 Haiyan Yang
Haiyan Yang Fang Cai2
Fang Cai2 Siyi Gan
Siyi Gan Liwen Wu
Liwen Wu- 1Department of Neurology, Hunan Children's Hospital, Changsha, China
- 2Department of Neurology, Chenzhou No. 1 People's Hospital, Chenzhou, China
- 3Department of Pediatrics, Xiangya Hospital, Central South University, Changsha, China
ISPD gene mutation-related diseases have high clinical and genetic heterogeneity, and no studies have yet reported any effective treatments. We describe six patients with dystroglycanopathies caused by ISPD gene mutations and analyze their genotypes and phenotypes to explore possible effective treatments. Our results confirm that the phenotype of limb-girdle muscular dystrophies can be easily misdiagnosed as Duchenne muscular dystrophy and that exon deletions of ISPD gene are relatively common. Moreover, low-dose prednisone therapy can improve patients' exercise ability and prolong survival and may be a promising new avenue for ISPD therapy.
Background
ISPD (MIM 614631, also named CDP-L-ribitol pyrophosphorylase A, CRPPA) gene, which maps to chromosome 7p21, has been associated with the loss of α-dystroglycan (α-DG) glycosylation (1–3). Defects in the post-translational modification of α-DG have been implicated in clinically distinct dystroglycanopathies that present as congenital muscular dystrophies with multisystem involvement, Walker–Warburg syndrome (MIM 614643), limb-girdle muscular dystrophies (LGMDs) (MIM 616052), or a spectrum of intermediate phenotypes (4). ISPD gene mutation-related diseases have high clinical and genetic heterogeneity (5). At present, there has been a lack of effective treatment for ISPD gene mutation-related diseases, and no studies have yet reported a possible effective treatment. Here, we report six patients with dystroglycanopathies caused by ISPD gene mutations and analyze their genotypes and phenotypes to explore possible effective treatments.
Case Presentation
Two siblings (case 1 and case 2) were reported in this family. The younger brother (case 1) was a 9-year-old male child who presented with a history of progressive weakness of the lower limbs since age 1.5 years. The results of laboratory testing revealed elevated creatine kinase (CK) (13,043.5 U/L) and elevated lactic acid (2.30 mmol/L) levels. Muscle biopsy showed myodystrophy changes. Muscle MRI showed different degrees of fatty infiltration with partial inflammation edema in each muscle group of the thigh (Figures 1A–D). The initial clinical diagnosis was considered Duchenne muscular dystrophy (DMD). The child has received prednisone (0.75 mg/kg/day) treatment since the age of 8, and the symptoms of muscle weakness have improved, along with a longer walking duration and less wrestling. After genetic diagnosis, the younger brother gradually stopped prednisone treatment plan. However, after prednisone reduction, the patient's motor function decreased, and the wrestling increased. The symptoms improved again when the child resumed prednisone therapy. The older sister (case 2) presented with a history of progressive weakness after birth and never gained the ability to walk independently. At the age of 3, a muscle biopsy showed myodystrophy changes; she did not receive special treatment and died at the age of 16.
