
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 06 October 2021
Sec. Pediatric Immunology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.703056
This article is part of the Research Topic Disorders Related to PI3K Hyper-activation View all 7 articles
 Antonio Marzollo1,2*
Antonio Marzollo1,2* Silvia Bresolin1,3
Silvia Bresolin1,3 Davide Colavito4
Davide Colavito4 Alice Cani3Paola Gaio5Luca Bosa5Claudia Mescoli6
Alice Cani3Paola Gaio5Luca Bosa5Claudia Mescoli6 Linda Rossini1
Linda Rossini1 Federica Barzaghi7
Federica Barzaghi7 Giorgio Perilongo5Alberta Leon4Alessandra Biffi1
Giorgio Perilongo5Alberta Leon4Alessandra Biffi1 Mara Cananzi5
Mara Cananzi5Nodular lymphoid hyperplasia (NLH) is a lymphoproliferative disease caused by non-clonal expansion of lymphoid cells in the gut mucosa. Little is known about the pathogenesis of NLH, which is often disregarded as an insignificant or para-physiologic phenomenon. We present the case of a girl with isolated diffuse NLH (extending from the stomach to the rectum) caused by activated PI3Kδ syndrome (APDS) due to the novel p.Glu525Gly variant in PIK3CD. The gain-of-function effect of the variant was confirmed by demonstration of over activation of the Akt/mTOR pathway in the patient's cells. APDS diagnosis led to treatment with sirolimus, which resulted in the complete remission of NLH and in the prevention of extra intestinal complications. In conclusion, we identify APDS as a novel cause of isolated NLH and suggest that patients with severe pan-enteric NLH should be screened for this disorder that may not be apparent on first-line immunological testing.
Lymphoid follicles are part of the gut-associated lymphoid tissue (GALT) and can be normally found in the mucosal and submucosal layers of the gastrointestinal tract, where they are mainly involved in immune surveillance and mucosal repair (1). Physiologically, they are predominantly located in the terminal ileum and in the anorectal region, while they are barely represented in the stomach and in the duodenum (1).
Intestinal nodular lymphoid hyperplasia (NLH) is a benign lymphoproliferative disease characterized by a diffuse or focal hyperplasia of lymphoid follicles along the intestine due to an accumulation of nonmalignant lymphoid cells in the gut mucosa (2). Upon endoscopy, NLH is defined as a cluster of ≥10 extruding lymphoid nodules, each at least 2 mm in diameter (3). It is mainly observed in the small intestine, less commonly in the large intestine, and rarely in the stomach or in the duodenum. Histologically, NLH is characterized by the presence of polymorphous hyperplastic lymphoid follicles with highly active germinal centers and well-defined lymphocyte mantles and is confined to the lamina propria and/or the superficial submucosa (2, 4). The exact epidemiology of NLH is unknown as published literature mainly includes case reports and small series of patients. NLH has been observed at any age, but it has most frequently been reported during childhood (4). Clinical manifestations include diarrhea, malabsorption, gastrointestinal bleeding, and abdominal pain, but many affected patients may be asymptomatic, and NLH can be an incidental finding (5, 6). Despite NLH being a non-clonal benign lesion, its presence has been reported as a risk factor for intestinal lymphoma (4). The pathogenesis of NLH is largely unknown, but several conditions have been associated with its development. These include viral (CMV, EBV, and HIV), bacterial (Helicobacter pylori, Yersinia enterocolitica), or parasitic (Giardia lamblia) infections, cow's milk protein allergy, familial Mediterranean fever (FMF), and other inborn errors of immunity (IEI) (3, 7). Among patients with IEI, NLH has been associated with common variable immune deficiency and selective IgA deficiency (2). To date, no genetically defined disease other than FMF has been shown to cause isolated NHL. We report here the case of a patient with activated PI3 kinase δ syndrome (APDS) due to a novel variant in PIK3CD presenting with isolated severe NHL.
To explore genetic causes of the dysregulation of the intestinal mucosal immune response, whole-exome sequencing was performed with an Agilent® clinical exome research kit and Illumina® sequencing technology, as previously described (8).
