
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 03 September 2021
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.715151
This article is part of the Research TopicInherited Protein Glycosylation Defects in Human DiseasesView all 15 articles
 Patryk Lipiński*
Patryk Lipiński* Anna Tylki-Szymańska
Anna Tylki-SzymańskaCongenital disorders of glycosylation (CDG) are a group of clinically heterogeneous disorders characterized by defects in the synthesis of glycans and their attachment to proteins and lipids. This manuscript aims to provide a classification of the clinical presentation, diagnostic methods, and treatment of CDG based on the literature review and our own experience (referral center in Poland). A diagnostic algorithm for CDG was also proposed. Isoelectric focusing (IEF) of serum transferrin (Tf) is still the method of choice for diagnosing N-glycosylation disorders associated with sialic acid deficiency. Nowadays, high-performance liquid chromatography, capillary zone electrophoresis, and mass spectrometry techniques are used, although they are not routinely available. Since next-generation sequencing became more widely available, an improvement in diagnostics has been observed, with more patients and novel CDG subtypes being reported. Early and accurate diagnosis of CDG is crucial for timely implementation of appropriate therapies and improving clinical outcomes. However, causative treatment is available only for few CDG types.
Congenital disorders of glycosylation (CDG), previously known as carbohydrate-deficient glycoprotein syndromes, constitute a group of inborn errors of metabolism (IEM) characterized by impaired synthesis and attachment of glycans to glycoproteins and glycolipids and impaired synthesis of glycosylphosphatidylinositol. Two main types of protein glycosylation are depicted, including N-glycosylation and O-glycosylation, while N-glycosylation is the most common type in the human body. So far, more than 150 CDG subtypes have been reported, while the phosphomannomutase-2 deficiency (PMM2-CDG) comprises the most common one (1–4).
Currently, CDG types are classified into defects in protein N-glycosylation, protein O-glycosylation, glycosphingolipid, and glycosylphosphatidylinositol (GPI) anchor glycosylation defects, and multiple glycosylation pathway defects (1–4).
CDG nomenclature is denoted by the affected gene name (non-italicized) followed by -CDG (i.e., PMM2-CDG, phosphomannomutase-2 deficiency) (1–4).
Most CDG types are autosomal recessive in inheritance, but autosomal dominant (i.e., EXT1/EXT2-CDG, GANAB-CDG, PRKCSH-CDG, POGLUT1-CDG, POFUT1-CDG) as well as X-linked (i.e., ALG13-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG) forms have also been described (1–4).
This manuscript aims to provide a classification of the clinical presentation, diagnostic methods, and treatment of CDG based on the literature review and our own experience (referral center in Poland). A diagnostic algorithm for CDG was also proposed.
CDG are usually multisystem diseases with neurological manifestation observed in most patients (5, 6). Like in other IEM, depending on the disease severity (age of symptom onset), mild to severe phenotypes could be observed.
Most CDG patients presenting with an early-onset neurovisceral phenotype have some signs and symptoms since birth. Thus, a detailed clinical analysis, including a physical examination (i.e., craniofacial dysmorphia, birth body length, weight, and head circumference) as well as family and pregnancy history (i.e., regarding non-immune hydrops fetalis, NIHF), is essential in the context of further biochemical and molecular analyses. NIHF was commonly reported in PMM2-CDG, ALG9-CDG, and ALG8-CDG, and the presence of NIHF is associated with poor outcomes (7).
Neurological signs and symptoms include psychomotor retardation, hypotonia, microcephaly, epileptic seizures, ataxia, peripheral neuropathy, and stroke-like episodes (5, 6). Deficiencies in several glycosylation pathways comprise the cause of epilepsy, while in some of them (i.e., ALG1-CDG, ALG3-CDG, ALG11-CDG, ALG13-CDG, DPM1-CDG, DPM2-CDG, MPDU1-CDG, DPAGT1-CDG, RFT1-CDG, PIGA-CDG, PIGW-CDG, and PIGQ-CDG) the severe epileptic encephalopathies have been described (8–23). Besides cerebellar and cerebral atrophy, most CDG patients with epilepsy do not have characteristic brain malformations. However, O-glycosylation disorders are associated with neuronal migration defects, including lissencephaly, polymicrogyria, schizencephaly, and neuronal heterotopia (24). The cerebellum is commonly affected in PMM2-CDG, dystroglycanopathies, and SRD5A3-CDG, while the course of cerebellar ataxia is not progressive (25–28). Several CDG types, especially dystroglycanopathies, are connected with congenital muscular dystrophy (29–33).
In the majority of CDG, liver involvement is observed as a part of multisystem phenotype, presenting with elevated serum transaminases (more often) and hepatomegaly (less often) in early infancy/childhood, while serum transaminases could normalize later in life (34–36). In the case of severe neurovisceral phenotype leading to premature death, severe liver involvement is observed as part of multiple organ failure (i.e., COG7-CDG, ALG3-CDG) (37). There is also a group of CDG, including MPI-CDG, CCDC115-CDG, and TMEM199-CDG, in which the disease is expressed mainly in the liver (no neurological manifestation) (38–43). There is no typical histologic pattern for liver disease in CDG; liver fibrosis, or even cirrhosis, was reported in PMM2-CDG, MPI-CDG, and TMEM199-CDG (44).
About 20% of CDG were reported to exhibit heart disease in the form of pericardial effusion, cardiomyopathy, arrhythmias, and structural abnormalities (45, 46). Structural (valvular and septal) defects are predominant in patients with GPI-anchor biosynthesis defects and COG-CDG (47–49). Pericardial effusions are characteristic features of PMM2-CDG, while dilated cardiomyopathy is typical for PGM1-CDG and DK1-CDG(50–52).
Recurrent and severe infections as a part of immunodeficiency phenotype were reported in ALG12-CDG, ATP6AP1-CDG, EXTL3-CDG, G6PC3-CDG, MOGS-CDG, PGM3-CDG, and SLC35C1-CDG (53–60).
Some types of CDG, including ALG3-CDG, ALG6-CDG, ALG9-CDG, ALG12-CDG, PGM3-CDG, CSGALNACT1-CDG, SLC35D1-CDG, and TMEM-165, were reported with well-defined skeletal dysplasia (61–69). In addition, some skeletal abnormalities are also unique for some types of CDG, including Schneckenbecken dysplasia in SLC35D1-CDG, brachytelephalangy in PIGV-CDG and PIGO-CDG, pseudodiastrophic dysplasia in ALG12-CDG, Gillessen-Kaesbach and Nishimura skeletal dysplasia in ALG9-CDG, and Desbuquois dysplasia in PGM3-CDG (48, 70–72).
