiological, preclinical, and clinical data into the model to predict drug exposure during pregnancy. These modeling strategies offer a promising approach to identify the drugs with PK changes during pregnancy to guide dose optimization in pregnancy, when there is lack of clinical data. PBPK modeling is also utilized to predict the fetal exposure of drugs and drug transfer via human milk following maternal exposure. This review focuses on the current status of the application of PBPK modeling to predict maternal and fetal exposure of drugs and thereby guide drug therapy during pregnancy.
Pregnant women take one to three medications on an average in addition to the routine iron and vitamin supplements recommended during pregnancy (1). Pregnant women take medications for acute illnesses such as nausea and vomiting, upper respiratory tract and urinary tract infections or for chronic conditions such as psychiatric disorders, HIV infection, epilepsy, organ transplantation, rheumatological conditions, or substance abuse disorder. Pharmacotherapy is also needed for pregnancy-induced conditions like hypertensive disorder, preterm labor and gestational diabetes (2). Pregnant women and their fetuses are orphan populations with regards to information on the safety and efficacy of drugs. Ninety eight percent of the drugs approved in the United States between 2000 and 2010 have insufficient data on drug dosing during pregnancy, while seventy percent of them have no data on drug use in pregnancy (3). Pregnant women are excluded from clinical studies due to ethical, fetal safety and medico-legal concerns. Therefore, there is limited data available on PK and PD of drugs used in pregnancy. Table 1 lists the issues and potential confounding factors contributing to the lack of PK and PD data in pregnancy. Current dosing recommendations in pregnancy are based on data obtained from non-pregnant population. In this context, modeling and simulation techniques like PopPK or PBPK can provide additional information regarding appropriate drug dosing in this special population. A summary of ideal studies that could be conducted during pregnancy and the next best alternative or alternate approaches that can be used when a clinical study is not practical to obtain necessary data, is presented in Table 2.

Table 1. Need for designed pharmacological studies performed during pregnancy, lactation and postpartum.
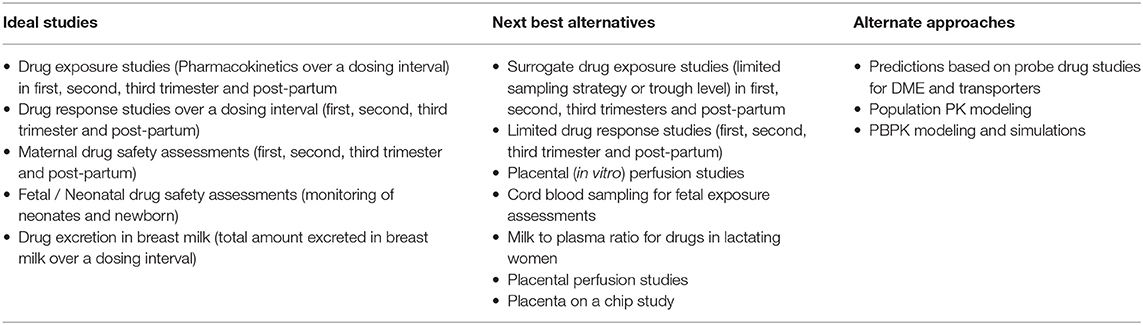
Table 2. Ideal studies in pregnancy and alternative approaches.
Several physiological changes occur during pregnancy that help support the growth and development of the fetus. The absorption, distribution, metabolism and excretion processes of drugs can be altered during pregnancy and may contribute to altered PK of drugs. Table 3 summarizes pregnancy mediated physiological changes that can impact PK processes. Reduced gastrointestinal motility and delayed gastric emptying time during pregnancy can reduce drug absorption. There is an increase in gastric pH during pregnancy which can lead to changes in absorption of acidic drugs due to increased ionization (10, 11). A systematic study evaluating the impact of changes in drug absorption on pharmacokinetics after intravenous vs. oral administration during pregnancy and postpartum is lacking. Several physiological changes may alter drug distribution such as increased plasma volume, maternal plasma protein dilution or organ volume variation (fat) (12–14). The expression and activity of certain CYP enzymes change during pregnancy which may lead to change in metabolism of selected substrates. The metabolism of drugs catalyzed by cytochrome P450 (CYP) isoenzymes CYP3A4, CYP2D6, CYP2C9 and certain uridine glucuronosyltransferases (UGT) isoenzymes UGT1A4 and UGT1A9 is increased during pregnancy (15) and the metabolism of CYP1A2 and CYP2C19 substrates is decreased during pregnancy (15, 16).
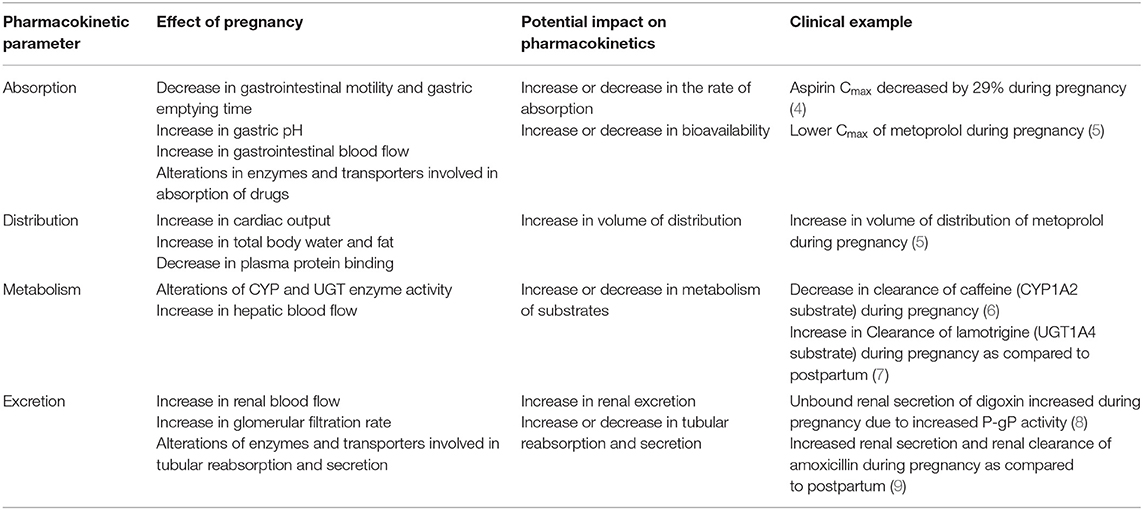
Table 3. Physiological changes and potential impact on PK of drugs.
Accumulating in-vivo and in-vitro data suggests that the increased levels of steroid hormones during pregnancy might be responsible for altered metabolism of certain substrates (17). For example, UGT1A1 up-regulation was seen in progesterone treated HEPG2 cells co-transfected with PXR as compared to control cells. An increase in the glucuronidation (UGT1A4) was observed in 17-beta estradiol treated HEPG2 cells co-transfected with ERα receptor (18). Progesterone treatment caused up-regulation of UGT1A in pregnant humanized UGT1A/ PXR mice as opposed to pregnant humanized UGT1A mice with PXR knockout suggesting the role of PXR activation leading to the up-regulation of UGT1A enzymes (19). The renal excretion of drugs is increased during pregnancy due to a 60–80% increase in renal blood flow and a 50% increase in glomerular filtration rate (20). To date there is limited data available elucidating the effect of pregnancy on intestinal, hepatic and renal transporters involved in the absorption, distribution, efflux, secretion and reabsorption of drugs.
Model-based approaches can provide some information regarding drug exposure and drug dosing in various patient populations when direct clinical data is not available. PBPK is a tool that can be used to predict drug exposure in such patient populations. This model-predicted data can be used to optimize drug dosing in special patient populations and can be further fine-tuned as more clinical data becomes available.