Human peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density gradient centrifugation, washed twice in Hank's BSS sterile solution (BioConcept, Allschwil, CH), and resuspended at 1 × 106 cells/ml with complete RPMI-1640 medium (Biochrom AG, Berlin, DE) containing 10% fetal bovine serum (FBS), 2 mM L-glutamine (Life Technologies, Carlsbad, USA), and penicillin/streptomycin antibiotics (100 U/ml, Life Technologies, Carlsbad, USA). Cells were stimulated with 1 μg/ml of anti-CD3 antibody (BD Biosciences, USA) and 100 IU/ml human IL-2 (Cell Guidance Systems, UK). Control cells were cultured without IL-2 and anti-CD3 antibody. After 3 days, stimulated T cells and control cells were washed and collected for lysis.
Proteins were extracted from activated T cells with T-PER and NaCl 5 M lysis buffer complete with protease and phosphatase inhibitor cocktail (Sigma-Aldrich, Darmstadt, DE). Protein was quantitated by Pierce BSA protein assay kit (Thermo Fisher Scientific, UK). Equal amounts of proteins (20 μg) were resolved using SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) Immobilon-p membranes (Merck-Millipore, Darmstadt, DE). Membranes were blocked with I-block™ (Thermo Fisher Scientific, Waltham, MA) for at least 1 h at room temperature and then were incubated overnight at 4°C under constant shaking with the primary antibody against p-Akt Ser 473 (Cell Signaling Technology, IT) or Akt (Cell Signaling Technology, IT). β-Actin (Sigma-Aldrich, Saint Louis, MO) was used as loading control. Membranes were next incubated with horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit antibodies (GE Healthcare, IT). All bands were visualized using ECL Select (GE Healthcare, IT), acquired with a GelDoc 2000 system (Bio-Rad, IT), and quantified by densitometry measurements using the using ImageJ software.
Fold change (FC) of pAkt/Akt was calculated in each sample as the ratio between pAkt and total Akt in stimulated cells and non-stimulated cells. Mean and SEM across three independent experiments were obtained. Multiple t-test was used to calculate the statistical difference between pAkt/Akt in stimulated (+) and control (–) cells in each sample.
The patient is the only daughter of non-consanguineous parents of Italian origin. At the age of 5 years, she had repeated episodes of hematochezia, which progressively evolved into chronic bloody mucous diarrhea lasting for over 4 weeks (9). When the girl was firstly evaluated at 5.5 years of age, physical examination was normal, and growth was regular. Her family and personal history were unremarkable, without any opportunistic or severe infection. Stool culture for bacteria and stool tests for viruses and parasites were negative. Fecal calprotectin showed repeatedly elevated results (>2,100 μg/g, normal value < 50 μg/g), while C-reactive protein and erythrocyte sedimentation rate were normal. Anti-neutrophil cytoplasmic antibodies (ANCA) and anti-Saccharomyces cerevisiae antibodies (ASCA) were negative. Sugar intestinal permeability was markedly altered (lactulose/mannitol ratio 0.09, normal value < 0.03). Upper and lower gastrointestinal digestive endoscopy showed numerous small nodules throughout the entire gastrointestinal tract from the stomach to the rectum (Figures 1A–F), whose histopathologic features were consistent with the diagnosis of NLH (Figures 1G–I). No signs of chronic intestinal inflammation or autoimmune enteropathy, such as enterocyte apoptosis, were observed in the multiple biopsies taken at endoscopy. Known infectious causes of NLH, namely, CMV, EBV, HIV, Yersinia enterocolitica, Helicobacter pylori, and Giardia lamblia infections, were excluded (3). Familial Mediterranean fever was ruled out by direct sequencing of the MEFV gene (7). A complete immunological evaluation was performed, showing substantial elevation of total IgG with normal first-line lymphocyte subset analysis and valid serological response to tetanus and diphtheria toxoids (Table 1). To exclude a cow's milk allergy (10), a trial of cow's milk protein-exclusion diet was attempted without improvement. Subsequently, the patient received a short course of steroids (oral prednisone at an initial dose of 1.5 mg/kg/day) with complete resolution of symptoms and normalization of calprotectin during treatment but rapid clinical and biochemical (i.e., calprotectin elevation) relapse upon discontinuation.