Some CDG also have unique characteristics in the form of a constellation of clinical symptoms), which may facilitate their recognition and shorten the diagnostic process, including:
• connective tissue involvement (cutis laxa in ATP6V0A2-CDG, COG7-CDG; inguinal hernias in ATP6AP1-CDG) (73–76);
• midline malformations, including palate/uvula cleft in PGM1-CDG; a constellation of congenital malformations, dilated cardiomyopathy, liver involvement, variable endocrine, and hematological abnormalities and no neurological disease in PGM1-CDG (38, 77);
• inverted nipples and abnormal fat distribution in PMM2-CDG; specific craniofacial dysmorphia in PMM2-CDG, including microcephaly, prominent forehead, flat nasal bridge, thin upper lip, and large ears; polycystic kidney disease and hyperinsulinemic hypoglycemia in PMM2-CDG due to a promotor defect (26, 78–80);
• cerebellar hypoplasia in PMM2-CDG, SRD5A3-CDG, dystroglycanopathies (25, 28);
• cerebellar ataxia and variable eye malformations, including optic disc hypoplasia and nystagmus in SRD5A3-CDG (28);
• achalasia and alacrima without adrenal insufficiency in GMPPA-CDG (81);
• severe immunodeficiency accompanied by a skeletal dysplasia in PGM3-CDG; immunodeficiency with the Bombay blood phenotype and severe growth and psychomotor retardation in leukocyte adhesion deficiency type II (known as SLC35C1-CDG) (57–59, 82, 83).
Since the introduction by Jaeken et al. (84), isoelectric focusing (IEF) of serum transferrin (Tf) is the method of choice for diagnosis of hypo-N-glycosylation disorders associated with sialic acid deficiency (1–4, 84, 85).
So far, several other laboratory techniques have been used for the separation and quantification of serum Tf isoforms, including high-performance liquid chromatography (HPLC), capillary zone electrophoresis (CZE), and mass spectrometry (MS) (86–94). Every diagnostic method has its own limitations. Both CZE and HPLC techniques are universal with low maintenance cost and suitable for CDG screening. An abnormal result should be further investigated by serum Tf IEF. In addition, serum Tf IEF is the most commonly used for diagnosis and monitoring of CDG, and thus considered as the reference method. Other techniques, including CZE and HPLC, can be adapted by the laboratories based on their equipment accessibility.
IEF of serum Tf from dried blood spot (DBS) samples was recently demonstrated as a reliable method for CDG screening (95, 96). DBS is firmly established in the analysis of various IEM, especially in the context of newborn screening programs across the world. However, there is a number of CDG for which there is no data regarding glycosylation abnormalities after birth and thus further studies are needed.
Based on our own experience (referral center in Poland), we create a diagnostic algorithm for CDG (Figure 1). We recommend to perform serum Tf IEF for an initial screening of glycosylation abnormalities in patients presenting with clinical and biochemical features listed in Table 1.
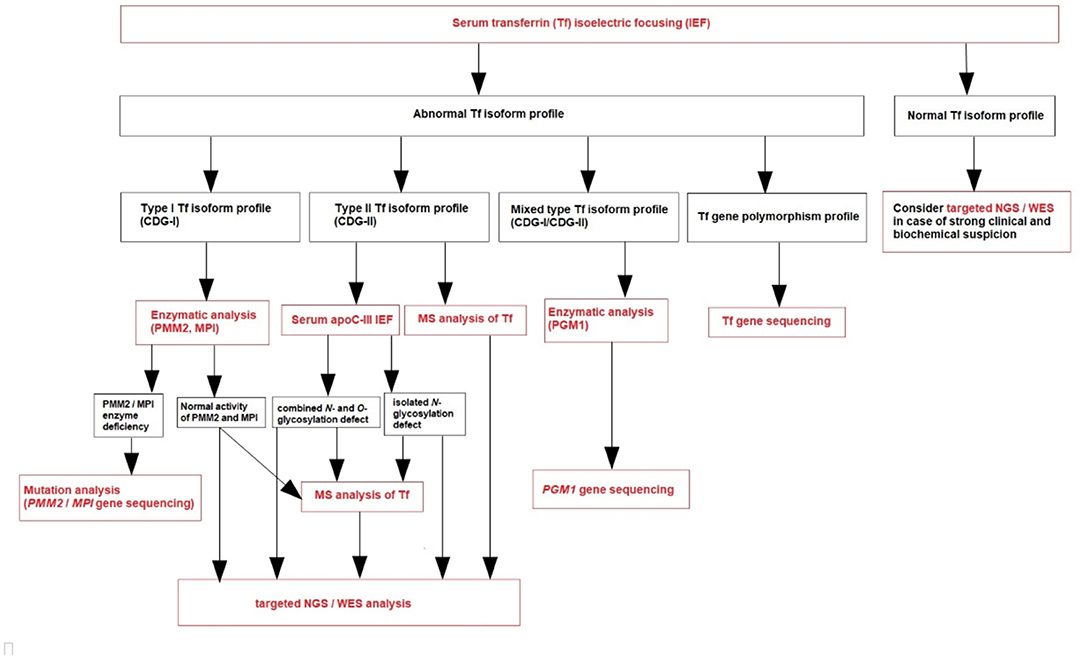
Figure 1. Diagnostic algorithm for CDG.

Table 1. Clinical and biochemical features requiring IEF of serum transferrin.
Transferrin (Tf) is a plasma iron transport protein with two asparagine N-glycosylation sites (Asn432 and Asn630), and the dominated isoform in healthy individuals is tetrasialo-Tf, while asialo- and monosialo-Tf isoforms are usually not detectable. A type 1 pattern (CDG-I) is associated with an increased asialo- and disialo-Tf, and decreased tetrasialo-Tf, indicating an assembly or transfer defect of the dolichol-linked glycan (85). A type 2 pattern (CDG-II) is associated with increased asialo-, monosialo-, disialo-, and trisialo-Tf, indicating a processing defect after glycan transfer in the ER or during Golgi glycosylation (85). PGM1-CDG presents features of the both types of serum Tf IEF patterns (mixed type, CDG-I/CDG-II).
During CDG diagnostic process, it is important to exclude secondary causes of N-hypoglycosylation as well as Tf gene polymorphisms. Several Tf gene polymorphisms (i.e., transferrin B2 presenting with an elevated pentasialo-Tf solely or transferrin C2 resulting in increased trisialo-Tf solely) are known to result in a shifted IEF pattern, caused by pI differences of the polypeptide chain.
It is also known that untreated patients with classic galactosemia (galactose-1-phosphate uridyltransferase deficiency) and fructosemia (fructose 1-phosphate aldolase deficiency) have secondarily an abnormal serum Tf isoform profile that could resemble CDG-I. For example, Bogdańska et al. reported a study on 19 pediatric patients with primary liver disease and increased secondary asialo-Tf and monosialo-Tf isoforms; none of the patients had an elevated level of trisialo-Tf isoform (97). On the other hand, Jansen et al. published a study about secondary glycosylation defects in 961 adult patients qualified for LTx or with chronic liver disease. It showed that 247 patients (26%) had hyposialylation of serum Tf, while the majority of them (70%) had an increase in the trisialo-Tf isoform (98).
