 Yajie Su1,2
Yajie Su1,2 Huijun Wang
Huijun Wang Wenhao Zhou
Wenhao Zhou Long Li
Long Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 01 October 2021
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.713458
This article is part of the Research Topic NGS Technologies of Rare Diseases Diagnosis View all 34 articles
MEGDEL syndrome and SATB2-associated syndrome (SAS) are both rare congenital disorders with poor prognoses caused by gene mutations. We present the case of a 2-day-old girl with an unexplained abnormal liver function, feeding problem, and dystonia. Using next-generation sequencing, we identified two novel mutations in SERAC1 and a mutation in SATB2. Now, she is 15 months old and has the characteristics of SAS, such as downslanting palpebral fissures and delayed primary dentition. Besides the typical phenotypes of MEGDEL syndrome, such as hypertonia, failure to thrive, deafness, and motor regression, she has progressive cholestasis and is prone to high serum lactate after rehabilitation training and hypoglycemia with low ketone under starving conditions. These phenotypes substantially differ from the transient liver function abnormalities and hypoglycemia reported in the literature.
MEGDEL syndrome (3-methylglutaconic acidemia, deafness, encephalopathy, and Leigh-like syndrome) is a rare specific mitochondrial disorder due to mutations in the SERAC1 gene (NM_032861), which is an autosomal recessive inherited disorder (1). The SERAC1 gene encodes for a phosphatidylglycerol remodeler that is essential for mitochondrial functions and intracellular cholesterol trafficking (2). The SATB2-associated syndrome (SAS) is a recently described syndrome caused by mutaion of SATB2 gene (NM_015265). It's an autosomal dominantly inherited disorder which characterized by developmental delay with absent or limited speech, behavioral problems, dysmorphic features, and craniofacial abnormalities, including palatal and dental abnormalities (3).
We describe a rare disease progression and a severe phenotype of progressive cholestasis of a girl from birth to 15 months, who was compound heterozygous for SERAC1 of MEGDEL syndrome and had a mutation in SATB2 of SATB2-associated syndrome.
A female infant was born at 38+ 5/7 weeks gestational age to a 38-year-old mother who had gestational anemia, which was supplemented with oral iron, and gestational diabetes, which was controlled with diet. The mother has a healthy 6-year-old girl but also had a spontaneous abortion and an artificial abortion due to embryo death. The mother's brother had a history of epilepsy and had passed away.
The infant's birth weight, head circumference, and length were 2,830 g (10–25th percentile), 33 cm (25–50th percentile), and 48 cm (10–25th percentile), respectively, with a normal Apgar score. However, she had mild blepharoptosis and there was a 3 cm cleft palate from the palate to the uvula. On day of life (DOL) 2, the infant vomited milk twice accompanied by weakness in sucking. A few hours later, she went into convulsions that involved rowing movements of the limbs. Her blood glucose was 0.7 mmol/L and blood gas analysis revealed anion gap metabolic acidosis (24.69 mmol/L). After providing glucose (8 mg/kg/min) and phenobarbital (10 mg/kg) injection treatment, the convulsions did not occur again, but she became somnolent, developed an unexplained abnormal liver function, and an aggravating increase in serum lactate (16.4 mmol/L). The infant was immediately transferred to our tertiary neonatal intensive care unit (NICU) center on DOL 3 for further newborn care.
The infant's serum lactate was normal on DOL 4 after giving intravenous fluids and nasogastric feeding. Blood glucose was normal on DOL 6 without intravenous fluids. The problems during hospitalization were poor neonatal sucking, reduced movements, and unexplained elevated liver enzymes (gamma-glutamyltransferase (GGT) 1,373 mmol/L, total bile acids (TBA) 58.2 μmol/L, aspartate aminotransferase (AST) 132 U/L,prothrombin time (PT) 46.1s, albumin 26.57 g/L) with normal stool color and abdominal ultrasound. In addition, magnetic resonance imaging (MRI) showed high signals on T2 and FLAIR, where the lenticular nucleus was swollen on both sides. There were multiple hyperintensity patches on the dorsal side of the brainstem and posterior limb of the internal capsule near the anterior and posterior horns of the right lateral ventricle (Figure 1). Evoked otoacoustic emission test was not passed on both ears.

Figure 1. Brain MRI at 10 days and 3 months of age. (A) T2W: Red boxes indicate a high signal of basal ganglia, swelling of bilateral lenticular nucleus; Red arrows indicate an abnormal signal point of posterior branch of right internal capsule. (B) T2W: Red boxes indicate white matter of occipital lobe has high signal, and the boundary of gray and white matter is not clear.