PBPK is a mechanistic approach that has been used in the drug development processes to determine safe and optimal doses to be used in clinical trials, estimate drug exposure in special populations and also to predict drug-drug interactions (21). It can be used as a viable alternative to generate clinical data in special patient populations. Regulatory agencies such as the US FDA and the European Medicines Agency have accepted the use of PBPK modeling to facilitate the decision-making process for conducting a clinical study in submissions for Investigational New Drug and New Drug Applications (22–24). PBPK models are multicompartmental models in which each compartment corresponds to one or more organ and is interconnected by the circulatory system. It integrates important physiological parameters (e.g., blood flow, enzyme and transporter abundance, cardiac output, glomerular filtration rate) and drug related parameters (blood-to-plasma ratio, plasma protein binding, permeability, solubility, in vitro metabolism or transport) which are known to influence drug PK and PD (25). Figure 1A represents an example of a minimal PBPK model (26) and Figure 1B represents an example of a PBPK model with each tissue/organ in the body being considered as a separate compartment (27). Pregnancy creates the need for additional compartments in the PBPK model. Figure 2 depicts the structure of pregnancy-PBPK (p-PBPK) model used in three different PBPK modeling software. The most important compartment in a p-PBPK model is the fetal unit. This is combined into a single “lumped” compartment known as the fetoplacental unit in the Simcyp and GastroPlus software. The fetoplacental unit incorporates the fetus, placenta, amniotic fluid, membranes and umbilical cord as depicted in Figures 2A,B. However, in the Open Systems Pharmacology software package, each of these units are considered discrete and accounted for separately, along with the inclusion of myometrium and endometrium, as seen in Figure 2C. Table 4 summarizes the physiological parameters that are considered in the Simcyp p-PBPK model.
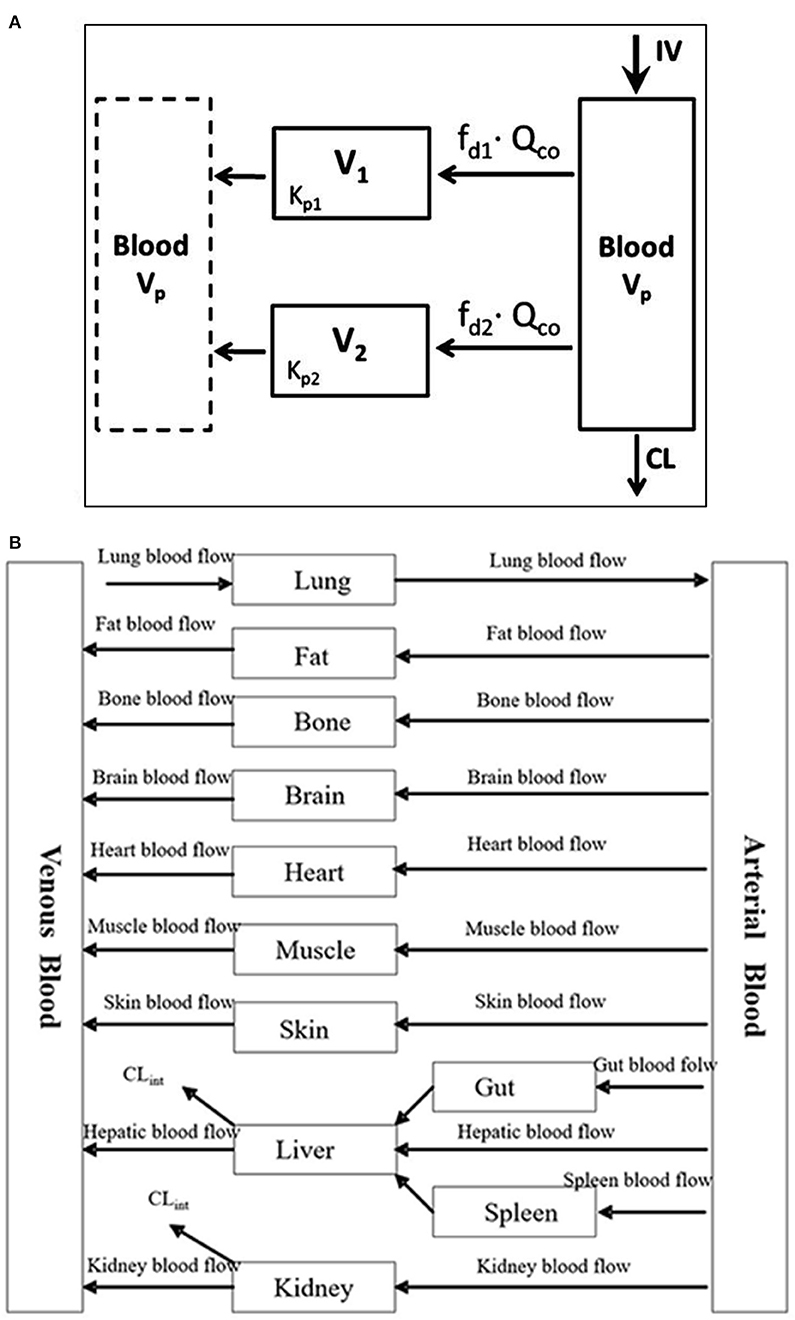
Figure 1. (A) Minimal PBPK model with two tissue compartments (26). (B) Example of a PBPK model (27).
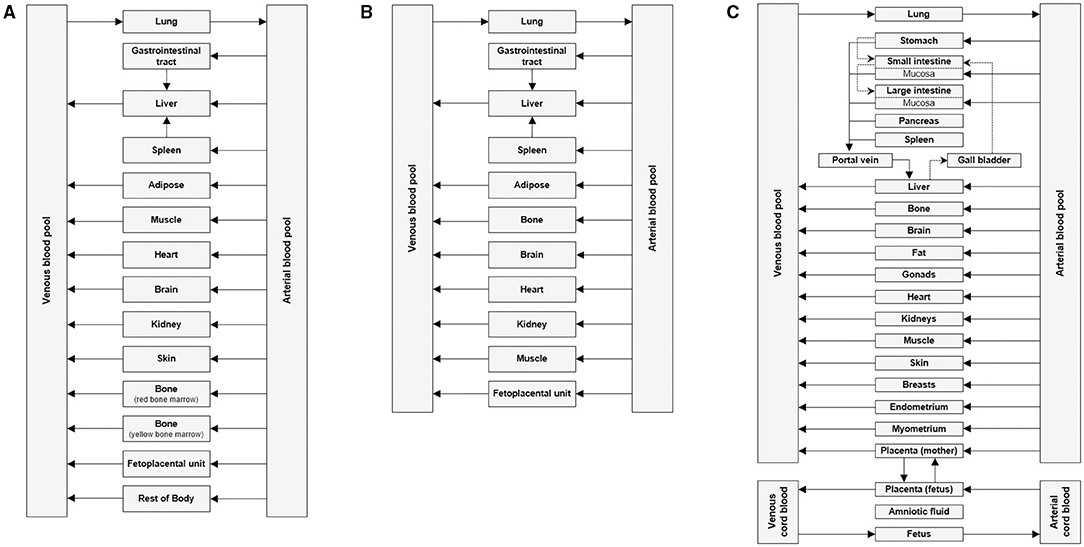
Figure 2. Basic structure of p-PBPK model in (A) Gatsroplus (B) SimCyp and (C) Open Systems Pharmacology (28).

Table 4. Physiological parameters that are modified for pregnancy prediction in Simcyp p-PBPK model.
Physiological changes during pregnancy are gestational age dependent. For example, the activity of UGT1A4 increased by 200% during the first and second trimesters and by 300% during the third trimester leading to increased clearance of lamotrigine (7). Similarly, the changes in organ blood flow, activity of certain metabolic enzymes and transporters are dependent on gestational age. The p-PBPK models incorporate these gestational age-related physiological changes into a normal PBPK model to simulate pregnant population. These p-PBPK models can then be used to predict the gestational age dependent pharmacokinetics of different drugs.
Several p-PBPK models have been developed and evaluated for antiretroviral, anti-malarial, psychoactive drugs, drugs used for the treatment of substance use disorder and environmental chemicals. Although these models have been able to predict the pharmacokinetics of certain drugs during pregnancy reasonably well, there are still several challenges that remain unresolved. There is no/limited information available to fully evaluate all the assumptions that are used in such models. There is paucity of data on combined effect of pregnancy and disease state (e.g., diabetes, malaria, hypertensive disorder) on gestational age-related changes in various physiological parameters and hence the predictions must be interpreted with caution. Data for drug elimination kinetics are typically scaled from in-vitro cell culture experiments and these experiments do not account for all the physiological changes which necessitates additional extrapolation factors to be incorporated. There is lack of information regarding changes in all drug metabolizing enzyme and transporter activity across gestational ages. Enzyme or transporter activity determined using probe drug data is specific to the trimester in which the study was conducted and cannot be extrapolated to other trimesters.
An exhaustive literature search was conducted using PubMed with the keywords PBPK and pregnancy. The results from the search with clinical observations are listed in Table 5 with specific examples discussed below.