Normal serum Tf IEF profile does not exclude CDG—we should consider targeted next-generation sequencing (NGS) or whole-exome sequencing (WES) in case of a strong clinical and biochemical suspicion. PMM2-CDG due to promotor defect and several other CDG, like SLC35A1-CDG, SLC35A3-CDG, SEC23B-CDG, and PGM3-CDG, could show normal N-glycosylation profile. What is more, serum Tf IEF could be normal as well as abnormal in several others CDG, like ALG13-CDG, SLC35A2-CDG, RTF1-CDG, and SRD5A3-CDG (1–4, 85).
After the diagnosis of CDG-I based on serum Tf IEF, phosphomannomutase-2 (PMM2) and phosphomannose isomerase (PMI) activity should be measured in fibroblasts or leukocytes in the proper clinical context (1–4, 85). PMM2-CDG has the best-defined clinical phenotype and is by far the most frequent N-glycan assembly defect (99). PMI activity should be measured in case of clinical and biochemical presentation mainly expressed by the liver. If the clinical phenotype is not typical for PMM2-CDG and MPI-CDG, and in the case of normal PMM2 and PMI activity, plasma N-glycan analysis by MS (total plasma and/or intact transferrin glycoprofiling) could be done (1–4, 85). Nowadays, this is replaced by next-generation sequencing (NGS), including targeted NGS (panel of genes known to be involved in CDG) or whole exome sequencing (WES) (Figure 1). However, since NGS became more widely available, an improvement in diagnostics has been observed, with more patients and novel CDG subtypes being reported. These molecular analyses could take more time to result and be more expensive than laboratory CDG screening, although the high-throughput methods (like MS) are not routinely available (like in our center) and require both a qualified staff and comprehensive equipment.
After the diagnosis of CDG-II, IEF of serum apolipoprotein C-III (apoC-III) is recommended to perform (Figure 1) to distinguish between an exclusive N-glycosylation defect and a combined disorder of N- and O-glycosylation (1–4, 85). This analysis was described by Wopereis et al. in 2003 as a screening method for defects in the biosynthesis of the core 1 mucin-type O-glycans (100). However, some patients could also have an abnormal biosynthesis of core 1 O-glycans, including those with hemolytic uremic syndrome due to Streptococcus pneumoniae. Therefore, other laboratory methods have been developed parallel to serum Tf IEF, including serum apoC-III CZE.
Protein-linked glycan analysis should next be performed in attempt to identify the defective step, or targeted NGS, or WES (94).
Molecular analysis is necessary to confirm the final diagnosis of CDG and predict the possible genotype-phenotype correlation. However, the combination of MS with clinical exome sequencing (especially WES) is helpful to identify new CDG defects.
An early diagnosis of CDG is crucial for the timely implementation of appropriate therapies. However, causative treatment is available only for few CDG types in the form of specific monosaccharide supplementation therapy (i.e., galactose for PGM1-CDG, fucose for SLC35C1-CDG, Mn2+ for TMEM165-CDG, or mannose for MPI-CDG) (101–103). For the majority of patients, only symptomatic treatment can be offered. The natural history for most CDG types is unknown (also due to lack of long-term follow-up); however, cerebellar ataxia in PMM2-CDG is not progressive, and patients could even slowly improve with age (26, 27).
Several therapeutic strategies were developed for PMM2-CDG, including mannose supplementation, inhibition of MPI, pharmacological chaperones, proteostasis regulators (celastrol), acetazolamide, and antisense therapy (104). To date, no causative treatment for PMM2-CDG exists. However, acetazolamide was reported to be well tolerated and effective for cerebellar syndrome (105). In addition, Taday et al. recently published a study on long-term oral mannose supplementation in 20 patients with PMM2-CDG (106). The therapy was tolerated well, and biochemical improvement was noted in the majority of patients.
Symptomatic treatment in PMM2-CDG includes:
• nutritional support (including percutaneous endoscopic gastrostomy placement) in failure-to-thrive patients;
• regular albumin infusions, octreotide therapy, or a diet rich in mid-chain fatty acids (MCTs) in protein-losing enteropathy;
• levothyroxine in the presence of decreased free thyroxine;
• fresh frozen plasma infusions to prevent bleeding episodes;
• pleural-pericardial window formation in recurrent pericardial effusion (26, 36).
The administration of mannose in MPI-CDG improves the clinical and biochemical outcome (including serum transferrin isoforms); however, patients can still develop progressive liver fibrosis (107–110). Mannose therapy in MPI-CDG was also discontinued in a few patients due to side effects (40). One reported patient with MPI-CDG required liver transplantation due to chronic liver disease with the development of hepatopulmonary syndrome (39).
Two patients with CCDC115-CDG underwent LTx; one rejected the transplant and died while the other is doing well, showing biochemical improvement of liver function tests and transferrin glycosylation profile (111).
Galactose therapy in PGM1-CDG is safe and associated with a significant improvement of N-glycosylation and clinical parameters (liver function tests, coagulation, blood glucose) (38, 77). Four patients with PGM1-CDG underwent heart transplantation, and all died due to cardiac disease-related complications. Three other patients reported by Tegtmeyer et al. were listed for heart transplantation (38).
Heart transplantation could be considered as a treatment option in other patients with cardiac involvement. It was performed in three patients with defects in dolichol synthesis (DOLK-CDG, DK1-CDG), despite supportive heart failure therapy (ACE inhibitors, β-blockers, and diuretics); one of them died unexpectedly 2 years after transplantation at the age of 16.5 years (51).
Besides PGM1-CDG, galactose supplementation showed promising results in SLC35A2-CDG, SLC39A8-CDG, and TMEM165-CDG (112–114).
Fucose supplementation in SLC35C1-CDG was reported to decrease infection rates, normalize neutrophil counts, and improve psychomotor development. However, it should be monitored carefully due to the risk of autoimmune and hemolytic reactions (115–117).
Uridine supplementation in CAD-CDG patients was reported to improve the clinical manifestation, including seizure cessation, cognitive and motor development, and normalization of biochemical parameters (118).
Hematopoietic stem cell transplantation was successfully applied to treat CDG with immunodeficiency in PGM3-CDG children (57).
PL and AT-S: project administration and writing—review and editing. AT-S: supervision. PL: writing—original draft. Both authors contributed to the article and approved the submitted version.