Blood tandem mass spectrometry (MS/MS) showed an increase in tyrosine 227.85 μM (25–225 μM). Urine gas chromatography-mass spectrometry (GC/MS) showed an increase in oxalate 57.78 (0–33), 3-methylglutaconic aciduria 4.35 (0–1), and 3-(4-hydroxyphenyl) lactic acid 1122.49 (0–20). Next-generation sequencing and Sanger sequencing detected two compound heterozygotes of SERAC1 (c.1502-1G>C(likely pathogenic) from the maternal side, c.227-228dupAT(pathogenic) from the paternal side) and a de novo variant of SATB2 (c.1166G>A) (likely pathogenic) (Figure 2). The quality control of WES data analysis and procedure of variant annotation refers to pipeline version 2 of Children's Hospital of Fudan University (4). We classified the pathogenicity of the identified variants according to the ACMG guideline, the details showed in (Supplementary Table 1).
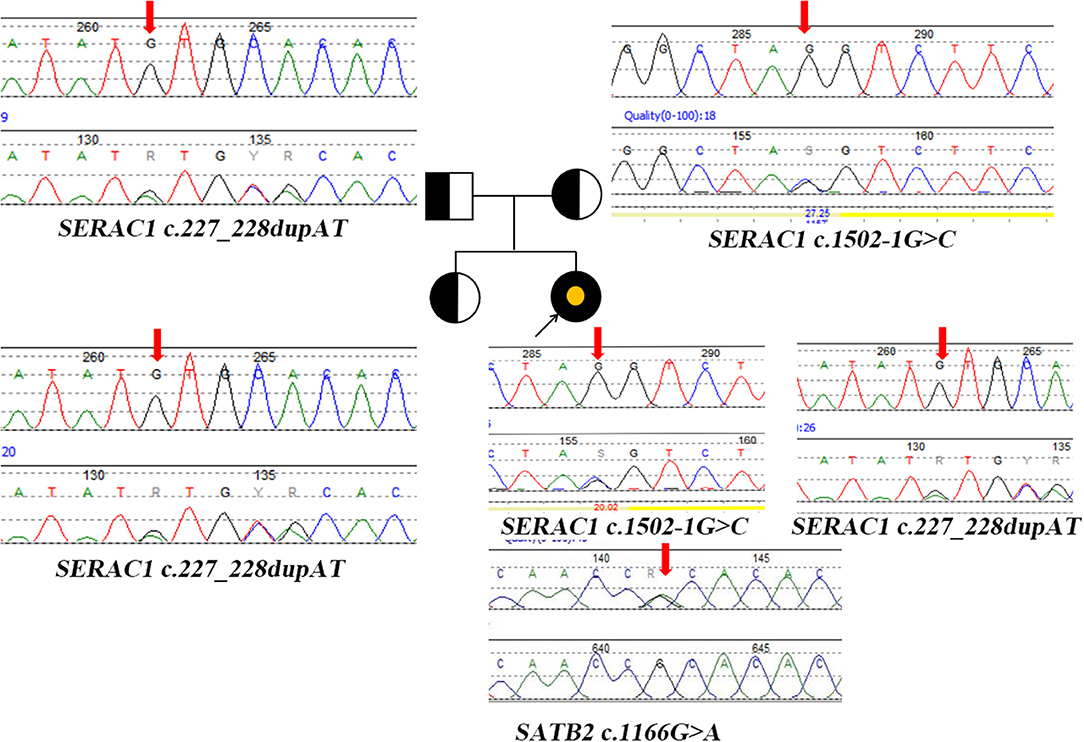
Figure 2. Pedigree diagram and genetic characterization of the family with SERAC1 and SATB2. Parents and sisters are heterozygous carriers of the SERAC1 gene; The yellow point represents the mutation of SATB2 gene. Heterozygous mutation (c.227-228dupAT) in SERAC1 was found in the patient, her father and her healthy sister. Heterozygous mutation (c.1502-1G>C) in SERAC1 was found in the patient and her mother. Heterozygous mutation (c.1166G>A) in SATB2 was only found in the patient; Red arrow indicate location of mutation.