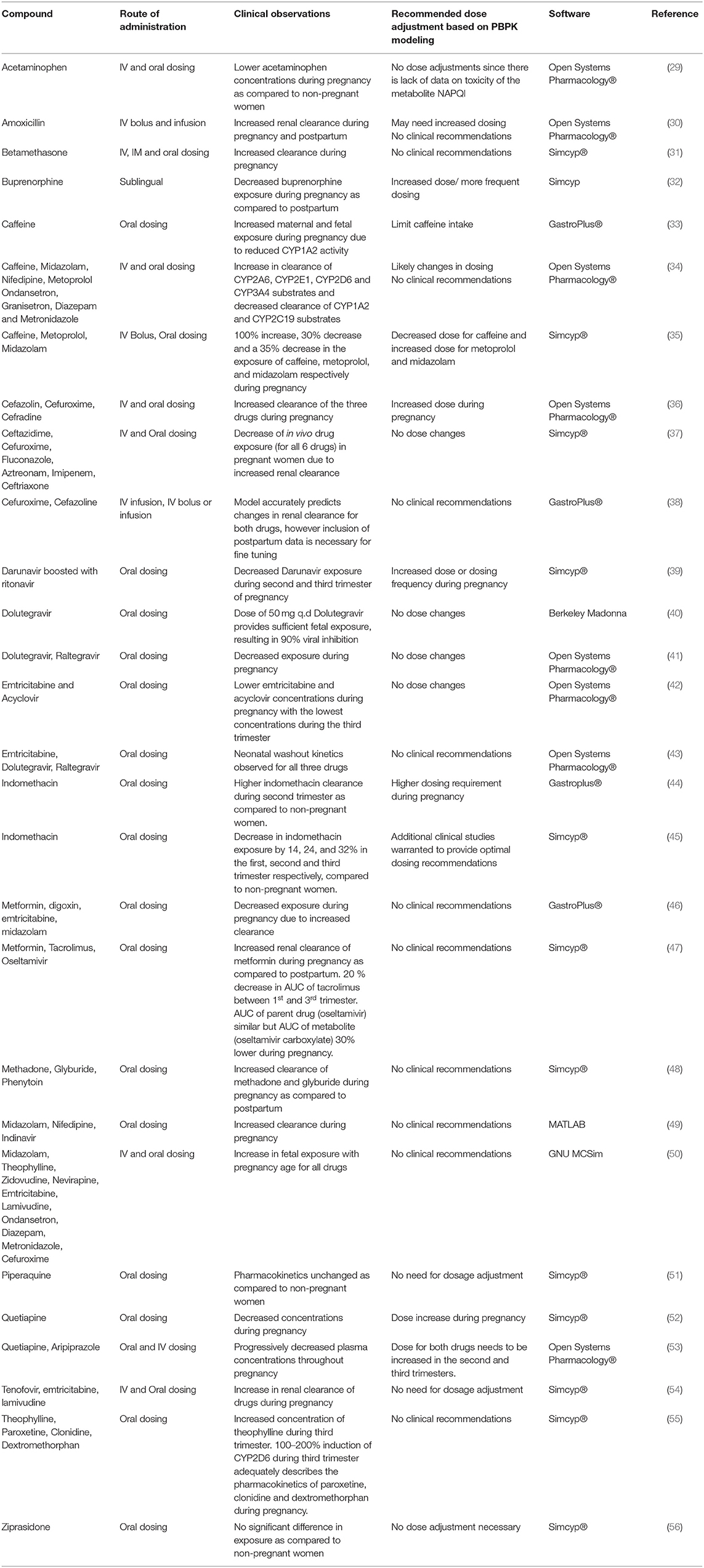
Table 5. Review of published p-PBPK models.
PBPK modeling has been used as a tool to guide and optimize drug dosing in pregnancy for several drugs and scenarios discussed below.
Ziprasidone is an antipsychotic drug used to treat schizophrenia and other psychiatric disorders. It is administered orally and is metabolized by CYP3A4 primarily in the liver. Biesdorf et al. established a PBPK model to predict drug exposure during pregnancy using the Simcyp inbuilt pregnancy population which includes gestational age-related changes in blood flow, glomerular filtration rate, plasma protein binding etc. Since the model used the pregnancy population in Simcyp, some physiological changes that were not very specific to the route of elimination of Ziprasidone were also incorporated. The model predicted exposures correlated well with the clinical data and exposure of ziprasidone during pregnancy at 6, 20, and 34 weeks of gestation. Since the exposure of ziprasidone during pregnancy was comparable to non-pregnant women, no dose adjustment is recommended during pregnancy for this drug (56). Ke et al. developed a PBPK model to evaluate maternal exposure of the antenatal corticosteroids dexamethasone and betamethasone which are primarily metabolized by CYP3A4. In this model, the fraction of dexamethasone metabolized by CYP3A4 was obtained from a clinical DDI study with itraconazole. However, for betamethasone, an in-vitro study was conducted to investigate the role of CYP3A4 in its metabolism. Ideally a clinical DDI study should be conducted to verify the fraction of betamethasone metabolized by CYP3A4 (31).
Quetiapine, an antipsychotic drug metabolized mainly by CYP3A4 and CYP2D6, shows decreased exposure during pregnancy possibly due to known increase in the activity of these two enzymes. A PBPK modeling approach was used to optimize the dosing regimen to target a predetermined therapeutic range (52). Though the model recommended a dose increase during pregnancy, information about the pregnancy mediated changes on PD is also needed to implement the recommended change in the dose during pregnancy.
PBPK models developed previously have been also modified/ refined to determine the exposure of substrates during pregnancy. Ke et al. refined a previously published PBPK model to include CYP3A4 activity changes during the third trimester based on data from the probe drug midazolam and used it to predict the exposures of nifedipine and indinavir in pregnancy. The site of CYP3A4 induction during the third trimester was proposed to be mainly the liver (49). However, subsequent models were not able to reproduce these findings. The model by De Sousa Mendes et al showed that a 90–100% CYP3A4 induction is required to capture the PK changes in third trimester for drugs metabolized by CYP3A4 (57). Whereas the model by Dallmann et al. using Open Systems Pharmacology suggested that a 60% induction in liver and intestine CYP3A4 is enough to describe the observed PK changes (58). There is still ongoing discussion regarding the magnitude and site of CYP3A4 induction in pregnancy and there are several shortcomings with using probe drug data for CYP3A4 assessments of CYP3A activity for other drugs. The models developed cannot be applied to predict the pharmacokinetics of other drugs and also for evaluating the pharmacokinetics across different trimesters.
Liu et al. developed a p-PBPK model for emtricitabine and acyclovir which are antiviral drugs primarily excreted unchanged in the urine by glomerular filtration and tubular secretion (42). The model had several limitations such as not accounting for potential changes in gastrointestinal absorption due to pregnancy. Additionally, since intravenous data for acyclovir in women was not available, observed drug concentrations were extrapolated based on PK data from men. A previously developed PopPK model for ganciclovir, a drug in the same class as acyclovir, has shown higher ganciclovir clearance in women than men after correcting for individual body surface area and glomerular filtration (59). Therefore, it is likely that there may be a significant underestimation of acyclovir as well in the model developed by Liu et al.
A limited number of PBPK models in the literature have evaluated the impact of genotype on pharmacokinetics of drugs during pregnancy. Efavirenz which is used for the treatment of human immunodeficiency virus (HIV) is metabolized by the highly polymorphic enzyme CYP2B6. Though both 400 mg and 600 mg doses show similar efficacy, a 400 mg dose is suggested to avoid dose related toxicities. However, there is limited data on the PK of 400 mg dose in pregnancy. p-PBPK model developed by Chetty et al. using Simcyp evaluated the pharmacokinetics after a reduced dose of 400 mg in CYP2B6 extensive metabolizers. The model predicted that approximately 57% of extensive metabolizers would show trough concentrations below the therapeutic target during third trimester, suggesting dose reduction during pregnancy may lead to therapeutic failure in extensive metabolizers (60). The utility of this model to predict drug exposure in rapid and ultra-rapid metabolizers during pregnancy remains unknown. Additionally, evidence suggests that race and ethnicity have an impact on CYP2B6 activity. The model by Chetty et al. has been developed and evaluated only for the Caucasian population and therefore the generalizability of the model to other populations is questionable. Models incorporating other inbuilt populations such as in Simcyp (e.g., Japanese, Chinese etc.) can be used to optimize drug dosing in the non-Caucasian populations (61).
p-PBPK models have also been extended to determine the PD effect of drugs used in pregnancy. Darakjian et al. developed a PBPK-PD model for caffeine in pregnancy. The PD model evaluated the effect of caffeine on phosphodiesterase enzyme (PE), cyclic adenosine monophosphate (cAMP) and epinephrine levels, which are factors associated with increased miscarriage risk. Increased caffeine plasma levels due to reduction in CYP1A2 activity during pregnancy led to greater inhibition of the PE enzyme, higher cAMP and greater increase of epinephrine levels which could increase the risk of pregnancy loss. Despite not being validated, the model was able to predict the increased concentration of caffeine in the fetoplacental compartment indicating its potential utility (33). Alqahtani et al. developed a PBPK-PD model to estimate concentrations of indomethacin in the second trimester of pregnancy and to support dose adjustment based on PD rationale in the pregnant population. Although the PBPK-PD model suggested a higher indomethacin dosing requirement during pregnancy, it cannot be directly used in clinical practice without further in-vivo validation (44).