The study was funded by the Children's Memorial Health Institute intramural grant S190/2020.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Jaeken J, Peanne R. What is new in CDG? J Inherit Metab Dis. (2017) 40:569–86. doi: 10.1007/s10545-017-0050-6
2. Francisco R, Marques-da-Silva D, Brasil S, Pascoal C, Dos Reis Ferreira V, Morava E, et al. The challenge of CDG diagnosis. Mol Genet Metab. (2019) 126:1–5. doi: 10.1016/j.ymgme.2018.11.003
3. Péanne R, de Lonlay P, Foulquier F, Kornak U, Lefeber DJ, Morava E, et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur J Med Genet. (2018) 61:643–63. doi: 10.1016/j.ejmg.2017.10.012
4. Ferreira CR, Altassan R, Marques-Da-Silva D, Francisco R, Jaeken J, Morava E. Recognizable phenotypes in CDG. J Inherit Metab Dis. (2018) 41:541–53. doi: 10.1007/s10545-018-0156-5
5. Makhamreh MM, Cottingham N, Ferreira CR, Berger S, Al-Kouatly HB. Nonimmune hydrops fetalis and congenital disorders of glycosylation: A systematic literature review. J Inherit Metab Dis. (2020) 43:223–33. doi: 10.1002/jimd.12162
6. Paprocka J, Jezela-Stanek A, Tylki-Szymańska A, Grunewald S. Congenital Disorders of Glycosylation from a Neurological Perspective. Brain Sci. (2021) 11:88. doi: 10.3390/brainsci11010088
7. Freeze HH, Eklund EA, Ng BG, Patterson MC. Neurological aspects of human glycosylation disorders. Annu Rev Neurosci. (2015) 38:105–25. doi: 10.1146/annurev-neuro-071714-034019
8. Fiumara A, Barone R, Del Campo G, Striano P. Jaeken J. Early-Onset Epileptic Encephalopathy in infants with different forms of Congenital Disorders of Glycosylation (CDG). Brain Dev. (2017) 39:366–67. doi: 10.1016/j.braindev.2016.11.008
9. Fiumara A, Barone R, Del Campo G, Striano P. Jaeken J. Electroclinical features of early-onset epileptic encephalopathies in congenital disorders of glycosylation (CDGs). JIMD Rep. (2016) 27:93–9. doi: 10.1007/8904_2015_497
10. Pereira AG, Bahi-Buisson N, Barnerias C, Boddaert N, Nabbout R, de Lonlay P, et al. Epileptic spasms in congenital disorders of glycosylation. Epileptic Disord. (2017) 19:15–23. doi: 10.1684/epd.2017.0901
11. Ng BG, Shiryaev SA, Rymen D, Eklund EA, Raymond K, Kircher M, et al. University of Washington Center for Mendelian Genomics, Matthijs G, Freeze HH. ALG1-CDG: clinical and molecular characterization of 39 unreported patients. Hum Mutat. (2016) 37:653–60.
12. Paketci C, Edem P, Hiz S, Sonmezler E, Soydemir D, Sarikaya Uzan G, et al. Successful treatment of intractable epilepsy with ketogenic diet therapy in twins with ALG3-CDG. Brain Dev. (2020) 42:539–45. doi: 10.1016/j.braindev.2020.04.008
13. Wu X, Rush JS, Karaoglu D, Krasnewich D, Lubinsky MS, Waechter CJ, et al. Deficiency of UDP-GlcNAc: Dolichol phosphate N-acetylglucosamine-1 Phosphate transferase (DPAGT1) causes a novel congenital disorder of glycosylation type Ij. Hum Mutat. (2003) 22:144–50. doi: 10.1002/humu.10239
14. Ng BG, Underhill HR, Palm L, Bengtson P, Rozet JM, Gerber S, et al. University of Washington Center for Mendelian Genomics, Freeze HH, Eklund EA. DPAGT1 deficiency with encephalopathy (DPAGT1-CDG): clinical and genetic description of 11 new patients. JIMD Rep. (2019) 44:85–92. doi: 10.1007/8904_2018_128
15. Barone R, Aiello C, Race V, Morava E, Foulquier F, Riemersma M, et al. DPM2-CDG: a muscular dystrophy-dystroglycanopathy syndrome with severe epilepsy. Ann Neurol. (2012) 72:550–8. doi: 10.1002/ana.23632
16. Ng BG, Eklund EA, Shiryaev SA, Dong YY, Abbott MA, Asteggiano C, et al. Predominant and novel de novo variants in 29 individuals with ALG13 deficiency: Clinical description, biomarker status, biochemical analysis, and treatment suggestions. J Inherit Metab Dis. (2020) 43:1333–48. doi: 10.1002/jimd.12290
17. Paprocka J, Jezela-Stanek A, Boguszewicz Ł, Sokół M, Lipiński P, Jamroz E, et al. The first metabolome analysis in children with epilepsy and ALG13-CDG resulting from c.320A>G variant. Children (Basel). (2021) 8:251. doi: 10.3390/children8030251
18. Galama WH. Verhaagen-van den Akker SLJ, Lefeber DJ, Feenstra I, Verrips A. ALG13-CDG with infantile spasms in a male patient due to a de novo ALG13 gene mutation. JIMD Rep. (2018) 40:11–6. doi: 10.1007/8904_2017_53
19. Chiyonobu T, Inoue N, Morimoto M, Kinoshita T, Murakami Y. Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with West syndrome and hyperphosphatasia with mental retardation syndrome. J Med Genet. (2014) 51:203–7. doi: 10.1136/jmedgenet-2013-102156
20. Bayat A, Knaus A, Pendziwiat M, Afenjar A, Barakat TS, Bosch F, et al. Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia. (2020) 61:1142–55. doi: 10.1111/epi.16545
21. Kato M, Saitsu H, Murakami Y, Kikuchi K, Watanabe S, Iai M, et al. Mutations cause early-onset epileptic encephalopathies and distinctive features. Neurology. (2014) 82:1587–96. doi: 10.1212/WNL.0000000000000389
22. Johnstone DL, Nguyen TTM, Zambonin J, Kernohan KD, St-Denis A, Baratang NV, et al. Early infantile epileptic encephalopathy due to biallelic pathogenic variants in PIGQ: Report of seven new subjects and review of the literature. J Inherit Metab Dis. (2020) 43:1321–32. doi: 10.1002/jimd.12278
23. Martin HC, Kim GE, Pagnamenta AT, Murakami Y, Carvill GL, Meyer E, et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet. (2014) 23:3200–11. doi: 10.1093/hmg/ddu030
24. Schiller S, Rosewich H, Grünewald S, Gärtner J. Inborn errors of metabolism leading to neuronal migration defects. J Inherit Metab Dis. (2020) 43:145–55. doi: 10.1002/jimd.12194
