 Fábio Carneiro1
Fábio Carneiro1 Júlia Duarte2Francisco Laranjeira3Sofia Barbosa-Gouveia4
Júlia Duarte2Francisco Laranjeira3Sofia Barbosa-Gouveia4 María-Luz Couce4*
María-Luz Couce4* Maria José Fonseca5
Maria José Fonseca5- 1Neurology Department, Hospital Garcia de Orta, Almada, Portugal
- 2Neuroradiology Department, Hospital Garcia de Orta, Almada, Portugal
- 3Biochemical Genetics Unit, Centro de Genética Médica Doutor Jacinto Magalhães, Porto, Portugal
- 4Unit of Diagnosis and Treatment of Congenital Metabolic Diseases, Hospital Clínico Universitario de Santiago de Compostela, Instituto de Investigación Sanitaria de Santiago de Compostela, Centro de Investigación Biomédica en Red de Enfermedades Raras, European Reference Network for Rare Hereditary Metabolic Disorders, Santiago de Compostela, Spain
- 5Child Development Centre Torrado da Silva, Hospital Garcia de Orta, Almada, Portugal
Pathogenic variants of the ADGRG1 gene are associated with bilateral frontoparietal polymicrogyria, defined radiologically by polymicrogyria with an anterior-posterior gradient, pontine and cerebellar hypoplasia and patchy white matter abnormalities. We report a novel homozygous ADGRG1 variant with atypical features. The patient presented at 8 months of age with motor delay, esotropia, hypotonia with hyporeflexia and subsequently developed refractory epilepsy. At the last assessment, aged 12 years, head control, sitting and language were not acquired. Magnetic resonance imaging revealed diffuse polymicrogyria with relative sparing of the anterior temporal lobes, without an anterior-posterior gradient, diffuse hypomyelination and pontine and cerebellar hypoplasia. A panel targeting brain morphogenesis defects yielded an unreported homozygous ADGRG1 nonsense variant (dbSNP rs746634404), present in the heterozygous state in both parents. We report a novel ADGRG1 variant associated with diffuse polymicrogyria without an identifiable anterior-posterior gradient, diffuse hypomyelination and a severe motor and cognitive phenotype. Our case highlights the phenotypic diversity of ADGRG1 pathogenic variants and the clinico-anatomical overlap between recognized polymicrogyria syndromes.
Introduction
Polymicrogyria (PMG) is a cortical malformation characterized by supernumerary, small gyri with abnormal cortical lamination. PMG syndromes are markedly heterogeneous in etiology and phenotype, including infectious or vascular causes as well as genetically defined multiple congenital anomaly syndromes or inborn errors of metabolism. PMG can also occur in isolation (unilateral or bilateral PMG syndromes), frequently with a genetic etiology (1). Bilateral PMG syndromes are further categorized according to the topographical distribution of the abnormal gyral pattern into bilateral frontoparietal (BFPP), bilateral generalized (BGP), bilateral perisylvian, bilateral posterior or bilateral parasagittal PMG (1).
Autosomal recessive pathogenic variants of GPR56 or ADGRG1 (OMIM*604110; henceforth designated as ADGRG1), a member of the family of G protein–coupled receptors (GPCRs), were found to be specifically associated with BFPP (2, 3). BFPP is radiologically defined by the presence of PMG with an anterior to posterior gradient, bilateral patchy white matter signal changes and brainstem and cerebellar hypoplasia or dysplasia (3). The clinical phenotype consists of early onset hypotonia with subsequent motor and cognitive developmental delay, seizures and eye abnormalities (strabismus and/or nystagmus) (3). We herein report a novel ADGRG1 variant causing a generalized PMG pattern and a severe clinical phenotype.
Case Presentation
The proband is a 12-year-old male, the second-child of consanguineous parents (first degree cousins). The family history is relevant for learning disabilities and motor delay in a paternal aunt and learning disabilities in a paternal uncle. Notably, the patient's older brother by 9 years has no history of developmental delay or learning difficulties.
Gestation and birth were uneventful and in the post-natal period the patient only presented neonatal jaundice (maximal bilirubin dosing 12.4 mg/dL) treated with phototherapy.