On DOL 17, the infant was discharged from hospital with a nasogastric tube. After symptomatic treatment, the high GGT at admission had dropped to 264 mmol/L and the PT returned to normal (16.2 s). She experienced slow weight gain and increasing TBA, which was unmanageable by drugs after discharge. When she was 3 months old, there was a continuous mild increase in methionine (Met) for 2 months. After providing Met-free formula milk, the level of Met reduced to normal, and her motor development improved. When she was 12 months old, she had a high fever of 39°C. Afterward, her gross and fine motor regressed to the level of 5 months. Now, she is 15 months old with four primary teeth and has completed a cleft palate repair surgery in order to improve her nutritional status. When she was discharged from the hospital in the first time, the GGT were maintained between 430 and 470 mmol/L, the AST were maintained between 62 and 87 U/L and albumin were maintained between 31.4 and 36.9 g/L. In addition, PT (15.1 s) and international normalized ratio (1.31) was slightly elevated at 12 months of age. Only bile acids were continuously increasing. Because she had normal stool color, normal skin color, and normal abdominal ultrasound, we excluded the extrahepatic biliary atresia and choledochal cyst and did not perform liver biopsy. She is susceptible to higher serum lactate after rehabilitation training and hypoglycemia with low ketone under starving conditions (Table 1).
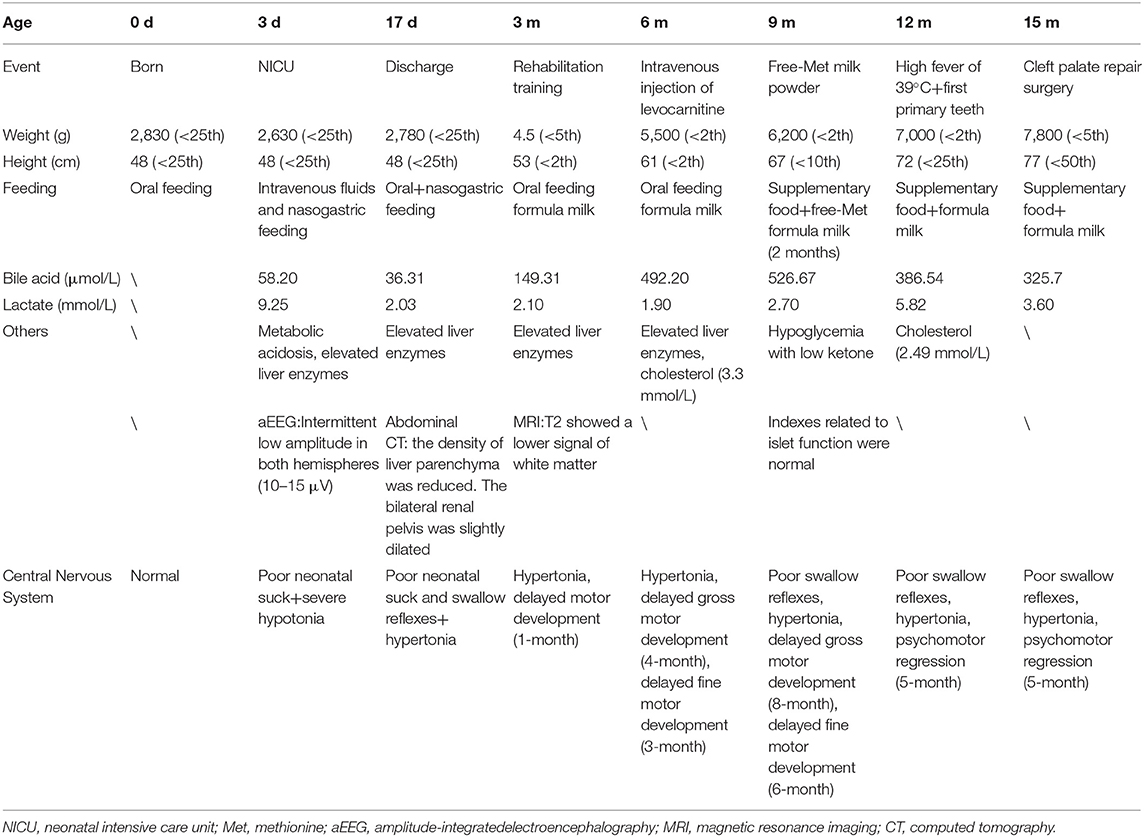
Table 1. Follow-up of the infant from birth to 15 months of age.