25. Barone R, Fiumara A, Jaeken J. Congenital disorders of glycosylation with emphasis on cerebellar involvement. Semin Neurol. (2014) 34:357–66. doi: 10.1055/s-0034-1387197
26. Altassan R, Péanne R, Jaeken J, Barone R, Bidet M, Borgel D, et al. International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow up. J Inherit Metab Dis. (2019) 42:5–28. doi: 10.1002/jimd.12024
27. Serrano M, de Diego V, Muchart J, Cuadras D, Felipe A, Macaya A, et al. Phosphomannomutase deficiency (PMM2-CDG): ataxia and cerebellar assessment. Orphanet J Rare Dis. (2015) 10:138. doi: 10.1186/s13023-015-0358-y
28. Morava E, Wevers RA, Cantagrel V, Hoefsloot LH, Al-Gazali L, Schoots J, et al. Novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain. (2010) 133:3210–20. doi: 10.1093/brain/awq261
29. Martin PT. The dystroglycanopathies: the new disorders of O-linked glycosylation. Semin Pediatr Neurol. (2005) 12:152–8. doi: 10.1016/j.spen.2005.10.003
30. Falsaperla R, Praticò AD, Ruggieri M, Parano E, Rizzo R, Corsello G, et al. Congenital muscular dystrophy: from muscle to brain. Ital J Pediatr. (2016) 42:78. doi: 10.1186/s13052-016-0289-9
31. Brancaccio A. A molecular overview of the primary dystroglycanopathies. J Cell Mol Med. (2019) 23:3058–62. doi: 10.1111/jcmm.14218
32. Hu P, Yuan L, Deng H. Molecular genetics of the POMT1-related muscular dystrophy-dystroglycanopathies. Mutat Res. (2018) 778:45–50. doi: 10.1016/j.mrrev.2018.09.002
33. Godfrey C, Foley AR, Clement E, Muntoni F. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev. (2011) 21:278–85. doi: 10.1016/j.gde.2011.02.001
34. Marques-da-Silva D, Dos Reis Ferreira V, Monticelli M, Janeiro P, Videira PA, Witters P, et al. Liver involvement in congenital disorders of glycosylation (CDG). A systematic review of the literature. J Inherit Metab Dis. (2017) 40:195–207. doi: 10.1007/s10545-016-0012-4
35. Starosta RT, Boyer S, Tahata S, Raymond K, Lee HE, Wolfe LA, et al. Liver manifestations in a cohort of 39 patients with congenital disorders of glycosylation: pin-pointing the characteristics of liver injury and proposing recommendations for follow-up. Orphanet J Rare Dis. (2021) 16:20. doi: 10.1186/s13023-020-01630-2
36. Bogdańska A, Lipiński P, Szymańska-Rozek P, Jezela-Stanek A, Rokicki D, Socha P, et al. Clinical, biochemical and molecular phenotype of congenital disorders of glycosylation: long-term follow-up. Orphanet J Rare Dis. (2021) 16:17. doi: 10.1186/s13023-020-01657-5
37. Spaapen LJ, Bakker JA, van der Meer SB, Sijstermans HJ, Steet RA, Wevers RA, et al. Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. J Inherit Metab Dis. (2005) 28:707–14. doi: 10.1007/s10545-005-0015-z
38. Tegtmeyer LC, Rust S, van Scherpenzeel M, Ng BG, Losfeld ME, Timal S, et al. Multiple phenotypes in phosphoglucomutase 1 deficiency. N Engl J Med. (2014) 370:533–42. doi: 10.1056/NEJMoa1206605
39. Janssen MC, de Kleine RH, van den Berg AP, Heijdra Y, van Scherpenzeel M, Lefeber DJ, et al. Successful liver transplantation and long-term follow-up in a patient with MPI-CDG. Pediatrics. (2014) 134:e279–83. doi: 10.1542/peds.2013-2732
40. Damen G, de Klerk H, Huijmans J, den Hollander J, Sinaasappel M. Gastrointestinal and other clinical manifestations in 17 children with congenital disorders of glycosylation type Ia, Ib, and Ic. J Pediatr Gastroenterol Nutr. (2004) 38:282–87. doi: 10.1097/00005176-200403000-00010
41. Girard M, Poujois A, Fabre M, Lacaille F, Debray D, Rio M, et al. CCDC115-CDG: A new rare and misleading inherited cause of liver disease. Mol Genet Metab. (2018) 124:228–35. doi: 10.1016/j.ymgme.2018.05.002
42. Vajro P, Zielinska K, Ng BG, Maccarana M, Bengtson P, Poeta M, et al. Three unreported cases of TMEM199-CDG, a rare genetic liver disease with abnormal glycosylation. Orphanet J Rare Dis. (2018) 13:4. doi: 10.1186/s13023-017-0757-3
43. Jansen JC, Timal S, van Scherpenzeel M, Michelakakis H, Vicogne D, Ashikov A, et al. TMEM199 deficiency is a disorder of Golgi homeostasis characterized by elevated aminotransferases, alkaline phosphatase, and cholesterol and abnormal glycosylation. Am J Hum Genet. (2016) 98:322–30. doi: 10.1016/j.ajhg.2015.12.011
44. de Koning TJ, Nikkels PG, Dorland L, Bekhof J, De Schrijver JE, van Hattum J, et al. Congenital hepatic fibrosis in 3 siblings with phosphomannose isomerase deficiency. Virchows Arch. (2000) 437:101–5. doi: 10.1007/s004280000185
45. Marques-da-Silva D, Francisco R, Webster D, Dos Reis Ferreira V, Jaeken J, Pulinilkunnil T. Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature. J Inherit Metab Dis. (2017) 40:657–72. doi: 10.1007/s10545-017-0066-y
46. Footitt EJ, Karimova A, Burch M, Yayeh T, Dupré T, Vuillaumier-Barrot S, et al. Cardiomyopathy in the congenital disorders of glycosylation (CDG): a case of late presentation and literature review. J Inherit Metab Dis. (2009) 32:S313–9. doi: 10.1007/s10545-009-1262-1
47. Knaus A, Pantel JT, Pendziwiat M, Hajjir N, Zhao M, Hsieh TC, et al. Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Med. (2018) 10:3. doi: 10.1186/s13073-017-0510-5
48. Carmody LC, Blau H, Danis D, Zhang XA, Gourdine JP, Vasilevsky N, et al. Significantly different clinical phenotypes associated with mutations in synthesis and transamidase+remodeling glycosylphosphatidylinositol (GPI)-anchor biosynthesis genes. Orphanet J Rare Dis. (2020) 15:40. doi: 10.1186/s13023-020-1313-0
49. D'Souza Z, Taher FS, Lupashin VV. Golgi inCOGnito: From vesicle tethering to human disease. Biochim Biophys Acta Gen Subj. (2020) 1864:129694. doi: 10.1016/j.bbagen.2020.129694