PBPK models can be potentially used to predict drug-drug interactions in pregnancy when it is difficult to conduct clinical studies in vulnerable populations. Piperaquine is an antimalarial drug used during pregnancy. Approximately 1 million pregnancies in sub-Saharan Africa are complicated with co-infection of human immunodeficiency virus (HIV) and malaria, however there is paucity of data on anti-HIV medication mediated exposure changes of piperaquine during pregnancy (62). Olafuyi et al. developed a PBPK model to predict the drug-drug interaction potential between piperaquine and anti-HIV drugs (ritonavir and efavirenz) for Thai, Papua New Guinean, and Sudanese populations. The model showed no change in piperaquine PK due to co-administration of anti-HIV drugs and indicated no need for a change in the dose (51).
The p-PBPK model becomes more complex upon addition of the fetoplacental unit since the model requires inclusion of placental transfer parameters, fetus and placental enzyme and transporter kinetics and blood flow to various additional anatomical units to predict exposure in fetus.
The in-vitro placental perfusion model is one of the tools used to study transplacental transfer of drugs (63). It can also be used to investigate the effect of exogenous and endogenous chemicals on maternal and fetal perfusion and transfer. It offers several advantages as the placental barrier is maintained and separate perfusion of the maternal and fetal side can be achieved. However, information about transplacental drug transfer and expression of enzymes and transporters during different stages of pregnancy cannot be obtained as the tissue for perfusion studies is normally available only at the time of delivery. The placenta is in a metabolically static state during these experiments as compared to the metabolically changing state during different stages of pregnancy (63, 64). Transplacental transfer parameters like diffusion, clearance index, elimination constant and placenta partition coefficient can be obtained from these experiments and incorporated in a PBPK model to predict fetal exposure later in pregnancy. The placental perfusion has been instrumental in developing PBPK models and has been used for predicting fetal exposure of dolutegravir, tenofovir, emtricitabine, and nevirapine (40, 57, 65).
Another approach is to incorporate data from in-vitro experiments using placental tissue, microsomes or human placental cell lines. Mian et al. developed a PBPK model to predict fetal exposure of acetaminophen. Different methods to estimate the placental transfer (ex vivo cotyledon perfusion experiments or scaling based on Caco-2 cell permeability experiments, physicochemical properties in MoBi) were incorporated in the model and the predictions show a comparable fetal exposure. Maturation of enzymes in the fetal liver was accounted for to determine the molar dose fraction of acetaminophen converted to N-acetyl-p-benzoquinone imine. The model incorporating the ex-vivo perfusion model data showed the best correlation with observed cord blood data for acetaminophen but may not hold true for all compounds (29). There is limited information available on placental enzymes and transporters in particular at various stages of pregnancy and further studies in this area would be helpful in developing IVIVE for placental clearance across various trimesters. Data obtained from primary placental cells, human choriocarcinoma cells or placenta-on-a-chip model may be more physiologically relevant to obtain transplacental parameters (66, 67). Protein abundance information for placental transporters which is available from recent reports can be incorporated into maternal-fetal PBPK models to further improve the model predictions (68).
Animal models offer another promising approach but differences in hemodynamics and placental structure can pose challenges in extrapolation of animal data to humans. The gestational age and the associated changes in physiology differ substantially between animals and humans requiring correction factors while extrapolating these data to humans. A PBPK model to predict fetal exposure of a brominated flame retardant, BDE-47 was developed by parameterizing the model with concentrations of BDE-47 from the literature and previous pharmacokinetic and toxicokinetic studies. This model was able to predict the fetal concentrations of BDE-47 in rats after maternal exposure within one standard deviation of the experimental data indicating its potential to be extrapolated to other species including humans after careful consideration of anatomical and physiological differences in placental structure and function (69).
Abduljalil et al. reviewed the literature for studies evaluating changes in fetal parameters (e. g., body weight, body surface area, body water, abdominal circumference, body fat) during fetal growth. This data was used to create mathematical algorithms to describe changes in these fetal parameters with gestational age which can potentially be added to the fetal PBPK model (70).
Transplacental transfer parameters from in silico models, in vitro and ex vivo studies have been incorporated into p-PBPK models. Codaccioni et al. developed p-PBPK model for ten compounds using four different models of placental exchange based on in vitro, ex vivo, and in silico information. The non-pregnant and pregnant as well as fetal PK simulations were compared with observed profiles at delivery for each of the ten compounds. A comparison of the model predictions across different trimesters of pregnancy yielded inconclusive results. These models can be optimized and potentially be used based on the purpose of the study and type of data and resources available (50). In the absence of clinical data to evaluate the fetal PBPK models, umbilical cord concentrations observed at delivery were used. Zhang et al. developed a maternal-fetal PBPK model which incorporated gestational age-related changes in fetal physiologic parameters such as fetal serum albumin, liver volume, uterus blood flow etc. Sensitivity analysis identified that a single time-point umbilical venous/ maternal plasma ratio is not reflective of the fetal exposure (71). The various gaps in knowledge for modeling maternal-fetal pharmacology are summarized in Table 6.
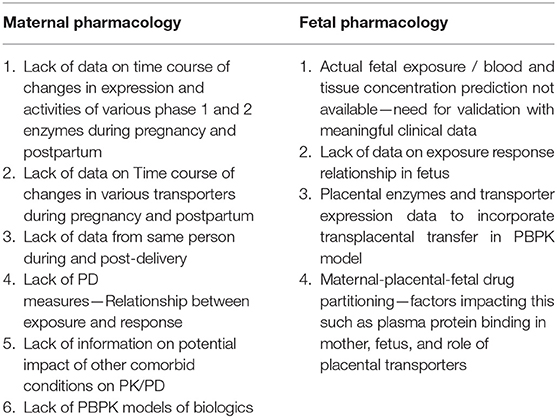
Table 6. Current gaps in modeling maternal-fetal pharmacology.
An exhaustive literature search was conducted using PubMed with the keywords PBPK and fetal exposure. The publications from the search describing the development and validation of p-PBPK models to determine fetal exposure are presented in Table 7.
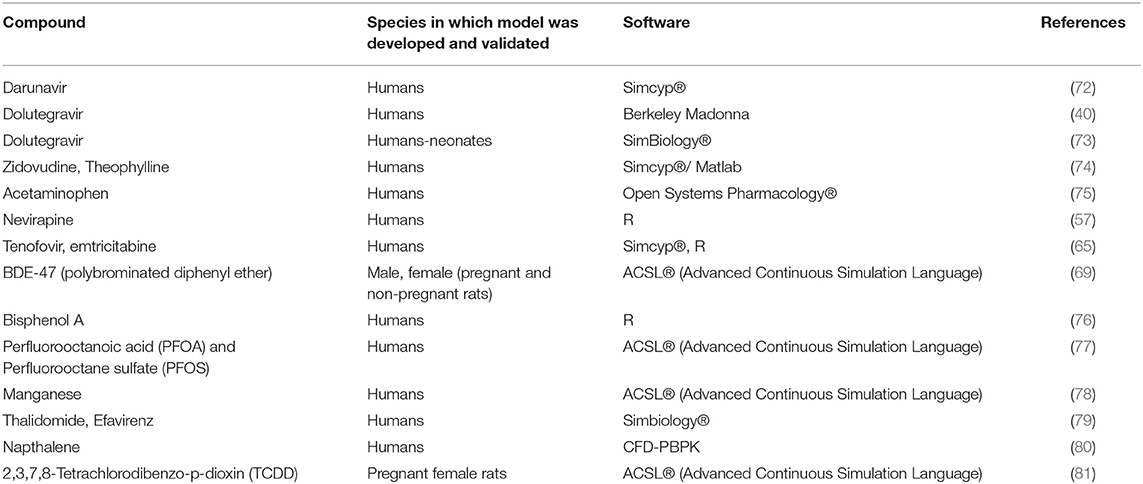
Table 7. List of published p-PBPK models to predict fetal exposure.
