
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 23 September 2021
Sec. Pediatric Urology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.729932
 Sriharsha Talluri1
Sriharsha Talluri1 Michael A. Goedde1Eran Rosenberg1,2Katie L. Canalichio1,2Dennis Peppas1,2
Michael A. Goedde1Eran Rosenberg1,2Katie L. Canalichio1,2Dennis Peppas1,2 Jeffrey T. White1,2*
Jeffrey T. White1,2*Prune belly syndrome (PBS) is a rare congenital disease that predominantly occurs in males and is identified by its classic triad of abdominal wall musculature deficiencies, cryptorchidism, and urinary tract abnormalities. However, numerous anomalies involving the kidneys, heart, lungs, and muscles have also been reported. A multitude of chromosomal abnormalities have been implicated in its pathogenesis. PBS can occur in association with trisomy 18 and 21. Gene duplications and deletions have also been reported; however, a definite cause of PBS is still unknown. We report the first PBS patient with a copy number variant in 16p11.2.
Prune belly syndrome (PBS) is a rare congenital disease occurring predominantly in males. It has also been referred to as Eagle-Barrett syndrome, triad syndrome, and Obrinsky's syndrome and is known by its classic triad of abdominal wall musculature deficiencies, cryptorchidism, and urinary tract abnormalities (1). Despite numerous case reports and reviews, there is a scarcity of information in the literature regarding definitive causes. Several theories have been surmised, including primary defect of intermediate and lateral plate mesoderm (2, 3), intrinsic defect of the urinary tract, and in utero urethral obstruction (4). While chromosomal abnormalities do occur in PBS, it is the exception, not the rule. We report a male patient with PBS and a 16p11.2 chromosomal duplication.
Urology was consulted for newborn male with urinary dribbling accompanied with a distended abdomen. He was born via repeat cesarean section prematurely at 35 weeks 3 days' gestation. The pregnancy was complicated by breech presentation and oligohydramnios. Prenatal ultrasound at 26 weeks showed dilated bilateral upper urinary tracts, thick-walled distended bladder with a “keyhole sign,” and dilated posterior and anterior urethra (see Figure 1). Upon delivery, he was intubated in the delivery room for respiratory distress, and placement of a urinary catheter was unsuccessful.
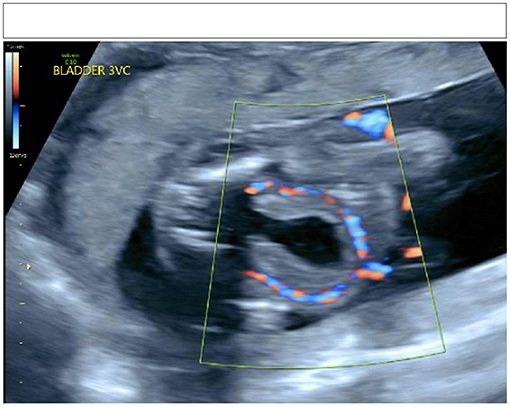
Figure 1. Keyhole sign on prenatal ultrasonography.
On examination, the patient's abdomen was distended with reduced abdominal wall musculature. There was tight phimosis of an uncircumcised phallus, and his scrotum was underdeveloped without palpable testes bilaterally.
Urinary catheterization was successful by urology with a six French Foley catheter. Shortly after birth, his creatinine was 0.5 mg/dl. He was extubated overnight on day of life (DOL) 1. Due to concern for urinary tract infection, the patient was started on antibiotic prophylaxis. His creatinine rose to a peak of 1.9 mg/dl and was trended until it reached a level of 1.1 mg/dl on DOL 16. It was not until 2 months of life that creatinine reached a nadir at 0.5 mg/dl. Renal ultrasound showed small kidneys with multiple cysts and massive bilateral hydroureter with mild hydronephrosis. Voiding cystourethrogram revealed absence of vesicoureteral reflux (VUR), poor emptying, and no apparent urethral obstruction.
Due to the presence of bilateral undescended testes, upper tract dilation, and abdominal abnormality, a diagnosis of incomplete PBS (Woodard classification 2/3) was made. Prior to discharge on DOL 16, the patient was started on clean intermittent catheterization.