50. Görlacher M, Panagiotou E, Himmelreich N, Hüllen A, Beedgen L, Dimitrov B, et al. Fatal outcome after heart surgery in PMM2-CDG due to a rare homozygous gene variant with double effects. Mol Genet Metab Rep. (2020) 25:100673. doi: 10.1016/j.ymgmr.2020.100673
51. Kapusta L, Zucker N, Frenckel G, Medalion B, Ben Gal T, Birk E, et al. From discrete dilated cardiomyopathy to successful cardiac transplantation in congenital disorders of glycosylation due to dolichol kinase deficiency (DK1-CDG). Heart Fail Rev. (2013) 18:187–96. doi: 10.1007/s10741-012-9302-6
52. Fernlund E, Andersson O, Ellegård R, Årstrand HK, Green H, Olsson H, et al. The congenital disorder of glycosylation in PGM1 (PGM1-CDG) can cause severe cardiomyopathy and unexpected sudden cardiac death in childhood. Forensic Sci Int Genet. (2019) 43:102111. doi: 10.1016/j.fsigen.2019.06.012
53. Pascoal C, Francisco R, Ferro T, Dos Reis Ferreira V, Jaeken J, Videira PA, et al. and immune response: From bedside to bench and back. J Inherit Metab Dis. (2020) 43:90–124. doi: 10.1002/jimd.12126
54. Monticelli M, Ferro T, Jaeken J, Dos Reis Ferreira V, Videira PA. Immunological aspects of congenital disorders of glycosylation (CDG): a review. J Inherit Metab Dis. (2016) 39:765–80. doi: 10.1007/s10545-016-9954-9
55. Sturiale L, Bianca S, Garozzo D, Terracciano A, Agolini E, Messina A, et al. ALG12-CDG: novel glycophenotype insights endorse the molecular defect. Glycoconj J. (2019) 36:461–72. doi: 10.1007/s10719-019-09890-2
56. Chang J, Block TM, Guo JT. Viral resistance of MOGS-CDG patients implies a broad-spectrum strategy against acute virus infections. Antivir Ther. (2015) 20:257–9. doi: 10.3851/IMP2907
57. Stray-Pedersen A, Backe PH, Sorte HS, Mørkrid L, Chokshi NY, Erichsen HC, et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet. (2014) 95:96–107. doi: 10.1016/j.ajhg.2014.05.007
58. Pacheco-Cuéllar G, Gauthier J, Désilets V, Lachance C, Lemire-Girard M, Rypens F, et al. Novel PGM3 mutation is associated with a severe phenotype of bone marrow failure, severe combined immunodeficiency, skeletal dysplasia, and congenital malformations. J Bone Miner Res. (2017) 32:1853–59. doi: 10.1002/jbmr.3173
59. Knapp KM, Luu R, Baerenfaenger M, Zijlstra F, Wessels HJCT, Jenkins D, et al. Biallelic variants in SLC35C1 as a cause of isolated short stature with intellectual disability. J Hum Genet. (2020) 65:743–50. doi: 10.1038/s10038-020-0764-4
60. Sadat MA, Moir S, Chun TW, Lusso P, Kaplan G, Wolfe L, et al. Glycosylation, hypogammaglobulinemia, and resistance to viral infections. N Engl J Med. (2014) 370:1615–25. doi: 10.1056/NEJMoa1302846
61. Murali C, Lu JT, Jain M, Liu DS, Lachman R, Gibbs RA, et al. Diagnosis of ALG12-CDG by exome sequencing in a case of severe skeletal dysplasia. Mol Genet Metab Rep. (2014) 1:213–9. doi: 10.1016/j.ymgmr.2014.04.004
62. Lepais L, Cheillan D, Frachon SC, Hays S, Matthijs G, Panagiotakaki E, et al. ALG3-CDG: Report of two siblings with antenatal features carrying homozygous p.Gly96Arg mutation. Am J Med Genet A. (2015) 167A:2748–54. doi: 10.1002/ajmg.a.37232
63. Tham E, Eklund EA, Hammarsjö A, Bengtson P, Geiberger S, Lagerstedt-Robinson K, et al. Novel phenotype in N-glycosylation disorders: Gillessen-Kaesbach-Nishimura skeletal dysplasia due to pathogenic variants in ALG9. Eur J Hum Genet. (2016) 24:198–207. doi: 10.1038/ejhg.2015.91
64. AlSubhi S, AlHashem A, AlAzami A, Tlili K, AlShahwan S, Lefeber D, et al. Further delineation of the ALG9-CDG phenotype. JIMD Rep. (2016) 27:107–12. doi: 10.1007/8904_2015_504
65. Sassi A, Lazaroski S, Wu G, Haslam SM, Fliegauf M, Mellouli F, et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J Allergy Clin Immunol. (2014) 133:1410–9. doi: 10.1016/j.jaci.2014.02.025
66. Vodopiutz J, Mizumoto S, Lausch E, Rossi A, Unger S, Janocha N, et al. Chondroitin Sulfate N-acetylgalactosaminyltransferase-1 (CSGalNAcT-1) Deficiency Results in a Mild Skeletal Dysplasia and Joint Laxity. Hum Mutat. (2017) 38:34–8. doi: 10.1002/humu.23070
67. Mizumoto S, Janecke AR, Sadeghpour A, Povysil G, McDonald MT, Unger S, et al. CSGALNACT1-congenital disorder of glycosylation: A mild skeletal dysplasia with advanced bone age. Hum Mutat. (2020) 41:655–67. doi: 10.1002/humu.23952
68. Zeevaert R, de Zegher F, Sturiale L, Garozzo D, Smet M, Moens M, et al. Bone dysplasia as a key feature in three patients with a novel congenital disorder of glycosylation (CDG) type ii due to a deep intronic splice mutation in TMEM165. JIMD Rep. (2013) 8:145–52. doi: 10.1007/8904_2012_172
69. Foulquier F, Amyere M, Jaeken J, Zeevaert R, Schollen E, Race V, et al. TMEM165 deficiency causes a congenital disorder of glycosylation. Am J Hum Genet. (2012) 91:15–26. doi: 10.1016/j.ajhg.2012.05.002
70. Rautengarten C, Quarrell OW, Stals K, Caswell RC, De Franco E, Baple E, et al. hypomorphic allele of SLC35D1 results in Schneckenbecken-like dysplasia. Hum Mol Genet. (2019) 28:3543–51. doi: 10.1093/hmg/ddz200
71. Horn D, Krawitz P, Mannhardt A, Korenke GC, Meinecke P. Hyperphosphatasia-mental retardation syndrome due to PIGV mutations: expanded clinical spectrum. Am J Med Genet A. (2011) 155A:1917–22. doi: 10.1002/ajmg.a.34102
72. Altassan R, Fox S, Poulin C, Buhas D. Hyperphosphatasia with mental retardation syndrome, expanded phenotype of PIGL related disorders. Mol Genet Metab Rep. (2018) 15:46–9. doi: 10.1016/j.ymgmr.2018.01.007
73. Dimitrov B, Himmelreich N, Hipgrave Ederveen AL, Lüchtenborg C, Okun JG, Breuer M, et al. Cutis laxa, exocrine pancreatic insufficiency and altered cellular metabolomics as additional symptoms in a new patient with ATP6AP1-CDG. Mol Genet Metab. (2018) 123:364–74. doi: 10.1016/j.ymgme.2018.01.008