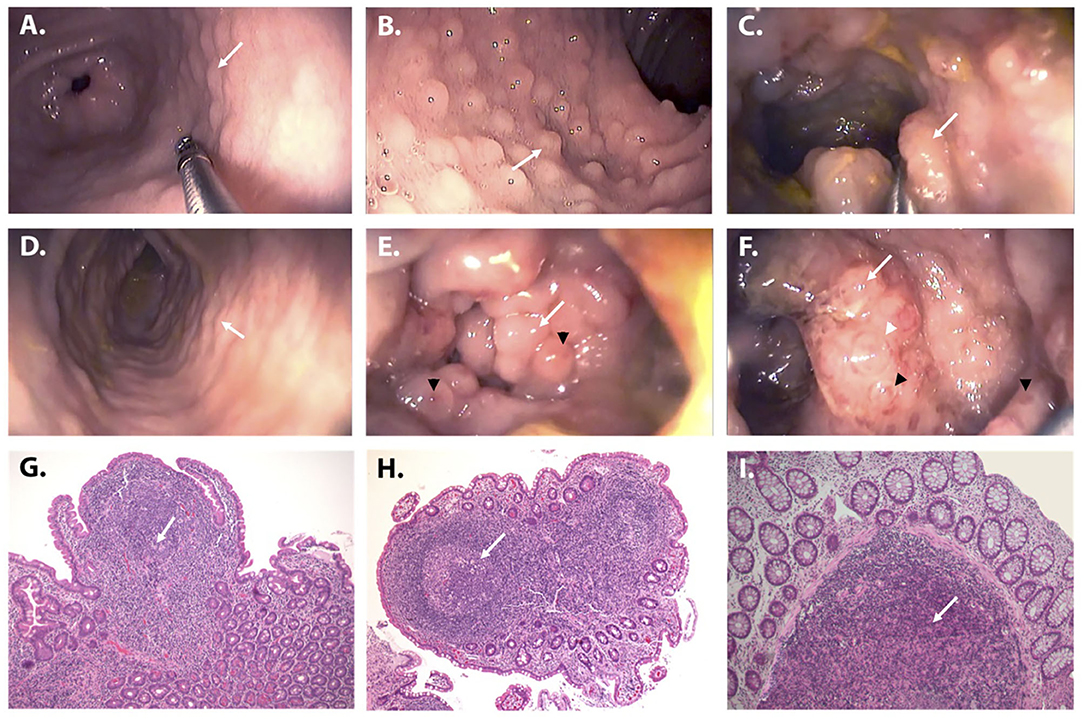
Figure 1. Endoscopic and histologic appearance of NLH in the patient. (A–F) Upper and lower gastrointestinal endoscopy demonstrating the presence of multiple nodules (from 0.2 to 1 cm in diameter) throughout the entire gastrointestinal tract (white arrows): stomach (A), duodenum (B), terminal ileum (C), ascending, transverse, and descending colon (representative image), (D) sigmoid colon (E), and rectum (F). The nodules were particularly prominent in the recto-sigmoid tract where they had a polypoid appearance, tended to obliterate the lumen, and presented a “red ring sign” around the base (white arrowhead) and mucosal ulcerations on the top (black arrowheads) (E,F). (G–I) Histologic examination of the mucosal samples collected along the gastrointestinal tract showed a diffuse hyperplasia of lymphoid follicles (white arrows), consistent with the diagnosis of NLH (H&E stain, ×10). The histologic examination (H&E, ×10) of the mucosal samples collected along the gastrointestinal tract showed a diffuse hyperplasia of lymphoid follicles (white arrows), without inflammation, granulomas, or other histological lesions.