Fetal drug exposure is normally important from a fetal safety perspective. From efficacy point of view while normally one is interested in maternal drug exposure, there are conditions where fetal exposure is also important to maximize efficacy. Darunavir, an anti-HIV drug, primarily metabolized by CYP3A4 is routinely administered with CYP3A4 inhibitor ritonavir to maintain higher plasma concentrations during pregnancy. p-PBPK model was developed by incorporating information from ex vivo human placental perfusion studies to simulate fetal exposure after different dosing regimens (72). The model was validated by comparing maternal, fetal and amniotic fluid concentrations. The fetal concentration was compared with the single time-point umbilical cord concentration obtained at delivery. The model was able to capture the observed clinical data thus indicating that the placental perfusion data can be successfully integrated into p-PBPK models to predict fetal blood concentration at term. This approach is especially beneficial in the case of anti-HIV drugs to ensure that the half-maximal effective concentration is achieved in the fetus and the mother.
Zhang et al. developed a model to predict the placental transfer of passively diffusing drugs. The transplacental transfer parameters for zidovudine and theophylline were obtained using midazolam as the calibrator. The model was validated using single time-point maternal plasma and umbilical cord concentrations and the model was able to successfully predict the concentrations observed in patients. However, this model can only be used for drugs that undergo passive diffusion across the placenta. The use of a more sensitive calibrator that can predict placental transfer of a wide range of drugs with different physiochemical properties can enhance the utility of this model to predict fetal exposure of other drugs (82).
PBPK modeling has also been used to predict the fetal exposure to environmental chemicals (83). Bisphenol A (BPA) is an environmental chemical ingested through dietary and non-dietary sources. It is rapidly converted to nontoxic conjugates BPA-glucuronide (BPAG) and BPA-sulfate (BPAS) via glucuronidation and sulfation pathways. Sharma et al. developed a PBPK model for predicting the fetal exposure of bisphenol A which was evaluated against the observed BPA concentrations in cord blood, fetus liver and amniotic fluid following exposure from maternal blood (76). Parametrization of glucuronidation in fetus was done by scaling of in-vitro adult hepatocyte data in the absence of data from fetal hepatocytes which could have been a valuable addition to the model. Additionally, incorporating information on conjugation and deconjugation of BPA in placenta and fetus could lead to better prediction of the fetal exposure using this model.
Physiologically based toxicokinetic (PBTK) models are mathematical models that integrate absorption, distribution, metabolism and excretion processes for chemicals in biological systems. These models can serve as a tool to inform health risk assessments. They are traditionally based on extrapolating simulations in animal model to predict human exposure. For instance, Gingrich et al. developed a pregnancy specific p-PBTK model to predict bisphenol A and bisphenol S exposures in fetus (84). The model was calibrated using pregnant sheep data and results were extrapolated to assess the risk in humans However, this latter step remains uncertain due to major differences in placental physiology and structure between the species. More recently, high throughput toxicokinetic (HTTK) modeling has been used as an alternative in which models are parametrized with in vitro data, structure-derived physicochemical properties (e.g., QSAR) or species specific physiological data for several chemicals (85).
Bunglawala et al. built a neonatal PBPK model for dolutegravir using pediatric clinical data with assumptions that solubility, body composition and transporter expression were similar to adults (73). However, development and age-related changes are known but were not accounted in the model. Further, the possibility of drug exposure through maternal breast milk or placenta was not considered, though it is known that dolutegravir readily crosses the placenta.
In contrast to the approach described above, Liu et al. developed a PBPK model to link prenatal and postnatal pharmacokinetics using previously published p-PBPK models for emtricitabine, dolutegravir and raltegravir (43). The total drug amounts in fetal compartments at term delivery were predicted and incorporated as initial conditions in the neonatal PBPK model to predict drug concentrations in neonatal elimination phase after birth. Emtricitabine is eliminated unchanged in the urine by glomerular filtration and active tubular secretion mediated by Organic Cation Transporter 2 (OCT2). The OCT2 ontogeny applied in this model is based on data obtained from one term newborn only (86). Hence, additional in-vitro and clinical data are needed to further incorporate the ontogeny of OCT2 in the neonatal PBPK models. Additionally, the model should be tested and verified with information from other compounds as well as coupled maternal-fetal-neonatal PBPK models to understand early neonatal pharmacokinetics.
Maternal milk is a rich source of nourishment and breast-feeding is encouraged by the U.S. Department of Health and Human Services due to the beneficial effects for the mother as well as the infant. Maternal factors such as age, parity, breastfeeding patterns, milk composition and volume and physicochemical properties of the drug such as protein binding, molecular weight and lipophilicity affect the amount of drug transferred into human milk. Clinical studies focusing on quantifying the human milk exposure of drugs are needed. In the absence of clinical data, PBPK models have attempted to quantify infant exposure through human milk by integrating a breast tissue compartment. Loccisano et al. successfully developed a PBPK model to determine exposure of PFOA and PFOS in fetus and in infant through milk by extrapolating a previously developed and evaluated model in rats (77). As additional information on drug elimination kinetics in fetus and infant becomes available, it could be incorporated in to the model for better prediction of drug exposure in neonates (87).
Two differing approaches implemented in the prediction of infant exposure using PBPK are based on the method of drug uptake into human milk from plasma. One approach considers diffusion from drug in plasma via the breast tissue as done in PBPK modeling for lactational transfer of methylmercury (88). The other approach considers the direct passage of drug into the breast milk without considering the breast tissue as in the PBPK model to determine the infant exposure of organic pollutants (89).
Merrill et al. developed a PBPK model to predict perchlorate and iodide kinetics and subsequent perchlorate induced inhibition of iodide uptake in lactating mothers. The model was parameterized using data from previous models in male rat, lactating rat and non-pregnant women. However, this model has not been evaluated for perchlorate kinetics in humans due to lack of available clinical data (90). Isoniazid exposure to infant through breast milk was predicted using a validated PBPK model which accounted for the polymorphic expression of isoniazid metabolizing enzyme, N-acetyltransferase 2 (fast and slow metabolizers). Drug exposure was highest in slow metabolizing infants of slow metabolizing mothers, but the observed levels were still less than the infant exposure limit which is 10% of the maternal dose. The model was developed using information from ICRP reports which are generated based on data mainly from Caucasian population and should be cautiously extrapolated to other populations (82).
There has been tremendous progress over the past few years in the use of PBPK modeling to predict maternal and fetal exposure of drugs. By integrating physiological data, preclinical data, and clinical data, PBPK can be used to predict maternal and fetal exposure and guide optimization of maternal dosing during pregnancy when pharmacokinetic studies cannot be readily performed. Even though validation of these models is challenging due to limited clinical data, in-vitro and ex-vivo experimental data can be utilized to help predict fetal exposure of drugs. PBPK modeling can also serve as a tool to guide drug dosing during breastfeeding based on drug transfer through human milk. In summary, PBPK modeling offers promise as a potential tool to predict maternal and fetal exposure of drugs and thereby guide therapy in this special population.
NC: conception or design of the work, acquisition, analysis or interpretation of data for the work, drafting the work, provide approval for publication of the content, and agree to be accountable for all aspects of the work. PD and IS: revising it critically for important intellectual content, provide approval for publication of the content, and agree to be accountable for all aspects of the work. SC and RV: conception or design of the work, revising it critically for important intellectual content, provide approval for publication of the content, and agree to be accountable for all aspects of the work. All authors contributed to the article and approved the submitted version.