Global development was reportedly normal until 6 months of age, when hypotonia and failure to achieve motor milestones were first noticed and the patient was referred to our Child Neurology outpatient clinic (Figure 1). On first assessment, at 8 months of age, the general examination showed mild dysmorphic features (almond eyes, long philtrum and low-set ears) without other relevant findings. Neurological examination revealed bilateral alternating esotropia and predominantly upper limb and axial hypotonia with diminished reflexes. We found a significant delay in psychomotor development, cephalic control was not present and the patient was unable to sit. There was poor social interaction, only few vocalizations and no hand manipulation. The initial magnetic resonance imaging (MRI) at 9 months revealed diffuse polymicrogyria without an identifiable anterior-posterior gradient, diffuse hypomyelination, thin corpus callosum, supratentorial ventriculomegaly and pontine and cerebellar hypoplasia, affecting both cerebellar vermis and hemispheres. There were no abnormalities on the electroencephalogram (EEG). The patient underwent an evaluation for chromosomal abnormalities, fragile-X mutation, imprinting disorders, mitochondrial DNA and ATPase 6 mutations, comprehensive blood, urine and cerebrospinal fluid (CSF) metabolic screening and muscle biopsy with assessment of respiratory chain enzymatic activity, which were all normal. There was no evidence of systemic disease on echocardiogram, liver, renal and spleen ultrasounds.
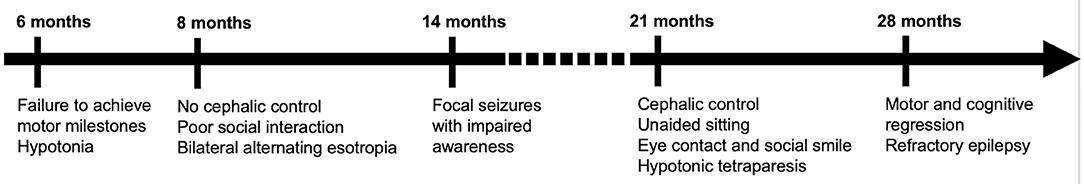
Figure 1. Timeline of symptoms and signs.
At 14 months, the patient developed focal seizures with impaired consciousness (behavioral arrest and eye deviation) initially responsive to valproate. The EEG showed predominantly frontal spikes and spike-waves, within a normal background.
The patient progressed very slowly and was able to maintain cephalic control and an unaided sitting position at 21 months. He was able to maintain eye contact, smile and produce elementary sounds, but language was not present.
At 2.5 years, however, there was significant motor and cognitive regression with loss of cephalic control and unsupported sitting ability and poorer interaction. Simultaneously, a gradual worsening of epilepsy was observed, with the appearance of multiple seizure types including epileptic spasms, atonic, myoclonic and tonic-clonic bilateral seizures. The EEG showed slow background activity with multifocal, anterior or centro-parietal predominant, spikes and spike-waves. The patient was subsequently treated with several anti-epileptics including vigabatrin, levetiracetam, topiramate, clonazepam, and perampanel, with partial control of seizures, but without significant motor and cognitive improvement. Serial neurological examinations consistently showed bilateral alternating esotropia, horizontal nystagmus and flaccid tetraparesis, without pyramidal or cerebellar signs. Visual and auditory evoked-potentials revealed prolonged conduction times bilaterally. Repeat MRI at 11 years of age (Figure 2) was globally consistent with the above-mentioned features.
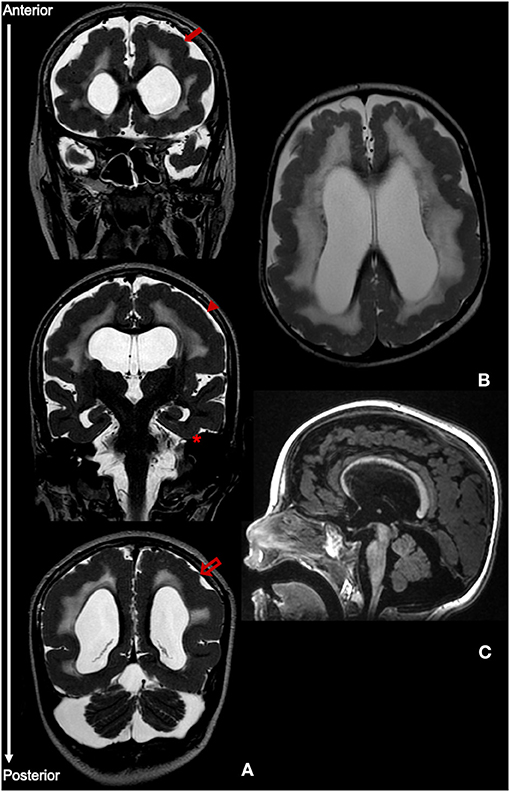
Figure 2. MRI findings at age 11 years. (A) Coronal T2-weighted images showing PMG without an anterior-posterior gradient of involvement, including frontal cortex (closed arrow), parietal cortex (arrowhead) and occipital cortex (open arrow). There is relative sparing of the temporal lobes (asterisk). (B) Axial T2 image displaying diffuse white matter hypomyelination. (C) Sagittal T1 image highlighting pontine and cerebellar hypoplasia.