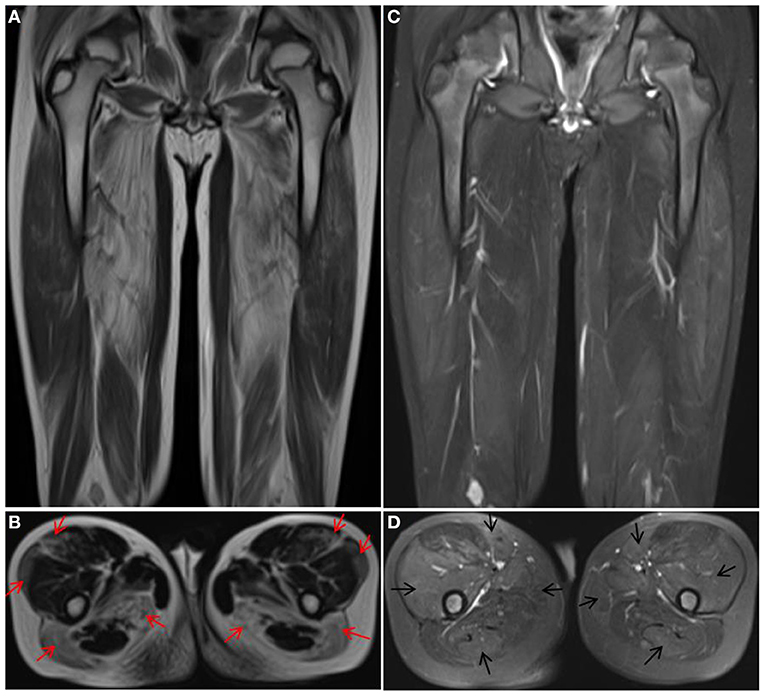
Figure 1. Muscle MRI of case 1. T1 (A,B) showed obvious fat infiltration in thigh muscles, most notably in the vastus lateralis, rectus femoris, biceps femoris, and semimembranosus muscles (red arrow), but the fat infiltration in the vastus intermedius, sartorius, gracilis, and semitendinosus muscles was mild. T2 (C,D) showed obvious inflammation edema in the muscles with mild fat infiltration (black arrow).
Case 3 was a 16-year-old male patient who presented with a history of progressive weakness of the lower limbs from childhood. The results of laboratory testing revealed elevated CK levels (7,431.5 U/L), and muscle biopsy showed myodystrophy changes. The initial clinical diagnosis was considered DMD. The patient has received prednisone treatment (20 mg, gradually increased to 35 mg, Qd, 10-day regimen followed by a pause for 20 days) since the age of 12, and the symptoms of muscle weakness have improved. The child has no obvious Gowers' sign and can jump 2–3 cm off the ground with both feet but can barely jump off the ground with one foot.
Case 4 was a 10-year-old male child who presented with a history of progressive weakness of the lower limbs since age 1.3 years and the loss of ambulation at age 10. The results of laboratory testing revealed elevated CK (6,329.6 U/L). Muscle biopsy showed myodystrophy changes. The child was not treated with any medication.
Two sisters were also identified in this family. The proband (case 5) was 5 years old, was female, and had normal development, but she had slight weakness when climbing and running. Her CK level was between 2,400 and 5,600 U/L. She had a 3-year-old sister (case 6) with the same motor development features who suffered from retinoblastoma, and her CK was between 1,000 and 8,800 U/L. Their mother was 32 years old, presenting with intolerance of movement and an elevated CK level of 600–4,500 U/L. Muscle biopsy of the proband revealed atrophy in parts of the muscle fibers, mild hyperplasia of connective tissue, negative dystrophin-N staining in most muscle fibers, and decreased expression of dystrophin-C and dystrophin-R. Except for the younger sister of the proband, who underwent ophthalmectomy, the proband was not treated with any medication.
Methods
Written informed consent was obtained from the patients and their parents. This study was approved by the Medical Ethics Committee of Hunan Children's Hospital.
Genetic testing included the use of the multiplex ligation-dependent probe amplification (MLPA) method to screen for DMD gene duplication and deletion and next-generation sequencing methods to screen for other genes that might cause elevated CK and muscle weakness. All experiments and analyses were performed according to our previous research methods (6).
Sequence variants were annotated using population and literature databases, including 1,000 Genomes, dbSNP, GnomAD, Clinvar, HGMD, and OMIM. Variant interpretation was performed according to the American College of Medical Genetics and Genomics (ACMG) guidelines (7).