74. Witters P, Breckpot J, Foulquier F, Preston G, Jaeken J, Morava E. Expanding the phenotype of metabolic cutis laxa with an additional disorder of N-linked protein glycosylation. Eur J Hum Genet. (2018) 26:618–21. doi: 10.1038/s41431-017-0044-8
75. Jansen EJ, Timal S, Ryan M, Ashikov A, van Scherpenzeel M, Graham LA, et al. ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nat Commun. (2016) 7:11600. doi: 10.1038/ncomms11600
76. Van Damme T, Gardeitchik T, Mohamed M, Guerrero-Castillo S, Freisinger P, Guillemyn B, et al. Mutations in ATP6V1E1 or ATP6V1A cause autosomal-recessive cutis laxa. Am J Hum Genet. (2017) 100:216–27. doi: 10.1016/j.ajhg.2016.12.010
77. Altassan R, Radenkovic S, Edmondson AC, Barone R, Brasil S, Cechova A, et al. International consensus guidelines for phosphoglucomutase 1 deficiency (PGM1-CDG): Diagnosis, follow-up, and management. J Inherit Metab Dis. (2021) 44:148–63. doi: 10.1002/jimd.12286
78. Schiff M, Roda C, Monin ML, Arion A, Barth M, Bednarek N, et al. Clinical, laboratory and molecular findings and long-term follow-up data in 96 French patients with PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) and review of the literature. J Med Genet. (2017) 54:843–51. doi: 10.1136/jmedgenet-2017-104903
79. Al-Maawali AA, Miller E, Schulze A, Yoon G, Blaser SI. Subcutaneous fat pads on body MRI-an early sign of congenital disorder of glycosylation PMM2-CDG (CDG1a). Pediatr Radiol. (2014) 44:222–25. doi: 10.1007/s00247-013-2782-2
80. Cabezas OR Flanagan SE Stanescu H García-Martínez E Caswell R Lango-Allen H. Polycystic Kidney Disease with Hyperinsulinemic Hypoglycemia Caused by a Promoter Mutation in Phosphomannomutase 2. J Am Soc Nephrol. (2017) 28:2529–39. doi: 10.1681/ASN.2016121312
81. Diaz J, Kane TD, Leon E. Evidence of GMPPA founder mutation in indigenous Guatemalan population associated with alacrima, achalasia, and mental retardation syndrome. Am J Med Genet A. (2020) 182:425–30. doi: 10.1002/ajmg.a.61476
82. Sturla L, Fruscione F, Noda K, Miyoshi E, Taniguchi N, Contini P, et al. Core fucosylation of N-linked glycans in leukocyte adhesion deficiency/congenital disorder of glycosylation IIc fibroblasts. Glycobiology. (2005) 15:924–34. doi: 10.1093/glycob/cwi081
83. Etzioni A, Sturla L, Antonellis A, Green ED, Gershoni-Baruch R, Berninsone PM, et al. Leukocyte adhesion deficiency (LAD) type II/carbohydrate deficient glycoprotein (CDG) IIc founder effect and genotype/phenotype correlation. Am J Med Genet. (2002) 110:131–5. doi: 10.1002/ajmg.10423
84. Jaeken J, van Eijk HG, van der Heul L, Corbeel L, Eeckels R, Eggermont E. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta. (1984) 144:245–47. doi: 10.1016/0009-8981(84)90059-7
85. Lefeber DJ, Morava E, Jaeken J. How to find and diagnose a CDG due to defective N-glycosylation. J Inherit Metab Dis. (2011) 34:849–52. doi: 10.1007/s10545-011-9370-0
86. Helander A, Eriksson G, Stibler H, Jeppsson JO. Interference of transferrin isoform types with carbohydrate-deficient transferrin quantification in the identification of alcohol abuse. Clin Chem. (2001) 47:1225–33. doi: 10.1093/clinchem/47.7.1225
87. Carchon HA, Chevigné R, Falmagne JB, Jaeken J. Diagnosis of congenital disorders of glycosylation by capillary zone electrophoresis of serum transferrin. Clin Chem. (2004) 50:101–11. doi: 10.1373/clinchem.2003.021568
88. Quintana E, Navarro-Sastre A, Hernández-Pérez JM, García-Villoria J, Montero R, Artuch R, et al. Screening for congenital disorders of glycosylation (CDG): transferrin HPLC versus isoelectric focusing (IEF). Clin Biochem. (2009) 42:408–15. doi: 10.1016/j.clinbiochem.2008.12.013
89. Babovic-Vuksanovic D, O'Brien JF. Laboratory diagnosis of congenital disorders of glycosylation type I by analysis of transferrin glycoforms. Mol Diagn Ther. (2007) 11:303–11. doi: 10.1007/BF03256251
90. Casetta B, Malvagia S, Funghini S, Martinelli D, Dionisi-Vici C, Barone R, et al. A new strategy implementing mass spectrometry in the diagnosis of congenital disorders of N-glycosylation (CDG). Clin Chem Lab Med. (2020) 59:165–71. doi: 10.1515/cclm-2020-0650
91. Messina A, Palmigiano A, Esposito F, Fiumara A, Bordugo A, Barone R, et al. For high throughput and isomeric N-glycan separation and characterization in Congenital Disorders Glycosylation and human diseases. Glycoconj J. (2021) 38:201–11. doi: 10.1007/s10719-020-09947-7
92. Abu Bakar N, Lefeber DJ, van Scherpenzeel M. Clinical glycomics for the diagnosis of congenital disorders of glycosylation. J Inherit Metab Dis. (2018) 41:499–513. doi: 10.1007/s10545-018-0144-9
93. Hipgrave Ederveen AL, de Haan N, Baerenfaenger M, Lefeber DJ, Wuhrer M. Dissecting total plasma and protein-specific glycosylation profiles in congenital disorders of glycosylation. Int J Mol Sci. (2020) 21:7635. doi: 10.3390/ijms21207635
94. Van Scherpenzeel M, Willems E, Lefeber DJ. Clinical diagnostics and therapy monitoring in the congenital disorders of glycosylation. Glycoconj J. (2016) 33:345–58. doi: 10.1007/s10719-015-9639-x
95. Bogdańska A, Kozłowski D, Pajdowska M, Lipiński P, Tylki-Szymańska A. Transferrin isoform analysis from dried blood spots and serum samples by gel isoelectric focusing for screening congenital disorders of glycosylation. Acta Biochim Pol. (2021) 68:139–42. doi: 10.18388/abp.2020_5576
96. Wolking AB, Park JH, Grüneberg M, Reunert J, Fingerhut R, Fobker M, et al. Transferrin glycosylation analysis from dried blood spot cards and capillary blood samples. J Chromatogr B Analyt Technol Biomed Life Sci. (2019) 1106–7:64–70. doi: 10.1016/j.jchromb.2019.01.004