Genetic analysis with a next-generation sequencing panel targeting brain morphogenesis defects yielded a homozygous ADGRG1 nonsense variant (rs746634404), caused by a nucleotide change [(NM_001145771.2: c.1504C>T) (NP_ 005673.3: p.Arg502Ter)] in exon 12. This variant is not present homozygously in GnomAD. Segregation analysis through Sanger sequencing confirmed the presence of the variant in the homozygous state in the patient and revealed that both parents were heterozygous carriers (Figure 3A). The variant is located in the seven-transmembrane (7TM) domain of the ADGRG1 protein, which binds to a heterotrimeric G-protein complex and initiates downstream signaling (Figures 3B,C). The reported allele is rare in the population [GnomAD frequency: f = 0.0000119; European (Non-Finnish): f = 0.00000881]. In silico analysis was performed using two different algorithms to predict the pathogenicity of the variant. DANN (4) (Deleterious Annotation of genetic variants using Neural Networks) is a pathogenicity scoring methodology based on neural networks, with values ranging from 0 to 1 (where 1 represents variants predicted to be most damaging). MutationTaster (5) employs a Bayes classifier to predict the disease potential of an alteration, based on evolutionary conservation, splice-site, mRNA, protein and regulatory features. The reported variant was predicted as “damaging” by DANN (DANN score: 0.9983) and as “disease causing” by MutationTaster. Finally, the level of evidence of pathogenicity was classified as “very strong” according to American College of Medical Genetics criteria (6), as the nonsense variant was identified in a gene for which loss-of-function is a known mechanism of disease.
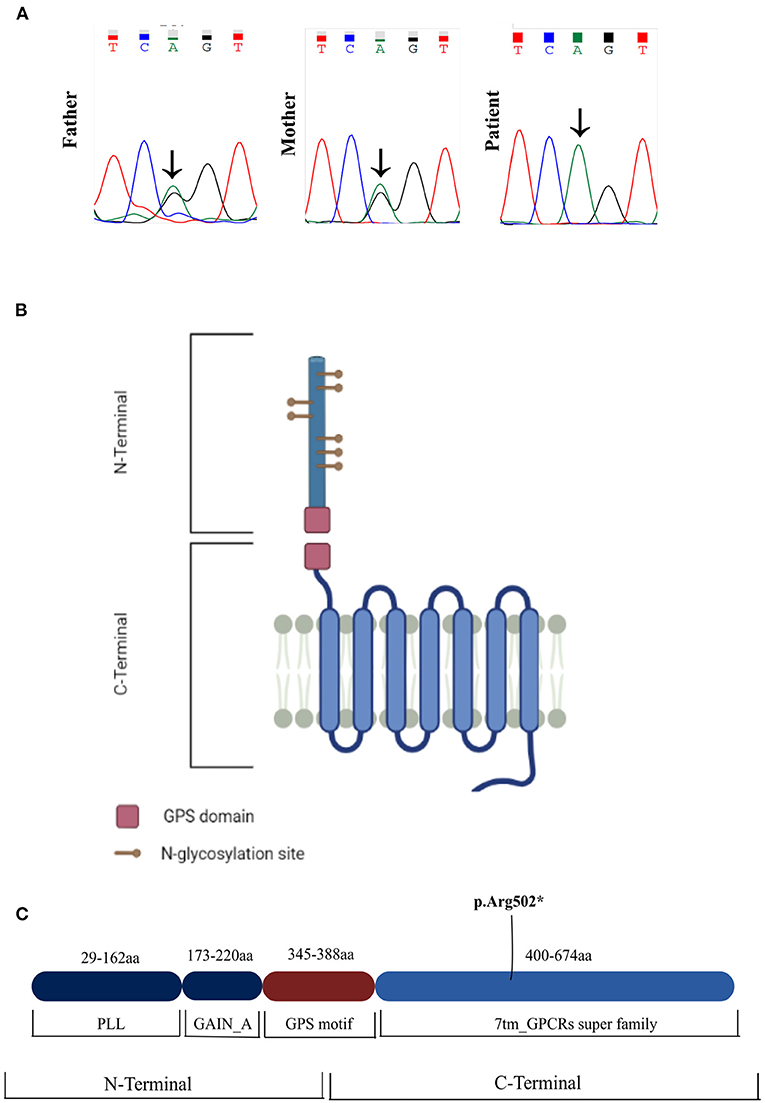
Figure 3. (A) Sanger sequencing chromatograms of the patient and parents. The patient is a homozygous carrier of the nonsense variant c.1504C>T. The variant was present in the heterozygous state in both parents. (B,C) Schematic of the ADGRG1 protein, showcasing the novel variant in the 7TM domain of the protein (p.Arg502*). 7TM, 7 transmembrane domain; GAIN, GPCR-Autoproteolysis-INducing domain; GPS, GPCR proteolysis site motif; PLL, Pentraxin/Laminin/neurexin/sex-hormone-binding-globulin-Like domain.