This work is partially funded by grants from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD, HD047905) through the Obstetric Pharmacology Research Center (OPRC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernandez-Diaz SS. National Birth Defects Prevention, Medication use during pregnancy, with particular focus on prescription drugs: 1976-2008. Am J Obstet Gynecol. (2011) 205:51 e1–8. doi: 10.1016/j.ajog.2011.02.029
2. Judith CF, Rubin D, Loffredo C. The Baltimore-Washington Infant Study Group, Use of prescription and non-prescription drugs in pregnancy. J Clin Epidemiol. (1993) 46:581–9. doi: 10.1016/0895-4356(93)90132-K
3. Adam MP, Polifka JE, Friedman JM. Evolving knowledge of the teratogenicity of medications in human pregnancy. Am J Med Genet C Semin Med Genet. (2011) 157C:175–82. doi: 10.1002/ajmg.c.30313
4. Shanmugalingam R, Wang X, Munch G, Fulcher I, Lee G, Chau K, et al. A pharmacokinetic assessment of optimal dosing, preparation, and chronotherapy of aspirin in pregnancy. Am J Obstet Gynecol. (2019) 221:255 e1–255 e9. doi: 10.1016/j.ajog.2019.04.027
5. Hogstedt S, Bo Lindberg MD, Peng DR, Regardh CG, Rane A. Pregnancy induced increase in metoprolol metabolism. Clin Pharmacol Ther. (1985) 37:688–92. doi: 10.1038/clpt.1985.114
6. H.R.a. Knutti CSR, Effect of the pregnancy on pharmacokinetics of caffeine, European Journal of Clinical Pharmacology (1981) 121–126. doi: 10.1007/BF00637512
7. N.D. Pennell PB, Stowe ZN, Helmers SL, Montgomery JQ, Henry TR. Impact of pregnancy and childbirth on metabolism of lamotrigine. Neurology. (2004) 62:292. doi: 10.1212/01.WNL.0000103286.47129.F8
8. Hebert MF, Easterling TR, Kirby B, Carr DB, Buchanan ML, Rutherford T, Thummel KE, Fishbein DP, Unadkat JD Effects of pregnancy on CYP3A and P-glycoprotein activities as measured by disposition of midazolam and digoxin: a University of Washington specialized center of research study. Clin Pharmacol Ther. (2008) 84:248–53. doi: 10.1038/clpt.2008.1
9. Andrew MA, Easterling TR, Carr DB, Shen D, Buchanan ML, Rutherford T, Bennett R, Vicini P, Hebert MF Amoxicillin pharmacokinetics in pregnant women: modeling and simulations of dosage strategies. Clin Pharmacol Ther. (2007) 81:547–56. doi: 10.1038/sj.clpt.6100126
10. Waldum HL Serum group 1 pepsinogens during pregnancy. Scand J Gastroent. (1980) 15:61–3. doi: 10.3109/00365528009181433
11. Vasicka LT, Bright ARH. Peptic ulcer and pregnancy, review of hormonal relationships and a report of one case of massive gastrointestinal hemorrhage. Obstet Gynecol Surv. (1957) 12:1–13. doi: 10.1097/00006254-195702000-00001
12. Dawes M, Chowienczyk PJ. Drugs in pregnancy. Pharmacokinetics in pregnancy. Best Pract Res Clin Obstet Gynaecol. (2001) 15:819–26. doi: 10.1053/beog.2001.0231
13. Cheung LT, CK Swaminathan R Urinary excretion of some enzymes and proteins during pregnancy. Clin Chem. (1989) 35:1978–80. doi: 10.1093/clinchem/35.9.1978
14. Erman A, Neri A, Sharoni R, Rabinov M, Kaplan B, Rosenfeld JB, et al. Enhanced urinary albumin excretion after 35 weeks of gestation and during labour in normal pregnancy. Scand J Clin Lab Invest. (1992) 52:409–13. doi: 10.3109/00365519209088376
15. Isoherranen N, Thummel KE. Drug metabolism and transport during pregnancy: how does drug disposition change during pregnancy and what are the mechanisms that cause such changes? Drug Metab Dispos. (2013) 41:256–62. doi: 10.1124/dmd.112.050245
16. Tracy TS, Venkataramanan R, Glover DD, Caritis SNH. National Institute for Child, U Human Development Network of Maternal-Fetal-Medicine, Temporal changes in drug metabolism (CYP1A2, CYP2D6 and CYP3A Activity) during pregnancy. Am J Obstet Gynecol. (2005) 192:633–9. doi: 10.1016/j.ajog.2004.08.030
17. Jeong H. Altered drug metabolism during pregnancy: hormonal regulation of drug-metabolizing enzymes. Expert Opin Drug Metab Toxicol. (2010) 6:689–99. doi: 10.1517/17425251003677755
18. Chen H, Yang K, Choi S, Fischer JH, Jeong H. Up-regulation of UDP-glucuronosyltransferase (UGT) 1A4 by 17beta-estradiol: a potential mechanism of increased lamotrigine elimination in pregnancy. Drug Metab Dispos. (2009) 37:1841–7. doi: 10.1124/dmd.109.026609
19. Chen S, Yueh MF, Evans RM, Tukey RH. Pregnane-x-receptor controls hepatic glucuronidation during pregnancy and neonatal development in humanized UGT1 mice. Hepatology. (2012) 56:658–67. doi: 10.1002/hep.25671
20. Davison JM, Dunlop W. Renal hemodynamics and tubular function normal human pregnancy. Kidney Int. (1980) 18:152–61. doi: 10.1038/ki.1980.124
21. Khalil F, Laer S. Physiologically based pharmacokinetic modeling: methodology, applications, and limitations with a focus on its role in pediatric drug development. J Biomed Biotechnol. (2011) 2011:907461. doi: 10.1155/2011/907461
22. Luzon E, Blake K, Cole S, Nordmark A, Versantvoort C, Berglund EG. Physiologically based pharmacokinetic modeling in regulatory decision-making at the European Medicines Agency. Clin Pharmacol Ther. (2017) 102:98–105. doi: 10.1002/cpt.539
23. Wagner C, Pan Y, Hsu V, Sinha V, Zhao P. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: an analysis of PBPK submissions to the US FDA. Clin Pharmacokinet. (2016) 55:475–83. doi: 10.1007/s40262-015-0330-y
24. Wagner C, Pan Y, Hsu V, Grillo JA, Zhang L, Reynolds KS, Sinha V, Zhao P. Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin Pharmacokinet. (2015) 54:117–27. doi: 10.1007/s40262-014-0188-4
25. Jones H, Rowland-Yeo K Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst Pharmacol. (2013) 2:e63. doi: 10.1038/psp.2013.41
26. Cao Y, Jusko WJ. Applications of minimal physiologically-based pharmacokinetic models. J Pharmacokinet Pharmacodyn. (2012) 39:711–23. doi: 10.1007/s10928-012-9280-2
27. Zhuang X, Lu C. PBPK modeling and simulation in drug research and development. Acta Pharm Sin B. (2016) 6:430–40. doi: 10.1016/j.apsb.2016.04.004
28. Dallmann A, Pfister M, van den Anker J, Eissing T. Physiologically based pharmacokinetic modeling in pregnancy: a systematic review of published models. Clin Pharmacol Ther. (2018) 104:1110–24. doi: 10.1002/cpt.1084
29. Mian P, van den Anker JN, van Calsteren K, Annaert P, Tibboel D, Pfister M, et al. Physiologically based pharmacokinetic modeling to characterize acetaminophen pharmacokinetics and N-Acetyl-p-Benzoquinone Imine (NAPQI) formation in non-pregnant and pregnant women. Clin Pharmacokinet. (2020) 59:97–110. doi: 10.1007/s40262-019-00799-5
30. Dallmann A, Himstedt A, Solodenko J, Ince I, Hempel G, Eissing T. Integration of physiological changes during the postpartum period into a PBPK framework and prediction of amoxicillin disposition before and shortly after delivery. J Pharmacokinet Pharmacodyn. (2020) 47:341–59. doi: 10.1007/s10928-020-09706-z
31. Ke AB, Milad MA Evaluation of maternal drug exposure following the administration of antenatal corticosteroids during late pregnancy using physiologically-based pharmacokinetic modeling. Clin Pharmacol Ther. (2019) 106:164–73. doi: 10.1002/cpt.1438
32. Zhang H, Kalluri HV, Bastian JR, Chen H, Alshabi A, Caritis SN, et al. Gestational changes in buprenorphine exposure: A physiologically-based pharmacokinetic analysis, Br J Clin Pharmacol. (2018) 84:2075–87. doi: 10.1111/bcp.13642
33. Darakjian LI, Kaddoumi A. Physiologically based pharmacokinetic/pharmacodynamic model for caffeine disposition in pregnancy. Mol Pharm. (2019) 16:1340–9. doi: 10.1021/acs.molpharmaceut.8b01276