When a newborn demonstrates an unexplained feeding problem and a high anion gap metabolic acidosis, doctors will change the direction of diagnosis and treatment to inherited metabolic diseases, especially because in this case, the infant had dystonia, an abnormal MRI, and increased liver enzymes and serum lactate. Even though the MS/MS presented an increased level of 3-methylglutaconic aciduria, we still cannot present a diagnosis because of the various phenotypes present in the neonatal period.
The infant carries two variants of SERAC1 (c.1502-1G>C from the maternal side, c.227-228dupAT from the paternal side), neither of which have been reported before (http://www.hgmd.cf.ac.uk). She has the typical phenotypes of 3-methylglutaconic acidemia, deafness, and Leigh-like syndrome (present with bilateral basal ganglia lesions on brain MRI) (5). She still has progressive cholestasis and is susceptible to high serum lactate after rehabilitation training and hypoglycemia with low ketone under starving conditions. These phenotypes are substantially different from the transient liver function abnormalities and hypoglycemia reported in the literature (6).
As we know, SERAC1 encodes for a phosphatidylglycerol remodeler that is essential for mitochondrial functions and intracellular cholesterol trafficking. Although there is no medicine that can completely treat the disease. When the diagnosis is clear, we should give baclofen (10 mg, bid), trihexyphenidyl hydrochloride (2 mg, bid) to improve her hypertonia and coenzyme Q10 (30 mg/d), L-carnitine(1 g,bid), vitamin B1 (100 mg/d), and vitamin E (100 mg/d) to improve mitochondrial functions. Due to the influence of pandemic of COVID-19, she didn't go to the hospital in time and was not treated until 5 months after birth. But her parents thought there was not obvious effect after treatment and did not adhere to the standard treatment. Here, we try to explain its function through the metabolic pathways (Figure 3). On one hand, mutations in SERAC1 cause leucine degradation barriers, which, in turn, cause an increase of 3-methylglutaconic aciduria; on the other hand, under glucose deficiency or starvation, fatty acids are oxidized in the liver to produce ketone bodies as energy for the brain. In the process of amino acid metabolism, fatty acid metabolism, and glycolysis, mitochondria are required to participate in the key production of Acetyl-CoA (7). Therefore, low ketone hypoglycemia not only occurs when fatty acid metabolism is abnormal (8) but also when mitochondrial function is abnormal. In the cholesterol metabolism pathway, impaired SERAC1 activity leads to deficiency of PG36:1 and increases free cholesterol (9), which is the main substance for the synthesis of bile acids.
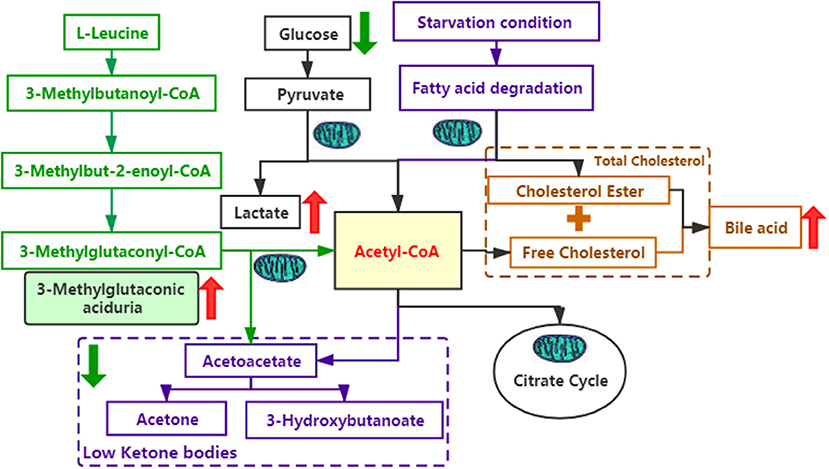
Figure 3. Several metabolic pathways involved in this case. The green font represents the leucine metabolic pathway; the purple font represents the fatty acid metabolic pathway; the black font represents the glucose metabolic pathway; and the yellow font represents the cholesterol metabolic pathway. The red arrows indicate an increase, whereas the green arrows indicate a decrease.
Many syndromes have the phenotype of developmental delay. We were unable to complete the intelligence test because of the patient's deafness and we could not distinguish whether the delayed psychomotor development was caused by mutations in SERAC1 or SATB2 or a combination of the two genes. However, the characteristics of downslanting palpebral fissures, cleft palate, and delayed primary dentition are consistent with phenotypes of SAS because SATB2 is expressed in upper-layer neurons, neural crest progenitors of the jaw, osteoblasts, odontoblasts, and other dental progenitor cells (10). We listed the phenotypes of the two diseases in Supplementary Table 2.