97. Bogdańska A, Lipiński P, Szymańska-Rozek P, Jankowska I, Socha P, Tylki-Szymańska A. Pediatric liver disease patients and secondary glycosylation abnormalities. Front Pediatr. (2021) 8:613224. doi: 10.3389/fped.2020.613224
98. Jansen JC, van Hoek B, Metselaar HJ, van den Berg AP, Zijlstra F, Huijben K, et al. Screening for abnormal glycosylation in a cohort of adult liver disease patients. J Inherit Metab Dis. (2020) 43:1310–20. doi: 10.1002/jimd.12273
99. Lipiński P, Bogdańska A, Tylki-Szymańska A. Congenital disorders of glycosylation: Prevalence, incidence and mutational spectrum in the Polish population. Mol Genet Metab Rep. (2021) 27:100726. doi: 10.1016/j.ymgmr.2021.100726
100. Wopereis S, Morava E, Grunewald S, Adamowicz M, Huijben KM, Lefeber DJ, et al. Patients with unsolved congenital disorders of glycosylation type II can be subdivided in six distinct biochemical groups. Glycobiology. (2005) 15:1312–19. doi: 10.1093/glycob/cwj017
101. Brasil S, Pascoal C, Francisco R. Marques-da-Silva D, Andreotti G, Videira PA, et al. CDG therapies: from bench to bedside. Int J Mol Sci. (2018) 19:1304. doi: 10.3390/ijms19051304
102. Verheijen J, Tahata S, Kozicz T, Witters P, Morava E. Therapeutic approaches in Congenital Disorders of Glycosylation (CDG) involving N-linked glycosylation: an update. Genet Med. (2020) 22:268–79. doi: 10.1038/s41436-019-0647-2
103. Witters P, Cassiman D, Morava E. Nutritional therapies in congenital disorders of glycosylation (CDG). Nutrients. (2017) 9:E1222. doi: 10.3390/nu9111222
104. Gámez A, Serrano M, Gallego D, Vilas A, Pérez B. New and potential strategies for the treatment of PMM2-CDG. Biochim Biophys Acta Gen Subj. (2020) 1864:129686. doi: 10.1016/j.bbagen.2020.129686
105. Martínez-Monseny AF, Bolasell M, Callejón-Póo L, Cuadras D, Freniche V, Itzep DC, et al. AZATAX: Acetazolamide safety and efficacy in cerebellar syndrome in PMM2 congenital disorder of glycosylation (PMM2-CDG). Ann Neurol. (2019) 85:740–51. doi: 10.1002/ana.25457
106. Taday R, Grüneberg M, DuChesne I, Reunert J, Marquardt T. Dietary mannose supplementation in phosphomannomutase 2 deficiency (PMM2-CDG). Orphanet J Rare Dis. (2020) 15:258. doi: 10.1186/s13023-020-01528-z
107. Harms HK, Zimmer KP, Kurnik K, Bertele-Harms RM, Weidinger S, Reiter K. Oral mannose therapy persistently corrects the severe clinical symptoms and biochemical abnormalities of phosphomannose isomerase deficiency. Acta Paediatr. (2002) 91:1065–72. doi: 10.1111/j.1651-2227.2002.tb00101.x
108. Girard M, Douillard C, Debray D, Lacaille F, Schiff M, Vuillaumier-Barrot S, et al. Long term outcome of MPI-CDG patients on D-mannose therapy. J Inherit Metab Dis. (2020) 43:1360–69. doi: 10.1002/jimd.12289
109. Čechová A, Altassan R, Borgel D, Bruneel A, Correia J, Girard M, et al. Consensus guideline for the diagnosis and management of mannose phosphate isomerase-congenital disorder of glycosylation. J Inherit Metab Dis. (2020) 43:671–93. doi: 10.1002/jimd.12241
110. Mention K, Lacaille F, Valayannopoulos V, Romano S, Kuster A, Cretz M, et al. Development of liver disease despite mannose treatment in two patients with CDG-Ib. Mol Genet Metab. (2008) 93:40–3. doi: 10.1016/j.ymgme.2007.08.126
111. Jansen JC, Cirak S, van Scherpenzeel M, Timal S, Reunert J, Rust S, et al. CCDC115 deficiency causes a disorder of golgi homeostasis with abnormal protein glycosylation. Am J Hum Genet. (2016) 98:310–21. doi: 10.1016/j.ajhg.2015.12.010
112. Morelle W, Potelle S, Witters P, Wong S, Climer L, Lupashin V, et al. galactose supplementation in patients with TMEM165-CDG rescues the glycosylation defects. J Clin Endocrinol Metab. (2017) 102:1375–86. doi: 10.1210/jc.2016-3443
113. Dörre K, Olczak M, Wada Y, Sosicka P, Grüneberg M, Reunert J, et al. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. J Inherit Metab Dis. (2015) 38:931–40. doi: 10.1007/s10545-015-9828-6
114. Ng BG, Buckingham KJ, Raymond K, Kircher M, Turner EH, He M, et al. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. Am J Hum Genet. (2013) 92:632–6. doi: 10.1016/j.ajhg.2013.03.012
115. Hidalgo A, Ma S, Peired AJ, Weiss LA, Cunningham-Rundles C, Frenette PS. Insights into leukocyte adhesion deficiency type 2 from a novel mutation in the GDP-fucose transporter gene. Blood. (2003) 101:1705–12. doi: 10.1182/blood-2002-09-2840
116. Lühn K, Wild MK, Eckhardt M, Gerardy-Schahn R, Vestweber D. The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat Genet. (2001) 28:69–72. doi: 10.1038/ng0501-69
117. Helmus Y, Denecke J, Yakubenia S, Robinson P, Lühn K, Watson DL, et al. Leukocyte adhesion deficiency II patients with a dual defect of the GDP-fucose transporter. Blood. (2006) 107:3959–66. doi: 10.1182/blood-2005-08-3334
Keywords: congenital disorders of glycosylation, clinical presentation, isoelectric focusing of serum transferrin, next-generation sequencing, treatment
Citation: Lipiński P and Tylki-Szymańska A (2021) Congenital Disorders of Glycosylation: What Clinicians Need to Know? Front. Pediatr. 9:715151. doi: 10.3389/fped.2021.715151
Received: 26 May 2021; Accepted: 10 August 2021;
Published: 03 September 2021.
Edited by:
Huali Shen, Fudan University, ChinaReviewed by:
Yoshinao Wada, Osaka Women's and Children's Hospital, JapanCopyright © 2021 Lipiński and Tylki-Szymańska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patryk Lipiński, cC5saXBpbnNraUBpcGN6ZC5wbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.