Discussion
We report a novel ADGRG1 truncating variant, presenting with atypically diffuse PMG and hypomyelination and a severe clinical phenotype.
At the time of publication, ADGRG1 pathogenic variants have been described in a total of 77 patients from 47 pedigrees and 34 different corresponding variants (2, 3, 7–20). In all reported cases, ADGRG1 variants were associated with a typical anatomical BFPP distribution, i.e., either a restricted frontoparietal PMG or an anterior-posterior gradient of cortical malformation. However, in our case a gradient of involvement was not identified. Complete or near-complete involvement of the entire cerebral cortex without any region of maximal involvement or any gradient of severity is the hallmark of BGP (21, 22). Relative sparing of the interhemispheric or temporal gyri has been described occasionally and is present in our patient. Considering other associated imaging features, patchy white matter signal changes or white matter volume reduction are most commonly associated with BFPP while more diffuse white matter involvement is present in more than 50% of BGP patients (22). The most distinguishing feature, however, is the presence of brainstem and cerebellar involvement in BFPP, also identified in our patient, and typically absent in BGP. Therefore, we believe that the anatomical distribution of cortical and non-cortical dysgenesis in our case mostly conforms to a severe and diffuse form of the BFPP syndrome.
Clinically, BFPP presents as a pseudomyopathic pattern, with hypotonia developing in the first year of life and occasionally being identified at birth (13). This initial clinical phenotype and imaging pattern is frequently suggestive of congenital muscular dystrophies of the “cobblestone complex” (13). The initial course of disease in our patient mimicked this pattern, but similarly to previously reported cases the muscle biopsy was normal and thus channeled the investigation away from this group of diseases. Following the pseudomyopathic presentation, the clinical course of BFPP is also considerably homogeneous in the cases reported in literature. Table 1 describes the frequency of clinical features associated with BFPP described in the reported cases.

Cognitive and motor delay are universal features, although their severity is variable between cases. While the majority of patients (79.3%) had severe cognitive impairment with language restricted to only a few words, motor impairment was reportedly milder. Indeed, most patients (81.6%) were able to walk, either with or without support, although gait is frequently described as “unsteady” or ataxic. Gait acquisition was delayed in almost all cases (13, 20). On the other hand, there was a total of nine cases without gait acquisition, originating from six different variants in seven pedigrees, of which five variants were either frameshift or nonsense (14, 20). In our case, also a nonsense variant, motor impairment was abnormally severe, since the patient was unable to walk, sit or achieve cephalic control. To our knowledge, there are only two reports sharing this severe phenotype. One is curiously of a Portuguese patient described by Santos-Silva et al. though the two cases do not share the same variant (14). The other report, by Cauley et al. describes a family with severe motor and cognitive delay, where the typical imaging phenotype of BFPP was present in association with Joubert-like features (7). The patients were found to have an additional pathogenic variant in KIAA0556 by exome sequencing, with both variants interacting to create a more severe and complex phenotype. Pyramidal signs and ataxia are also common (75.9 and 92.6%), although pyramidal signs were absent in our patient and ataxia was impossible to ascertain due to the severity of motor impairment. Other common clinical manifestations in BFPP and present in this case are epilepsy (88.2% of patients described in the literature, being drug-refractory in 60% of those) and oculomotor dysfunction (92.1% of reported cases, most commonly strabismus). In short, the clinical picture present in our case is in accordance to BFPP, although the severity of motor impairment is atypical.