Results
The detection for DMD gene from peripheral blood of the siblings (case 1 and case 2) was negative, including duplication and deletions, point mutations, and intron region mutations. Moreover, the possible RNA mutations of DMD gene were negative by detecting the muscle tissue of the siblings. These results were puzzling. Therefore, in September 2020, we reanalyzed the previous second-generation sequencing data and found that the siblings (case 1 and case 2) had ISPD c.1114_1116delGTT (p.V372del) mutation and exon 6–9 deletion (Figures 2A,B). The siblings (case 1 and case 2) were eventually diagnosed with LGMDs. The DMD gene duplication deletion, point mutation, and intron mutation test results of case 3 were all negative. Further next-generation sequencing revealed ISPD c.1114_1116delGTT (p.V372del) mutation, and the patient was eventually diagnosed as LGMDs. Case 4 had ISPD c.1114_1116delGTT (p.V372del) and POMT1 c.979G>A (p.V327I) mutations. The sisters (case 5 and case 6) had ISPD c.984G>T (p.Q328H) and c.550C>T (p.R184X) compound heterozygous mutations and DMD c.130dupC mutation. Further details can be found in Table 1.
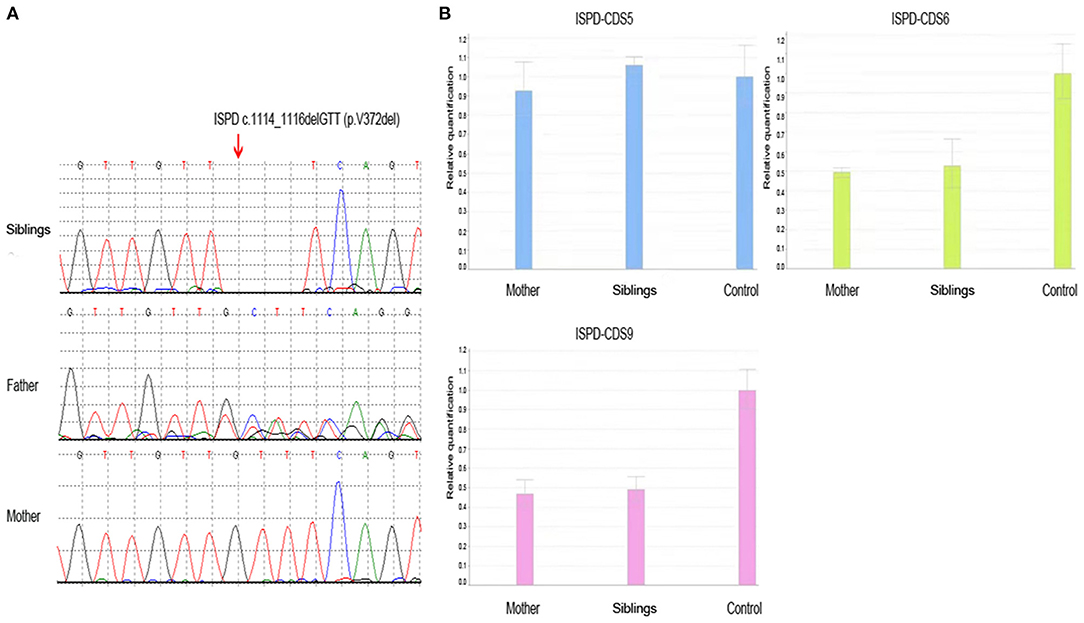
Figure 2. The result of whole-exome sequencing of the siblings. (A) The siblings had ISPD c.1114_1116delGTT (p.V372del) mutation, which was inherited from the father, while the mother did not carry the mutation. (B) The siblings had ISPD exon 6–9 deletion, which was inherited from the mother.