34. Dallmann A, Ince I, Coboeken K, Eissing T, Hempel G. A physiologically based pharmacokinetic model for pregnant women to predict the pharmacokinetics of drugs metabolized via several enzymatic pathways. Clin Pharmacokinet. (2018) 57:749–68. doi: 10.1007/s40262-017-0594-5
35. Gaohua L, Abduljalil K, Jamei M, Johnson TN, Rostami-Hodjegan A. A pregnancy physiologically based pharmacokinetic (p-PBPK) model for disposition of drugs metabolized by CYP1A2, CYP2D6 and CYP3A4. Br J Clin Pharmacol. (2012) 74:873–85. doi: 10.1111/j.1365-2125.2012.04363.x
36. Dallmann A, Ince I, Solodenko J, Meyer M, Willmann S, Eissing T, et al. Physiologically based pharmacokinetic modeling of renally cleared drugs in pregnant women. Clin Pharmacokinet. (2017) 56:1525–41. doi: 10.1007/s40262-017-0538-0
37. Song L, Yu Z, Xu Y, Li X, Liu X, Liu D, et al. Preliminary physiologically based pharmacokinetic modeling of renally cleared drugs in Chinese pregnant women. Biopharm Drug Dispos. (2020) 41:248–67. doi: 10.1002/bdd.2243
38. Szeto KX, Le Merdy M, Dupont B, Bolger MB, Lukacova V PBPK Modeling approach to predict the behavior of drugs cleared by kidney in pregnant subjects and fetus. The AAPS J. (2021) 23:89. doi: 10.1208/s12248-021-00603-y
39. Colbers A, Greupink R, Litjens C, Burger D, Russel FG. Physiologically based modelling of darunavir/ritonavir pharmacokinetics during pregnancy. Clin Pharmacokinet. (2016) 55:381–96. doi: 10.1007/s40262-015-0325-8
40. Freriksen JJM, Schalkwijk S, Colbers AP, Abduljalil K, Russel FGM, Burger DM, et al. Assessment of maternal and fetal dolutegravir exposure by integrating ex vivo placental perfusion data and physiologically-based pharmacokinetic modeling. Clin Pharmacol Ther. (2020) 107:1352–61. doi: 10.1002/cpt.1748
41. Liu XI, Momper JD, Rakhmanina NY, Green DJ, Burckart GJ, Cressey TR, et al. Prediction of maternal and fetal pharmacokinetics of dolutegravir and raltegravir using physiologically based pharmacokinetic modeling, Clin Pharmacokinet. (2020) 59:1433–50. doi: 10.1007/s40262-020-00897-9
42. Liu XI, Momper JD, Rakhmanina N, van den Anker JN, Green DJ, Burckart GJ. Physiologically based pharmacokinetic models to predict maternal pharmacokinetics and fetal exposure to emtricitabine and acyclovir. J Clin Pharmacol. (2020) 60:240–55. doi: 10.1002/jcph.1515
43. Liu XI, Momper JD, Rakhmanina NY, Green DJ, Burckart GJ, Cressey TR, et al. Physiologically based pharmacokinetic modeling framework to predict neonatal pharmacokinetics of transplacentally acquired emtricitabine, dolutegravir, and raltegravir. Clin Pharmacokinet. (2021) 60:795–809. doi: 10.1007/s40262-020-00977-w
44. Subbian S, Alqahtani S, Kaddoumi A. Development of physiologically based pharmacokinetic/pharmacodynamic model for indomethacin disposition in pregnancy. Plos ONE. (2015) 10:e0139762. doi: 10.1371/journal.pone.0139762
45. Pillai VC, Shah M, Rytting E, Nanovskaya TN, Wang X, Clark SM, et al. Prediction of maternal and fetal pharmacokinetics of indomethacin in pregnancy. Br J Clin Pharmacol. (2021). doi: 10.1111/bcp.14960
46. Xia B, Heimbach T, Gollen R, Nanavati C, He H A simplified PBPK modeling approach for prediction of pharmacokinetics of four primarily renally excreted and CYP3A metabolized compounds during pregnancy. AAPS J. (2013) 15:1012–24. doi: 10.1208/s12248-013-9505-3
47. Jogiraju VK, Avvari S, Gollen R, Taft DR. Application of physiologically based pharmacokinetic modeling to predict drug disposition in pregnant populations. Biopharm Drug Dispos. (2017) 38:426–38. doi: 10.1002/bdd.2081
48. Ke AB. Expansion of a PBPK model to predict disposition in pregnant women of drugs cleared via multiple enzymes including CYP2B6, CYP2C9 and CYP2C19. British Journal of Clinical Pharmacology. (2013) 77:554–70. doi: 10.1111/bcp.12207
49. Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. A PBPK model to predict disposition of CYP3A-metabolized drugs in pregnant women: verification and discerning the site of CYP3A induction. CPT Pharmacometrics Syst Pharmacol. (2012) 1:e3. doi: 10.1038/psp.2012.2
50. Codaccioni M, Brochot C. Assessing the impacts on fetal dosimetry of the modelling of the placental transfers of xenobiotics in a pregnancy physiologically based pharmacokinetic model. Toxicol Appl Pharmacol. (2020) 409:115318. doi: 10.1016/j.taap.2020.115318
51. Olafuyi O, Coleman M, Badhan RKS The application of physiologically based pharmacokinetic modelling to assess the impact of antiretroviral-mediated drug-drug interactions on piperaquine antimalarial therapy during pregnancy. Biopharm Drug Dispos. (2017) 38:464–78. doi: 10.1002/bdd.2087
52. Badhan RKS, Macfarlane H. Quetiapine dose optimisation during gestation: a pharmacokinetic modelling study. J Pharm Pharmacol. (2020) 72:670–81. doi: 10.1111/jphp.13236
53. Zheng L, Tang S, Tang R, Xu M, Jiang X, Wang L. Dose adjustment of quetiapine and aripiprazole for pregnant women using physiologically based pharmacokinetic modeling and simulation. Clin Pharmacokinet. (2021) 60:623–35. doi: 10.1007/s40262-020-00962-3
54. De Sousa Mendes M, Hirt D, Urien S, Valade E, Bouazza N, Foissac F, et al. Physiologically-based pharmacokinetic modeling of renally excreted antiretroviral drugs in pregnant women. Br J Clin Pharmacol. (2015) 80:1031–41. doi: 10.1111/bcp.12685
55. Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Isoherranen N, Unadkat JD. A physiologically based pharmacokinetic model to predict disposition of CYP2D6 and CYP1A2 metabolized drugs in pregnant women. Drug Metab Dispos. (2013) 41:801–13. doi: 10.1124/dmd.112.050161
56. Biesdorf C, Martins FS, Sy SKB, Diniz A. Physiologically-based pharmacokinetics of ziprasidone in pregnant women. Br J Clin Pharmacol. (2019) 85:914–23. doi: 10.1111/bcp.13872
57. De Sousa Mendes M, Lui G, Zheng Y, Pressiat C, Hirt D, Valade E, et al. A physiologically-based pharmacokinetic model to predict human fetal exposure for a drug metabolized by several CYP450 pathways. Clin Pharmacokinet. (2017) 56:537–50. doi: 10.1007/s40262-016-0457-5
58. Dallmann A, Solodenko J, Ince I, Eissing T. Applied concepts in PBPK. Modeling: how to extend an open systems pharmacology model to the special population of pregnant women. CPT Pharmacometrics Syst Pharmacol. (2018) 7:419–31. doi: 10.1002/psp4.12300
59. Perrottet N, Csajka C, Pascual M, Manuel O, Lamoth F, Meylan P, et al. Population pharmacokinetics of ganciclovir in solid-organ transplant recipients receiving oral valganciclovir. Antimicrob Agents Chemother. (2009) 53:3017–23. doi: 10.1128/AAC.00836-08
60. Chetty M, Danckwerts MP, Julsing A Prediction of the exposure to a 400-mg daily dose of efavirenz in pregnancy: is this dose adequate in extensive metabolisers of CYP2B6? Eur J Clin Pharmacol. (2020) 76:1143–1150. doi: 10.1007/s00228-020-02890-4
61. Ilic K, Hawke RL, Thirumaran RK, Schuetz EG, Hull JH, Kashuba AD, et al. The influence of sex, ethnicity, and CYP2B6 genotype on bupropion metabolism as an index of hepatic CYP2B6 activity in humans. Drug Metab Dispos. (2013) 41:575–81. doi: 10.1124/dmd.112.048108
62. Uneke CJ, Ogbonna A Malaria and HIV co-infection in pregnancy in sub-Saharan Africa: impact of treatment using antimalarial and antiretroviral agents. Trans R Soc Trop Med Hyg. (2009) 103:761–7. doi: 10.1016/j.trstmh.2008.06.017
63. M.K.a.Golan A. In-vitro models using human placenta to study fetal exposure of drugs. Clinical Medicine: Reproductive Health. (2008) 15–24. doi: 10.4137/CMRH.S974