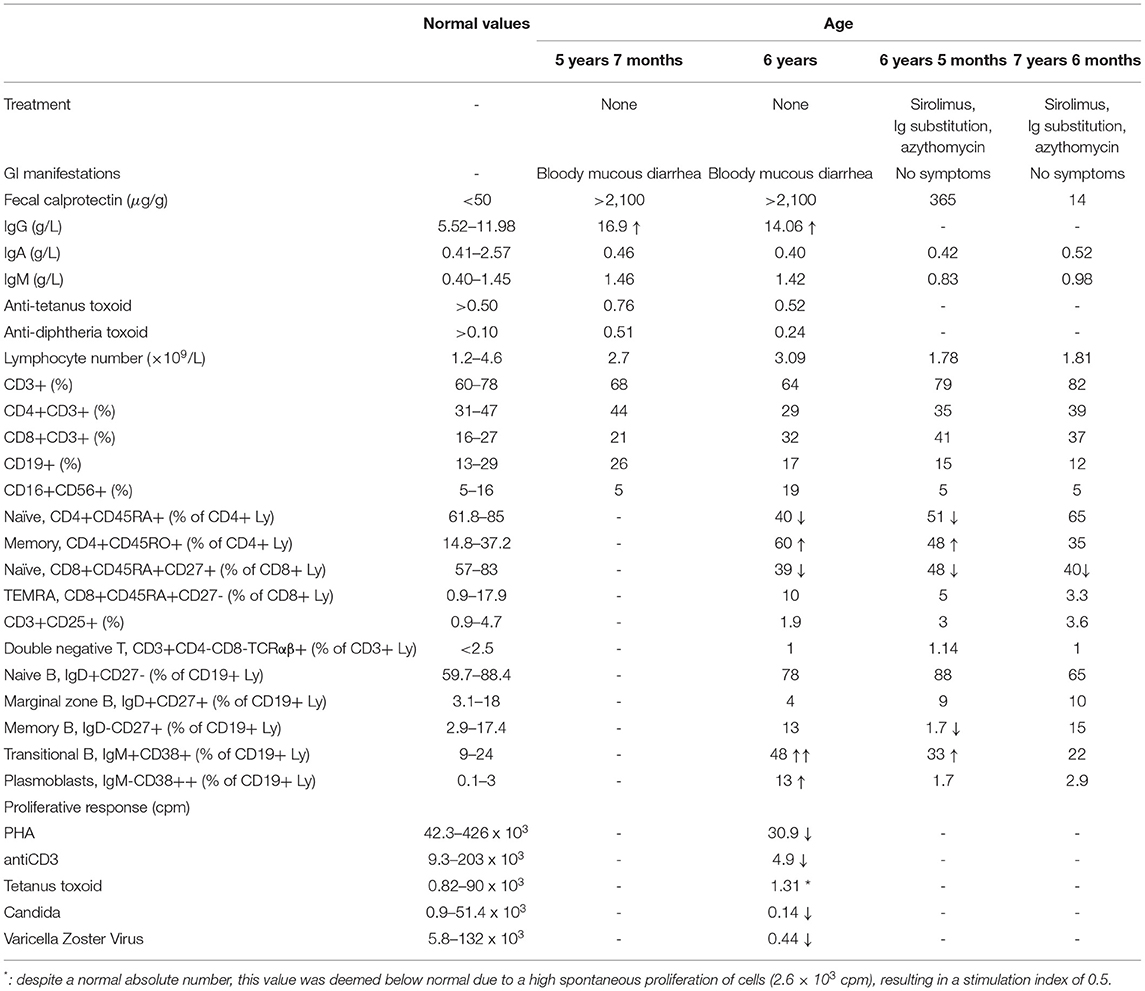
Table 1. Evolution of clinical symptoms, fecal calprotectin, and immunological parameters over time.