Could this severe phenotype be more in line with BGP? In the study by Leventer et al. (22), BGP patients were more likely to present with global developmental delay, intellectual disability, spastic quadriplegia, seizure onset at younger age, and hearing and cortical visual impairment, when compared to other PMG syndromes. Interestingly, the reported severity of cognitive and motor delay was also variable, with mild impairment in some cases, although severe phenotypes similar to our patient were reported (21). Thus, there seems to be considerable clinical overlap between BFPP and BGP in both the type and severity of clinical manifestations.
Several hypotheses can be proposed to explain the atypical clinical features in our case. First, they may result from the unique molecular characteristics of the gene product associated with the pathogenic variant. Physiologically, the ADGRG1 protein is a GPCR that binds extracellular matrix proteins (most notably collagen type III) and undergoes agonist-induced conformational changes. This, in turn, promotes release of the α subunit of the heterotrimeric G-protein complex from the 7TM domain of ADGRG1 and initiates downstream signaling via interaction with various cytosolic effector proteins. Even though other pathogenic variants of the surrounding 7TM region (such as the L487P, E496K, and R565W variants) do not present this particular phenotype (8, 12, 18), it is possible that subtle variations in the mutated protein impair different downstream signaling cascades, thereby resulting in varied phenotypic expressions. In our particular case, the severity of the clinical phenotype can also be interpreted in the context of an epileptic encephalopathy since there was motor and cognitive regression occurring simultaneously with epilepsy decompensation. Another hypothesis is the co-existence of additional pathogenic variants of genes involved in brain morphogenesis, as highlighted by the recent report from Cauley et al.
Conclusions
We report a novel ADGRG1 truncating variant associated with generalized PMG without an anterior-posterior gradient, diffuse hypomyelination and a severe motor phenotype. Clinical and radiological features of BFPP were also present, highlighting the clinical-anatomical overlap between recognized PMG syndromes and the phenotypic diversity of ADGRG1 pathogenic variants.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Clinical Research of Galicia (2015/410). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
FC and MF contributed to conception and design of the study. SB-G and M-LC performed the genetic experiments and data analysis. FC wrote the first draft of the manuscript. FC and SB-G designed the figures. All authors contributed to clinical care of the patient, manuscript revision, read, and approved the submitted version.
Funding
This work was supported by Foundation of Health Research Institute of Santiago de Compostela, Spain.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Sofia de Melo Araújo for assistance with proofreading and language editing.
References
1. Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain. (2012) 135:1348–69. doi: 10.1093/brain/aws019
2. Chang BS, Piao X, Bodell A, Basel-Vanagaite L, Straussberg R, Dobyns WB, et al. Bilateral frontoparietal polymicrogyria: clinical and radiological features in 10 families with linkage to chromosome 16. Ann Neurol. (2003) 53:596–606. doi: 10.1002/ana.10520
3. Piao X, Chang BS, Bodell A, Woods K, Benzeev B, Topcu M, et al. Genotype-phenotype analysis of human frontoparietal polymicrogyria syndromes. Ann Neurol. (2005) 58:680–7. doi: 10.1002/ana.20616
4. Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. (2015) 31:761–3. doi: 10.1093/bioinformatics/btu703
5. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11:361–2. doi: 10.1038/nmeth.2890
6. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
7. Cauley ES, Hamed A, Mohamed IN, Elseed M, Martinez S, Yahia A, et al. Overlap of polymicrogyria, hydrocephalus, and Joubert syndrome in a family with novel truncating mutations in ADGRG1/GPR56 and KIAA0556. Neurogenetics. (2019) 20:91–8. doi: 10.1007/s10048-019-00577-2