We followed-up the case for 15 months and presented a complete process of disease development. These findings extended the phenotype spectrum of the two diseases and provided new insights into such rare diseases. Although medicine of muscle relaxant and antioxidant play roles in symptomatic treatment, the child was not given timely and standardized treatment, which might aggravate her delayed motor development. In addition, MEGDEL syndrome has the phenotype of cholestasis, but we cannot determine whether it is a severe phenotype of MEGDEL syndrome or the superposition of the two rare syndromes.
We identified two novel variants of SERAC1 and a de novo variant of SATB2 in a newborn. The case showed typical and specific phenotypes of these two rare diseases, especially the progressive cholestasis and high serum lactate after rehabilitation training and hypoglycemia with low ketone under starving conditions. The article presented a serious phenotype and disease progression, and summarized the metabolic pathways of abnormal indicators in the infant. We hope to enlighten our pediatric colleagues by providing more information on such rare diseases.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by The People's Hospital of Xinjiang Uygur Autonomous Region (#2017008). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
YS and HZ conceptualized and designed the study, drafted the initial manuscript, and reviewed and revised the manuscript. HW, JY, and BW collected data, carried out the initial analyses, and reviewed and revised the manuscript. LL and WZ designed the data collection instruments, coordinated and supervised data collection, and critically reviewed the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Thank you to the family of the patient to provide the information.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.713458/full#supplementary-material
1. Finsterer J, Scorza FA, Fiorini AC, Scorza CA. MEGDEL syndrome. Pediatr Neurol. (2020) 110:25–9. doi: 10.1016/j.pediatrneurol.2020.03.009
2. Wortmann SB, Vaz FM, Gardeitchik T, Vissers LE, Renkema GH, Schuurs-Hoeijmakers JH, et al. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat Genet. (2012) 44:797–802. doi: 10.1038/ng.2325
3. Zarate YA, Fish JL. SATB2-associated syndrome: mechanisms, phenotype, and practical recommendations. Am J Med Genet A. (2017) 173:327–37. doi: 10.1002/ajmg.a.38022
4. Lin Y, Xinran D, Xiaomin P, Xiang C, Bingbing W, Huijun W, et al. Evaluation of turn around time and diagnostic accuracy of the next generation sequencing data analysis pipeline version 2 of Children's Hospital of Fudan University. Chinese J Evidence-Based Pediatr. (2018) 2:118–23. doi: 10.3969/j.issn.1673-5501.2018.02.008
5. Gerards M, Sallevelt SC, Smeets HJ. Leigh syndrome: resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol Genet Metab. (2016) 117:300–12. doi: 10.1016/j.ymgme.2015.12.004
6. Wortmann SB, de Brouwer APM, Wevers RA, Morava E. SERAC1 deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al., editors. GeneReviews(®). Seattle, WA: University of Washington; Initial Posting (2020).
7. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. (2015) 21:805–21. doi: 10.1016/j.cmet.2015.05.014
8. Talley JT, Mohiuddin SS. Biochemistry, Fatty Acid Oxidation. StatPearls. Treasure Island, FL: StatPearls Publishing (2021).
9. Roeben B, Schüle R, Ruf S, Bender B, Alhaddad B, Benkert T, et al. SERAC1 deficiency causes complicated HSP: evidence from a novel splice mutation in a large family. J Med Genet. (2018) 55:39–47. doi: 10.1136/jmedgenet-2017-104622
Keywords: MEGDEL syndrome, SATB2-associated syndrome, phenotype, next-generation sequencing, follow-up
Citation: Su Y, Zhang H, Wang H, Wu B, Yang J, Zhou W and Li L (2021) Case Report: Progressive Cholestasis: Severe Phenotype of MEGDEL Syndrome With SATB2-Associated Syndrome. Front. Pediatr. 9:713458. doi: 10.3389/fped.2021.713458
Received: 23 May 2021; Accepted: 03 September 2021;
Published: 01 October 2021.
Edited by:
Emiliano González Vioque, University Clinical Hospital of Santiago, SpainReviewed by:
Nida Mirza, Sri Aurobindo Institute of Medical Science, IndiaCopyright © 2021 Su, Zhang, Wang, Wu, Yang, Zhou and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenhao Zhou, emhvdXdlbmhhb0BmdWRhbi5lZHUuY24=; Long Li, bGlsb25nNjVAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.