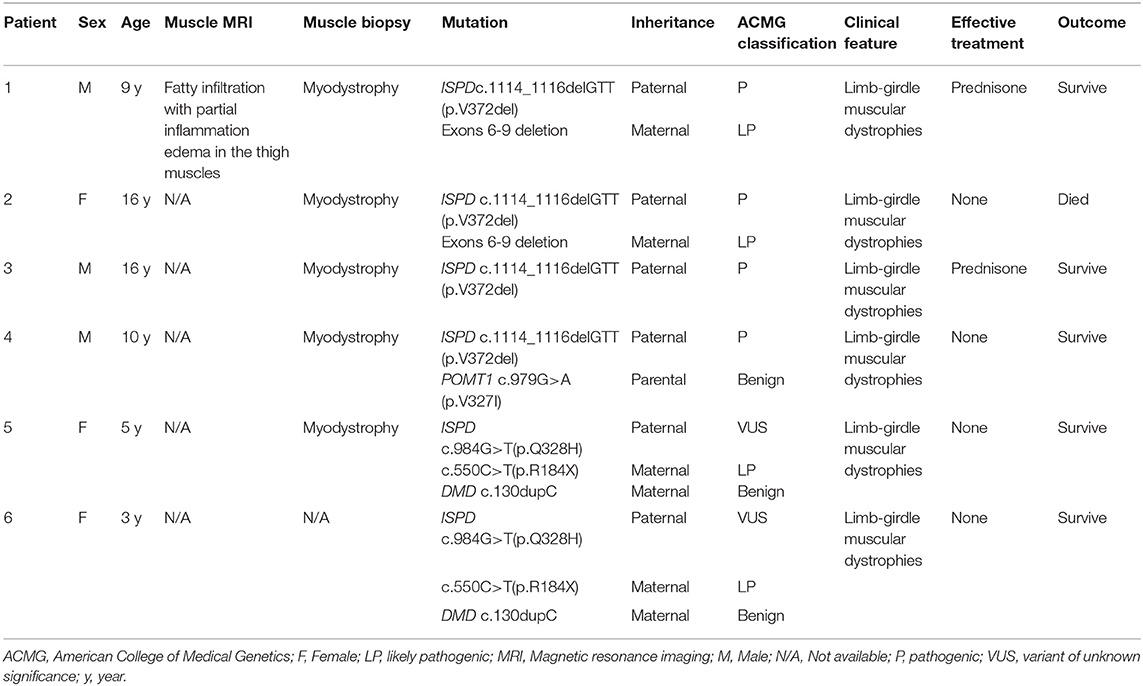
Table 1. Genotype and phenotype analysis of the 6 patients with ISPD mutation.
Discussion
Relevant case reports from PubMed database since its establishment until December 2020 were searched with ISPD gene as the key word, and the variants and clinical phenotypes of ISPD gene were summarized. Currently, a total of 58 patients with ISPD gene mutations have been reported in nine literatures (2–5, 8–12) (Table 2). The reported clinical phenotypes include intrauterine growth restriction, gastroschisis, Walker–Warburg syndrome, LGMDs, cobblestone lissencephaly, and congenital muscular dystrophy with developmental delay. For LGMDs due to ISPD mutations, onset occurs in late childhood, adolescence, or adulthood (8). The main clinical manifestations of LGMDs include proximal muscle involvement of the upper and lower extremities, elevated CK, gastrocnemius hypertrophy, and Gowers' sign (13). Muscle biopsy and muscle MRI revealed changes indicative of myodystrophy, which is difficult to distinguish from DMD; and thus, these cases can be easily misdiagnosed (14).