Though the incidence of PBS has been estimated to be ~3.8 per 100,000 live births (5), case reports and reviews of this rare disease are quite numerous. Much of the literature has shown that there is a higher incidence among the African American population (5, 6). Additionally, an infrequent presentation without cryptorchidism has been seen in females (6–8).
The presentation of those with PBS can be quite variable. The abdominal exam can range from mildly protuberant without “pruning” to grossly thin and wrinkled abdominal wall (9). Observed urinary tract abnormalities include VUR, hydroureteronephrosis, hypospadias, and patent urachus (6). Renal dysplasia occurs in roughly 5% of patients. VUR occurs in 75% of patients accompanied with immense ureteral dilation as stroma replaces smooth muscle. The bladder is massively enlarged with properties of delayed sensation but usually normal compliance. The posterior urethra is dilated due to prostatic hypoplasia, while the anterior urethra can range from atretic to a megalourethra. Aside from this classic triad of PBS, multiple sources have reported numerous additional features that occurred in patients with PBS, some of which include chronic kidney disease, pulmonary hypoplasia, cardiac anomalies, musculoskeletal anomalies, constipation, and malrotation (6, 9–12). Though the Woodard classification system was initially developed to stratify PBS patients into different management groups (13), the vast range of clinical presentations of PBS also lead to a more phenotype-driven organization. The categories utilized by this system are isolated PBS, PBS-plus, and syndromic PBS. Isolated PBS includes those with the classic triad of abdominal wall musculature deformities, abnormalities of the urinary system, and cryptorchidism. PBS-plus incorporates those with additional anomalies that do not involve the genitourinary system and cannot be attributed to a defined syndrome. Finally, syndromic PBS indicates that the triad of PBS coincides with other known syndromes, such as Duchenne muscular dystrophy, Pierre Robin syndrome, and VACTERL (9).
A multitude of chromosomal abnormalities have been implicated in the pathogenesis of PBS. PBS can occur in association with trisomy 18 and 21 (14–16). Weber et al. performed exon capture and massively parallel sequencing and identified a homozygous frameshift mutation in CHRM3 gene on chromosome 1q43 (17). Boghossian et al. identified a few copy number variants in patients with PBS; these included a 4q22 duplication that overlaps BMPR1B gene, duplications of STIM1 gene, duplication of NOG gene, and a deletion that involved MYOCD gene (3). Murray et al. documented the deletion of HNF-1β gene on chromosome 17 in a patient with PBS (18). Iqbal et al. reported on PBS patients with mutations of X-linked filamin A gene (19).
We report the first documented PBS patient with a copy number variant in 16p11.2. The genetic report showed heterogeneous copy number increase of 839.5 kb involving chromosome 16p11.2. This duplication overlaps the chromosome 16p11.2 deletion syndrome 220-kb critical region (OMIM 613444) and encompasses SH2B1 gene (OMIM 608937). A list of duplicated genes is displayed in Table 1. Patients with a deletion of this region present with developmental delay, learning disability, behavioral problems, dysmorphology, and obesity; however, the clinical significance of duplication is not clear at this time. One paper by Sampson et al. reported three patients with a 16p11.2 microdeletion. Each of these patients also presented with congenital anomalies of the kidney and urinary tract (CAKUT) as well as Hirschsprung disease (20). This SH2B1 gene is also associated with leptin signaling in the brain and insulin signaling. Another patient within our medical system is an 8-year-old male with a 16p11.2 microdeletion with a history of Klinefelter syndrome and bilateral retractile testicles. Though these abnormalities are connected to deletion rather than duplication of this chromosomal segment, it does point toward a strong connection between 16p11.2 and urinary development.
In this report, we present the first documented case report of a patient with PBS and a concurrent gene duplication of the 16p11.2 gene segment. As no single gene mutation has been identified as the definite cause of PBS, gene analysis of additional patients with this condition is needed to compare this patient's report.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Written informed consent was obtained from the participants' parents for the publication of this case report.
All authors have made significant contributions to the manuscript including design, drafting, revising, and approved the final manuscript and its submission to Frontiers.