64. Conings S, Amant F, Annaert P, Van Calsteren K. Integration and validation of the ex vivo human placenta perfusion model. J Pharmacol Toxicol Methods. (2017) 88:25–31. doi: 10.1016/j.vascn.2017.05.002
65. De Sousa Mendes M, Hirt D, Vinot C, Valade E, Lui G, Pressiat C, et al. Prediction of human fetal pharmacokinetics using ex vivo human placenta perfusion studies and physiologically based models. Br J Clin Pharmacol. (2016) 81:646–57. doi: 10.1111/bcp.12815
66. Evseenko DA, Paxton JW, Keelan JA. ABC drug transporter expression and functional activity in trophoblast-like cell lines and differentiating primary trophoblast. Am J Physiol Regul Integr Comp Physiol. (2006) 290:R1357–65. doi: 10.1152/ajpregu.00630.2005
67. Pemathilaka RL, Reynolds DE, Hashemi NN. Drug transport across the human placenta: review of placenta-on-a-chip and previous approaches. Interface Focus. (2019) 9:20190031. doi: 10.1098/rsfs.2019.0031
68. Anoshchenko O, Prasad B, Neradugomma NK, Wang J, Mao Q, Unadkat JD. Gestational age-dependent abundance of human placental transporters as determined by quantitative targeted proteomics. Drug Metab Dispos. (2020) 48:735–41. doi: 10.1124/dmd.120.000067
69. Emond C, Raymer JH, Studabaker WB, Garner CE, Birnbaum LS. A physiologically based pharmacokinetic model for developmental exposure to BDE-47 in rats. Toxicol Appl Pharmacol. (2010) 242:290–8. doi: 10.1016/j.taap.2009.10.019
70. Abduljalil K, Johnson TN, Rostami-Hodjegan A Fetal physiologically-based pharmacokinetic models: systems information on fetal biometry and gross composition. Clin Pharmacokinet. (2018) 57:1149–71. doi: 10.1007/s40262-017-0618-1
71. Zhang Z, Imperial MZ, Patilea-Vrana GI, Wedagedera J, Gaohua L, Unadkat JD Development of a Novel Maternal-Fetal Physiologically Based Pharmacokinetic Model I: Insights into Factors that Determine Fetal Drug Exposure through Simulations and Sensitivity Analyses. Drug Metab Dispos. (2017) 45:920–38. doi: 10.1124/dmd.117.075192
72. Schalkwijk S, Buaben AO, Freriksen JJM, Colbers AP, Burger DM, Greupink R. Russel FGM, prediction of fetal darunavir exposure by integrating human ex-vivo placental transfer and physiologically based pharmacokinetic modeling. Clin Pharmacokinet. (2018) 57:705–16. doi: 10.1007/s40262-017-0583-8
73. Bunglawala F, Rajoli RKR, Mirochnick M, Owen A, Siccardi M. Prediction of dolutegravir pharmacokinetics and dose optimization in neonates via physiologically based pharmacokinetic (PBPK) modelling. J Antimicrob Chemother. (2020) 75:640–7. doi: 10.1093/jac/dkz506
74. Zhang Z, Unadkat JD. Development of a novel maternal-fetal physiologically based pharmacokinetic model II: Verification of the model for passive placental permeability drugs. Drug Metab Dispos. (2017) 45:939–46. doi: 10.1124/dmd.116.073957
75. Mian P, Allegaert K, Conings S, Annaert P, Tibboel D, Pfister M, van Calsteren K, et al. Integration of placental transfer in a fetal-maternal physiologically based pharmacokinetic model to characterize acetaminophen exposure and metabolic clearance in the fetus. Clin Pharmacokinet. (2020) 59:911–25. doi: 10.1007/s40262-020-00861-7
76. Sharma RP, Schuhmacher M, Kumar V. The development of a pregnancy PBPK Model for Bisphenol A and its evaluation with the available biomonitoring data. Sci Total Environ. (2018) 624:55–68. doi: 10.1016/j.scitotenv.2017.12.023
77. Loccisano AE, Longnecker MP, Campbell JL Jr, Andersen ME, Clewell HJ 3rd. Development of PBPK models for PFOA and PFOS for human pregnancy and lactation life stages. J Toxicol Environ Health A. (2013) 76:25–57. doi: 10.1080/15287394.2012.722523
78. Yoon M, Schroeter JD, Nong A, Taylor MD, Dorman DC, Andersen ME, et al. Physiologically based pharmacokinetic modeling of fetal and neonatal manganese exposure in humans: describing manganese homeostasis during development. Toxicol Sci. (2011) 122:297–316. doi: 10.1093/toxsci/kfr141
79. Atoyebi SA, Rajoli RKR, Adejuyigbe E, Owen A, Bolaji O, Siccardi M, et al. Using mechanistic physiologically-based pharmacokinetic models to assess prenatal drug exposure: Thalidomide versus efavirenz as case studies. Eur J Pharm Sci. (2019) 140:105068. doi: 10.1016/j.ejps.2019.105068
80. Dustin Kapraun F, Schlosser PM, Nylander-French LA, Kim D, Yost EE, Druwe IL. A PBPK model for napthalene with inhalation and skin routes of exposure. Toxicological Sciences. (2020) 177:377–91. doi: 10.1093/toxsci/kfaa117
81. Emond C, Birnbaum LS, DeVito MJ. Physiologically based pharmacokinetic model for developmental exposures to TCDD in the rat. Toxicol Sci. (2004) 80:115–33. doi: 10.1093/toxsci/kfh117
82. Garessus EDG, Mielke H, Gundert-Remy U. Exposure of infants to isoniazid via breast milk after maternal drug intake of recommended doses is clinically insignificant irrespective of metaboliser status. A physiologically-based pharmacokinetic (PBPK) modelling approach to estimate drug exposure of infants via breast-feeding. Front Pharmacol. (2019) 10:5. doi: 10.3389/fphar.2019.00005
83. Brochot C, Casas M, Manzano-Salgado C, Zeman FA, Schettgen T, Vrijheid M, Bois FY Prediction of maternal and foetal exposures to perfluoroalkyl compounds in a Spanish birth cohort using toxicokinetic modelling. Toxicol Appl Pharmacol. (2019) 379:114640. doi: 10.1016/j.taap.2019.114640
84. Gingrich J, Filipovic D, Conolly R, Bhattacharya S, Veiga-Lopez A. Pregnancy-specific physiologically-based toxicokinetic models for bisphenol A and bisphenol S. Environ Int. (2021) 147:106301. doi: 10.1016/j.envint.2020.106301
85. Pearce RG, Setzer RW, Strope CL, Wambaugh JF, Sipes NS. httk: R package for high-throughput toxicokinetics. J Stat Softw. (2017) 79:1–26. doi: 10.18637/jss.v079.i04
86. Cheung KWK, van Groen BD, Spaans E, van Borselen MD, de Bruijn A, Simons-Oosterhuis Y, et al. A comprehensive analysis of ontogeny of renal drug transporters: mRNA analyses, quantitative proteomics, and localization. Clin Pharmacol Ther. (2019) 106:1083–92. doi: 10.1002/cpt.1516
87. Clewell RA, Gearhart JM. Pharmacokinetics of toxic chemicals in breast milk: Use of PBPK models to predict infant exposure. Environmental Health Perspectives. (2002) 110:A333–A337. doi: 10.1289/ehp.021100333
88. Ou L, Wang H, Chen C, Chen L, Zhang W, Wang X. Physiologically based pharmacokinetic (PBPK) modeling of human lactational transfer of methylmercury in China. Environ Int. (2018) 115:180–7. doi: 10.1016/j.envint.2018.03.018
89. Verner MA, Ayotte P, Muckle G, Charbonneau M, Haddad S. A physiologically based pharmacokinetic model for the assessment of infant exposure to persistent organic pollutants in epidemiologic studies. Environ Health Perspect. (2009) 117:481–7. doi: 10.1289/ehp.0800047
Keywords: maternal, fetal, pharmacology, pregnancy, PBPK
Citation: Chaphekar N, Dodeja P, Shaik IH, Caritis S and Venkataramanan R (2021) Maternal-Fetal Pharmacology of Drugs: A Review of Current Status of the Application of Physiologically Based Pharmacokinetic Models. Front. Pediatr. 9:733823. doi: 10.3389/fped.2021.733823
Received: 30 June 2021; Accepted: 16 September 2021;
Published: 03 November 2021.
Edited by:
André Dallmann, Bayer, GermanyReviewed by:
Marc Codaccioni, Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM), FranceCopyright © 2021 Chaphekar, Dodeja, Shaik, Caritis and Venkataramanan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raman Venkataramanan, cnZAcGl0dC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.