The observation of severe pan-enteric NLH in a young child together with the exclusion of intestinal infections and the presence of chronic-recurring symptoms elicited the diagnostic suspicion of an inherited immunological defect despite normal first-line tests. Given the absence of a guiding-diagnostic element, a high-throughput genetic test was performed to explore genetic causes of immune dysregulation and immune deficiency. Whole-exome sequencing with phenotype-driven analysis was performed, focusing on genes primarily implicated in immunological diseases (11, 12), revealing the presence of the heterozygous variant p.Glu525Gly (c.1574A>G) in the PIK3CD gene. Sanger sequencing confirmed the presence of this variant, and segregation analysis demonstrated its de novo occurrence (Figures 2A,B). The variation affects the same codon of two previously reported PIK3CD variants causing APDS, namely, c.1573G>A, p.(Glu525Lys) and c.1573A>C, p.(Glu525Ala) (Figure 2C) (13, 14). It has not been identified previously, and it is absent from the largest allele frequency databases (gnomAD, EVS, and 1000 Genomes Project). PolyPhen-2, SIFT, CADD-Phred, and MutationTaster prediction algorithms indicate with high confidence a deleterious effect on the resulting protein. Conservation tools, such as phyloP, GERP, and PhastCons, indicate that the DNA region harboring the variant is highly evolutionary conserved.
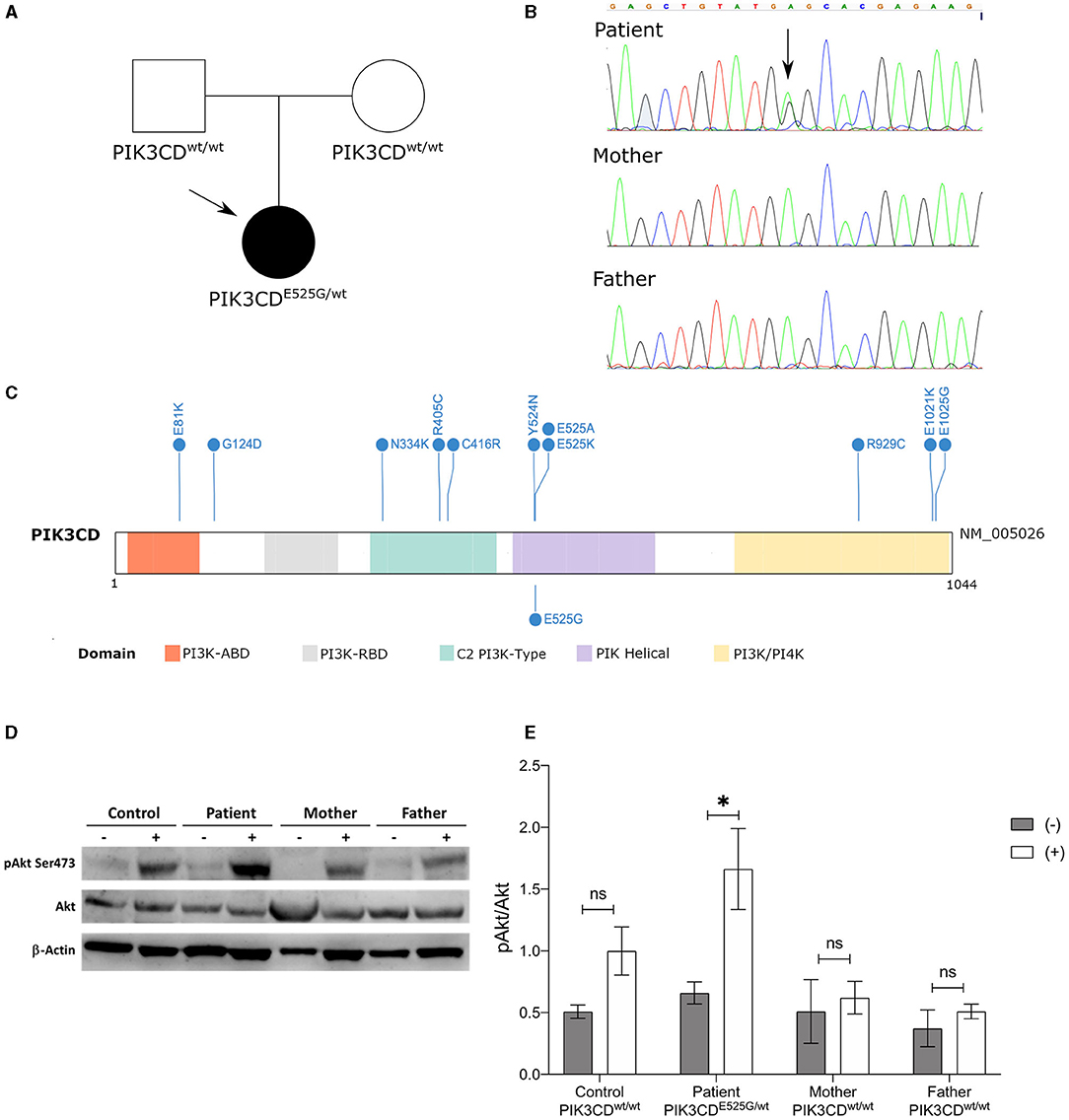
Figure 2. Genetic and functional characterization. (A) Pedigree of the family indicating the de novo occurrence of the variant. (B) Sanger electropherograms. (C) PIK3CD protein domain plot showing known activating variants (above) (15) and the variant described here (below). (D) Immunoblot of stimulated (+) and non-stimulated (–) T cells derived from PBMCs collected from the patient, her mother and father, and a normal control (CTRL). Expression of Akt and p-Akt (S473) is shown. β-Actin was employed as loading control. (E) Fold change of phosphorylation of p-Akt (S473)/Akt in activated T cells (+) and non-activated cells (–) from the patient, parents, and healthy control. Bars are represented as mean ± SEM. *p < 0.05; ns: not significant.