Patient care funding made available by individual. Funding for publication made available by Norton Healthcare.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Eagle JF Jr, Barrett GS. Congenital deficiency of abdominal musculature with associated genitourinary abnormalities: a syndrome. Report of 9 cases. Pediatrics. (1950) 6:721–36.
2. Stephens FD, Gupta D. Pathogenesis of the prune belly syndrome. J Urol. (1994) 152:2328–31. doi: 10.1016/S0022-5347(17)31669-5
3. Boghossian NS, Sicko RJ, Giannakou A, Dimopoulos A, Caggana M, Tsai MY, et al. Rare copy number variants identified in prune belly syndrome. Eur J Med Genet. (2018) 61:145–51. doi: 10.1016/j.ejmg.2017.11.008
4. Moerman P, Fryns JP, Goddeeris P, Lauweryns JM. Pathogenesis of the prune-belly syndrome: a functional urethral obstruction caused by prostatic hypoplasia. Pediatrics. (1984) 73:470–5.
5. Routh JC, Huang L, Retik AB, Nelson CP. Contemporary epidemiology and characterization of newborn males with prune belly syndrome. Urology. (2010) 76:44–8. doi: 10.1016/j.urology.2009.12.072
6. White JT, Sheth KR, Bilgutay AN, Roth DR, Austin PF, Gonzales ET, et al. Vesicoamniotic shunting improves outcomes in a subset of prune belly syndrome patients at a single tertiary center. Front Pediatr. (2018) 6:180. doi: 10.3389/fped.2018.00180
7. Reinberg Y, Shapiro E, Manivel JC, Manley CB, Pettinato G, Gonzalez R. Prune belly syndrome in females: a triad of abdominal musculature deficiency and anomalies of the urinary and genital systems. J Pediatr. (1991) 118:395–8. doi: 10.1016/S0022-3476(05)82153-5
8. Druschel CM. A Descriptive study of prune belly in New York State, 1983 to 1989. Archiv Pediatr Adolesc Med. (1995) 149:70–6.
9. Wong DG, Arevalo MK, Passoni NM, Iqbal NS, Jascur T, Kern AJ, et al. Phenotypic severity scoring system and categorisation for prune belly syndrome: application to a pilot cohort of 50 living patients. BJU Int. (2019) 123:130–9. doi: 10.1111/bju.14524
10. Ewig JM, Griscom NT, Wohl ME. The effect of the absence of abdominal muscles on pulmonary function and exercise. Am J Respir Crit Care Med. (1996) 153:1314–21. doi: 10.1164/ajrccm.153.4.8616560
11. Seidel NE, Arlen AM, Smith EA, Kirsch AJ. Clinical manifestations and management of prune-belly syndrome in a large contemporary pediatric population. Urology. (2015) 85:211–5. doi: 10.1016/j.urology.2014.09.029
12. Grimsby GM, Harrison SM, Granberg CF, Bernstein IH, Baker LA. Impact and frequency of extra-genitourinary manifestations of prune belly syndrome. J Pediatr Urol. (2015) 11:280.e281–06. doi: 10.1016/j.jpurol.2015.06.005
14. Frydman M, Magenis RE, Mohandas TK, Kaback MM, Opitz JM. Chromosome abnormalities in infants with prune belly anomaly: association with trisomy 18. Am J Med Genet. (1983) 15:145–8. doi: 10.1002/ajmg.1320150120
15. Nivelon-Chevallier A, Feldman JP, Justrabo E, Turc-Carel C. Trisomy 18 and prune belly syndrome. J Genet Hum. (1985) 33:469–74.