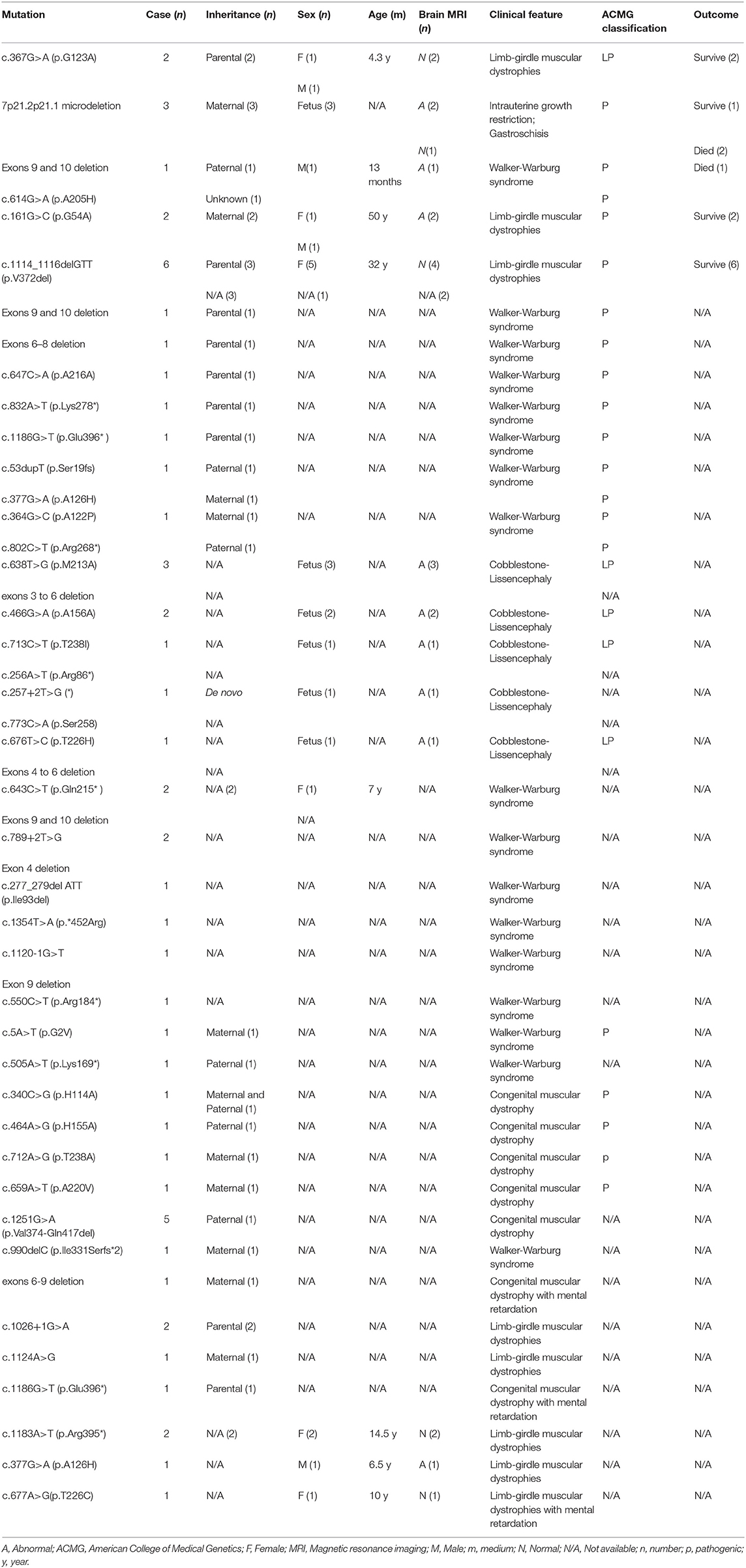
Table 2. Genotype and phenotype analysis of the ISPD gene reported in literature.
At present, 48 variants of ISPD have been reported, of which point mutations account for 70.8% (34/48) and exon deletions account for 29.2% (14/48) (2–5, 8–12) (Table 2). Collectively, the literature and our experience with these patients indicate that exon deletion of ISPD gene is common. These microdeletions can be clinically difficult to detect by conventional second-generation sequencing methods. Therefore, for patients with LGMDs clinically excluded from DMD, in addition to routine screening for ISPD point mutations, we must carefully consider the possibility of ISPD exon deletion, which should be investigated during data analysis.