To confirm the pathogenic role of the p.Glu525Gly variant, extended immunological phenotyping was performed along with in vitro functional tests. Detailed immune subset analysis demonstrated reduced CD4+CD45RA+ and CD8+CD45RA+CD27+ naïve T helper cells and elevated CD19+IgM+CD38+ transitional B cells (Table 1). T-cell proliferation was impaired upon stimulation with mitogens (PHA and anti-CD3), tetanus toxoid, Candida, and varicella zoster virus antigens. Given that PI3Kδ-activating mutations responsible for APDS induce an over activation of the Akt/mTOR pathway (13, 16), the quantity and the activation of AKT (pAkt-Ser473) were tested in the patient, in her parents (none of them carrying the p.Glu525Gly variant), and in a healthy control. After PBMC stimulation with anti-CD3 antibody and IL-2, significant hyperphosphorylation in the S473 residue of the AKT was observed in the patient as compared to her parents and the healthy control (Figures 2D,E), proving that the p.Glu525Gly variant behaves as a gain-of-function and causes PI3Kδ hyperactivation (13). Thus, the girl received a diagnosis of APDS.
In light of this, notwithstanding the absence of respiratory symptoms, a chest CT scan was performed, showing multiple small nodules in the lung parenchyma and mediastinal lymphadenopathies. Treatment with sirolimus (initial dose 2 mg/m2 aiming at levels of 4–12 ng/ml, with an optimal target of 9 ng/ml) was then initiated, leading to the gradual resolution of gastrointestinal symptoms and to the normalization of fecal calprotectin in one year (Table 1). Due to the occurrence of few episodes of respiratory infection, including one episode of pneumonia and the notion of a disturbed immunoglobulin efficacy despite normal levels in APDS patients (17), immunoglobulin substitution and azithromycin prophylaxis (10 mg/kg/day, 3 days/week) were started with prompt interruption of recurrent infections. At the last evaluation, the patient was 7.5 years old and asymptomatic. During the 18 months of treatment, she did not experience any clinically relevant infection or autoimmune disorder. Moreover, the numbers of naïve T cells and transitional B cells returned to normal values (Table 1). At the age of 7.5 years, the patient was diagnosed with childhood absence epilepsy, which was deemed unrelated to APDS, based on the absence of an association of the two disorders in the literature. Brain magnetic resonance imaging at the onset of epilepsy was unremarkable.