16. Metwalley KA, Farghalley HS, Abd-Elsayed AA. Prune belly syndrome in an Egyptian infant with Down syndrome: a case report. J Med Case Rep. (2008) 2:322. doi: 10.1186/1752-1947-2-322
17. Weber S, Mir S, Schlingmann KP, Nürnberg G, Becker C, Kara PE, et al. Gene locus ambiguity in posterior urethral valves/prune-belly syndrome. Pediatr Nephrol. (2005) 20:1036–42. doi: 10.1007/s00467-005-1977-7
18. Murray PJ, Thomas K, Mulgrew CJ, Ellard S, Edghill EL, Bingham C. Whole gene deletion of the hepatocyte nuclear factor-1β gene in a patient with the prune-belly syndrome. Nephrol Dial Transpl. (2008) 23:2412–5. doi: 10.1093/ndt/gfn169
19. Iqbal NS, Jascur TA, Harrison SM, Edwards AB, Smith LT, Choi ES, et al. Prune belly syndrome in surviving males can be caused by Hemizygous missense mutations in the X-linked Filamin A gene. BMC Med Genet. (2020) 21:38. doi: 10.1186/s12881-020-0973-x
20. Sampson MG, Coughlin CR 2nd, Kaplan P, Conlin LK, Meyers KEC, Zackai EH, et al. Evidence for a recurrent microdeletion at chromosome 16p11.2 associated with congenital anomalies of the kidney and urinary tract (CAKUT) and Hirschsprung disease. Am J Med Genet A. (2010) 152a:2618–22. doi: 10.1002/ajmg.a.33628
21. Chowdhury UR, Samant RS, Fodstad O, Shevde LA. Emerging role of nuclear protein 1 (NUPR1) in cancer biology. Cancer Metastasis Rev. (2009) 28:225–32. doi: 10.1007/s10555-009-9183-x
22. Logie L, Van Aalten L, Knebel A, Force T, Hastie CJ, MacLauchlan H, et al. Rab-GTPase binding effector protein 2 (RABEP2) is a primed substrate for Glycogen Synthase kinase-3 (GSK3). Sci Rep. (2017) 7:17682. doi: 10.1038/s41598-017-17087-6
23. Odermatt A, Taschner PE, Khanna VK, Busch HF, Karpati G, Jablecki CK, et al. Mutations in the gene-encoding SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+ ATPase, are associated with Brody disease. Nat Genet. (1996) 14:191–4. doi: 10.1038/ng1096-191
24. Figueroa KP, Pulst SM. Identification and expression of the gene for human ataxin-2-related protein on chromosome 16. Exp Neurol. (2003) 184:669–78. doi: 10.1016/S0014-4886(03)00287-5
25. Mirza M, Vainshtein A, DiRonza A, Chandrachud U, Haslett LJ, Palmieri M, et al. The CLN3 gene and protein: what we know. Mol Genet Genomic Med. (2019) 7:e859. doi: 10.1002/mgg3.859
26. Fujita Y, Ezura Y, Bujo H, Nakajima T, Takahashi K, Kamimura K, et al. Association of nucleotide variations in the apolipoprotein B48 receptor gene (APOB48R) with hypercholesterolemia. J Hum Genet. (2005) 50:203–9. doi: 10.1007/s10038-005-0240-1
27. Rousseau F, Basset L, Froger J, Dinguirard N, Chevalier S, Gascan H. IL-27 structural analysis demonstrates similarities with ciliary neurotrophic factor (CNTF) and leads to the identification of antagonistic variants. Proc Natl Acad Sci USA. (2010) 107:19420–5. doi: 10.1073/pnas.1005793107
28. Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, et al. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinform. (2016) 54:1.30.1–1.30.33. doi: 10.1002/cpbi.5
Keywords: prune belly syndrome, pediatric urology, chromosomal duplication, cryptorchidism, vesicoureteral reflux
Citation: Talluri S, Goedde MA, Rosenberg E, Canalichio KL, Peppas D and White JT (2021) Case Report: Novel Copy Number Variant 16p11.2 Duplication Associated With Prune Belly Syndrome. Front. Pediatr. 9:729932. doi: 10.3389/fped.2021.729932
Received: 24 June 2021; Accepted: 04 August 2021;
Published: 23 September 2021.
Edited by:
Ezekiel E. Young, University at Buffalo, United StatesReviewed by:
Sujit Kumar Chowdhary, Indraprastha Apollo Hospital, IndiaCopyright © 2021 Talluri, Goedde, Rosenberg, Canalichio, Peppas and White. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey T. White, amVmZnJleS53aGl0ZUBub3J0b25oZWFsdGhjYXJlLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.