Among our patients, ISPD mutations were found to coexist with gene mutations of another myopathy. Compared with the other two male patients in this cohort who had only ISPD gene mutations, the male patient who had ISPD and POMT1 gene mutations developed muscle weakness more quickly and lost walking ability at an earlier age. In the sisters with ISPD and DMD gene mutations, the degree of CK elevation was higher than that of their mother without synergistic ISPD pathogenic genes, and the symptoms of exercise intolerance appeared at an earlier age than their mother's. Therefore, whether ISPD mutation is synergistic with DMD or POMT1 gene mutation should be carefully considered. These conditions increase the difficulty and complexity of prenatal diagnosis, so clinicians must be vigilant.
In our study, two patients (case 1 and case 3) were clinically diagnosed with DMD before genetic diagnosis and were offered the currently recommended low-dose prednisone therapy for DMD (15), which was unexpectedly found to be effective in improving the patient's motor function, prolonging the walking time and reducing the frequency of wrestling. The motor function of the two patients was improved compared with that before and after treatment and that of untreated case 4. After the genetic diagnosis, we also attempted to reduce and discontinue the hormone treatment for case 1. After the hormone treatment was discontinued, the patient's muscle weakness returned, and the frequency of wrestling increased significantly. After the hormone treatment was resumed, the symptoms improved again. Muscle MRI in case 1 showed significant inflammation edema changes before muscle atrophy, which further suggested that prednisone therapy could alleviate symptoms after improving inflammation edema.
In conclusion, the genotypes and phenotypes of ISPD gene mutations represent a wide spectrum, and the phenotype of LGMDs is easily misdiagnosed as DMD. Exon deletions of ISPD gene are relatively common, so caution must be taken in the selection of gene testing methods and data analysis to avoid false-negative results. ISPD and other gene mutations coexist in some patients, so the possibility of synergistic pathogenesis needs to be confirmed by further large sample studies and functional verification. Our findings indicate that low-dose hormone therapy can improve patients' exercise ability and prolong survival and may be a promising new avenue for ISPD therapy.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of Hunan Children's Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
HY conducted the literature review and drafted the manuscript. FC, HL, SG, and TX made substantial contributions to the conception and interpretation of data. LW was responsible for revising the manuscript critically and has given final approval of the version to be published. All authors read and approved the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81671297). This work was supported by Provincial Technology Innovation Guidance Program Clinical Medical Technology Innovation Guidance (2017SK50704).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We wish to thank the patients and their families for the participation in this study.
References
1. Cirak S, Herrmann R, Ruthland P, Brokmann K, Korinthenberg R, Nurunberg P, et al. P097 A new locus for an autosomal recessive congenital muscular dystrophy without brain involvement maps to chromosome 7p21. Eur J Paediatr Neurol. (2009) 13(Suppl. 1):S51. doi: 10.1016/S1090-3798(09)70155-2
2. Willer T, Lee H, Lommel M, Yoshida-Moriguchi T, de Bernabe DB, Venzke D, et al. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat Genet. (2012) 44:575–80. doi: 10.1038/ng.2252