8. Sawal H, Harripaul R, Mikhailov A, Vleuten K, Naeem F, Nasr T, et al. Three mutations in the bilateral frontoparietal polymicrogyria gene GPR56 in Pakistani intellectual disability families. J Pediatr Genet. (2018) 07:060–6. doi: 10.1055/s-0037-1612591
9. Piao X, Basel-Vanagaite L, Straussberg R, Grant PE, Pugh EW, Doheny K, et al. An autosomal recessive form of bilateral frontoparietal polymicrogyria maps to chromosome 16q12.2-21. Am J Hum Genet. (2002) 70:1028–33. doi: 10.1086/339552
10. Piao X, Hill SS, Bodell A, Chang BS, Basel-Vanagaite L, Straussberg R, et al. G protein-coupled receptor-dependent development of human frontal cortex. Science (80-). (2004) 303:2033–6. doi: 10.1126/science.1092780
11. Harbord MG, Boyd S, Hall-Craggs MA, Kendall B, McShane MA, Baraitser M. Ataxia, developmental delay and an extensive neuronal migration abnormality in 2 siblings. Neuropediatrics. (1990) 21:218–21. doi: 10.1055/s-2008-1071501
12. Luo R, Yang HM, Jin Z, Halley DJJ, Chang BS, MacPherson L, et al. A novel GPR56 mutation causes bilateral frontoparietal polymicrogyria. Pediatr Neurol. (2011) 45:49–53. doi: 10.1016/j.pediatrneurol.2011.02.004
13. Bahi-Buisson N, Poirier K, Boddaert N, Fallet-Bianco C, Specchio N, Bertini E, et al. GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex. Brain. (2010) 133:3194–209. doi: 10.1093/brain/awq259
14. Santos-Silva R, Passas A, Rocha C, Figueiredo R, Mendes-Ribeiro J, Fernandes S, et al. Bilateral frontoparietal polymicrogyria: a novel GPR56 mutation and an unusual phenotype. Neuropediatrics. (2015) 46:134–8. doi: 10.1055/s-0034-1399754
15. Borgatti R, Marelli S, Bernardini L, Novelli A, Cavallini A, Tonelli A, et al. Bilateral frontoparietal polymicrogyria (BFPP) syndrome secondary to a 16q12.1-q21 chromosome deletion involving GPR56 gene. Clin Genet. (2009) 76:573–6. doi: 10.1111/j.1399-0004.2009.01262.x
16. Fujii Y, Ishikawa N, Kobayashi Y, Kobayashi M, Kato M. Compound heterozygosity in GPR56 with bilateral frontoparietal polymicrogyria. Brain Dev. (2014) 36:528–31. doi: 10.1016/j.braindev.2013.07.015
17. Desai NA, Udani V. GPR56 -related polymicrogyria. J Child Neurol. (2015) 30:1819–23. doi: 10.1177/0883073815583335
18. Parrini E, Ferrari AR, Dorn T, Walsh CA, Guerrini R. Bilateral frontoparietal polymicrogyria, Lennox-Gastaut syndrome, and GPR56 gene mutations. Epilepsia. (2009) 50:1344–53. doi: 10.1111/j.1528-1167.2008.01787.x
19. Öncü-Öner T, Ünalp A, Porsuk-Doru I, Agilkaya S, Güleryüz H, Saraç A, Ergüner B, et al. Gpr56 homozygous nonsense mutation p.r271* associated with phenotypic variability in bilateral frontoparietal polymicrogyria. Turk J Pediatr. (2018) 60:229. doi: 10.24953/turkjped.2018.03.001
20. Quattrocchi CC, Zanni G, Napolitano A, Longo D, Cordelli DM, Barresi S, et al. Conventional magnetic resonance imaging and diffusion tensor imaging studies in children with novel GPR56 mutations: further delineation of a cobblestone-like phenotype. Neurogenetics. (2013) 14:77–83. doi: 10.1007/s10048-012-0352-7
21. Chang BS, Piao X, Giannini C, Cascino GD, Scheffer I, Woods CG, et al. Bilateral generalized polymicrogyria (BGP): a distinct syndrome of cortical malformation. Neurology. (2004) 62:1722–8. doi: 10.1212/01.WNL.0000125187.52952.E9
Keywords: ADGRG1, bilateral generalized polymicrogyria, GPR56, polymicrogyria, bilateral frontoparietal polymicrogyria, case report
Citation: Carneiro F, Duarte J, Laranjeira F, Barbosa-Gouveia S, Couce M-L and Fonseca MJ (2021) Case Report: Diffuse Polymicrogyria Associated With a Novel ADGRG1 Variant. Front. Pediatr. 9:728077. doi: 10.3389/fped.2021.728077
Received: 20 June 2021; Accepted: 04 August 2021;
Published: 27 August 2021.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Vishal Sondhi, Armed Forces Medical College, Pune, IndiaHsi-Hsien Lin, Chang Gung University, Taiwan
Sultan Cingöz, Dokuz Eylul University, Turkey
Copyright © 2021 Carneiro, Duarte, Laranjeira, Barbosa-Gouveia, Couce and Fonseca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María-Luz Couce, bWFyaWEubHV6LmNvdWNlLnBpY29Ac2VyZ2FzLmVz