3. Roscioli T, Kamsteeg EJ, Buysse K, Maystadt I, van Reeuwijk J, van den Elzen C, et al. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat Genet. (2012) 44:581–5. doi: 10.1038/ng.2253
4. Tasca G, Moro F, Aiello C, Cassandrini D, Fiorillo C, Bertini E, et al. Limb-girdle muscular dystrophy with alpha-dystroglycan deficiency and mutations in the ISPD gene. Neurology. (2013) 80:963–5. doi: 10.1212/WNL.0b013e3182840cbc
5. Cirak S, Foley AR, Herrmann R, Willer T, Yau S, Stevens E, et al. ISPD gene mutations are a common cause of congenital and limb-girdle muscular dystrophies. Brain. (2013) 136:269–81. doi: 10.1093/brain/aws312
6. Yang H, Yin F, Gan S, Pan Z, Xiao T, Kessi M, et al. The study of genetic susceptibility and mitochondrial dysfunction in mesial temporal lobe epilepsy. Mol Neurobiol. (2020) 57:3920–30. doi: 10.1007/s12035-020-01993-4
7. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
8. Song D, Fu X, Ge L, Chang X, Wei C, Liu J, et al. A splice site mutation c.1251G>A of ISPD gene is a common cause of congenital muscular dystrophy in Chinese patients. Clin Genet. (2020) 97:789–90. doi: 10.1111/cge.13695
9. Vuillaumier-Barrot S, Bouchet-Seraphin C, Chelbi M, Devisme L, Quentin S, Gazal S, et al. Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly. Am J Hum Genet. (2012) 91:1135–43. doi: 10.1016/j.ajhg.2012.10.009
10. Czeschik JC, Hehr U, Hartmann B, Lüdecke HJ, Rosenbaum T, Schweiger B, et al. 160 kb deletion in ISPD unmasking a recessive mutation in a patient with Walker-Warburg syndrome. Eur J Med Genet. (2013) 56:689–94. doi: 10.1016/j.ejmg.2013.09.014
11. Baranello G, Saredi S, Sansanelli S, Savadori P, Canioni E, Chipparini L, et al. A novel homozygous ISPD gene mutation causing phenotype variability in a consanguineous family. Neuromuscul Disord. (2015) 25:55–9. doi: 10.1016/j.nmd.2014.08.007
12. Trkova M, Krutilkova V, Smetanova D, Becvarova V, Hlavova E, Jencikova N, et al. ISPD gene homozygous deletion identified by SNP array confirms prenatal manifestation of Walker-Warburg syndrome. Eur J Med Genet. (2015) 58:372–5. doi: 10.1016/j.ejmg.2015.05.004
13. Sparks SE, Quijano-Roy S, Harper A, Rutkowski A, Gordon E, Hoffman EP, et al. Congenital muscular dystrophy overview - retired chapter, for historical reference only. (1993).
14. Alcantara-Ortigoza MA, Reyna-Fabian ME, Gonzalez-Del Angel A, Estandia-Ortega B, Bermúdez-López C, Cruz-Miranda GM, et al. Predominance of dystrophinopathy genotypes in mexican male patients presenting as muscular dystrophy with a normal multiplex polymerase chain reaction DMD gene result: a study including targeted next-generation sequencing. Genes (Basel). (2019) 10:856. doi: 10.3390/genes10110856
15. Vazquez-Cardenas NA, Ibarra-Hernandez F, Lopez-Hernandez LB, Escobar-Cedillo RE, Ruano-Calderón LA, Gómez-Díaz B, et al. Diagnosis and treatment with steroids for patients with Duchenne muscular dystrophy: experience and recommendations for Mexico. Administracion del Patrimonio de la Beneficencia Publica Asociacion de Distrofia Muscular de Occidente. Rev Neurol. (2013) 57:455–62. doi: 10.33588/rn.5710.2013380
Keywords: ISPD gene, dystroglycanopathies, limb-girdle muscular dystrophies, Duchenne muscular dystrophy, pediatric
Citation: Yang H, Cai F, Liao H, Gan S, Xiao T and Wu L (2021) Case Report: ISPD Gene Mutation Leads to Dystroglycanopathies: Genotypic Phenotype Analysis and Treatment Exploration. Front. Pediatr. 9:710553. doi: 10.3389/fped.2021.710553
Received: 16 May 2021; Accepted: 21 July 2021;
Published: 18 August 2021.
Edited by:
Juergen Brunner, Innsbruck Medical University, AustriaReviewed by:
Stephanie Efthymiou, University College London, United KingdomAleksandra Jezela-Stanek, National Institute of Tuberculosis and Lung Diseases, Poland
Copyright © 2021 Yang, Cai, Liao, Gan, Xiao and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liwen Wu, d3VsaXdlbjIwMjBAc2luYS5jb20=