APDS (OMIM # 615513) is an inborn error of immunity caused by autosomal dominant gain-of-function variants in the PIK3CD or PIK3R1 genes, which encode for the catalytic and regulatory subunits of the phosphoinositide 3-kinase δ complex (13, 16, 18). The variants underlying APDS cause hyperactivation of the PI3K/AKT/mTOR/S6 kinase signaling cascade, which controls cell growth, proliferation, and metabolism (19, 20). Since PIK3CD and PIK3R1 are mainly expressed in lymphocytes, the most prominent physiological alterations of this condition are found in the immune system. APDS causes enhanced T-cell senescence with reduced naïve T cells and impaired B-cell responses (21). Follicular helper T cells (Tfh) are increased, resulting in the expansion of germinal centers in peripheral lymphoid organs (22). These immunological mechanisms underlie the clinical phenotype of patients with APDS, characterized by recurrent bacterial and viral infections, autoimmune disorders, severe lymphadenopathy, and increased risk of lymphoma, similar to other IEIs associated with immune dysregulation (23, 24). Gastrointestinal manifestations are reported in up to 50% of patients and include a broad spectrum of disorders (colitis, autoimmune enteropathy, and NLH) (17, 25). However, gastrointestinal involvement is typically not isolated and usually occurs later in the course of the disease (15).
APDS is usually suspected when patients develop recurrent pulmonary infections, lymphadenopathy, or hepatosplenomegaly in their first decade of life, and not in patients with isolated NLH. The diagnosis of APDS can be challenging, since first-level immunological tests (lymphocyte count, immunoglobulin levels, and basic lymphocyte subsets) can be normal, and gastroenterologists may have limited knowledge of recently identified IEIs. The diagnosis of APDS is further hurdled by the need to functionally prove the gain-of-function effect of novel variants in PIK3CD since it cannot be predicted by computational methods alone. Indeed, each novel variant in PIK3CD requires a specific functional validation to confirm its pathogenicity (26). Our report further extends the list of variants associated with this disorder and may be valuable for centers without access to functional testing with a rapid turnaround time.
The recognition of APDS as a cause of NLH had important consequences on the management of our patient. First, it allowed the establishment of targeted therapy for APDS (i.e., sirolimus), which, even if not usually employed for the treatment of NLH, led to the complete resolution of gastrointestinal symptoms (25). Second, it prompted us to investigate and detect the extraintestinal complications of the disease (i.e., subclinical pulmonary involvement). Third, it allowed the initiation of immunoglobulin substitution and azithromycin administration to prevent respiratory infections and bronchiectasis, which are a frequent complication of APDS (17). The diagnosis of APDS will also provide a future benefit for the patient as it will expand the therapeutic options when novel specific treatments for APDS (e.g., specific PI3K inhibitors such as leniolisib or seletalisib) become available (27, 28).
In conclusion, the presence of isolated NLH, especially if severe, widespread through the gastrointestinal tract or pan-enteric, should prompt the suspicion of an inborn error of immunity such as APDS, even in the absence of other clinical signs of immune deficiency and with normal first-line immunological testing. In such circumstances, broad immunological investigations, including multigene panel analysis, should be performed to warrant the prompt institution of a targeted potentially life-saving treatment.
The data presented in the study are deposited in the ClinVar repository, accession number SCV001810147.1
Ethical review and approval was not required for the study on human participants in accordance with local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants or their legal guardian/next of kin. Written informed consent was obtained from the participants or their legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
AM, PG, LB, LR, FB, AB, GP, and MC provided direct clinical care for the patient. SB and AC performed functional testing on the PIK3CD variant. DC and AL performed whole exome sequencing and variant interpretation. CM performed histopathological examination. AM, SB, and MC wrote the manuscript. All authors reviewed and approved the manuscript.
The work was funded by a grant from Fondazione Città della Speranza ONLUS (http://cittadellasperanza.org/), Associazione di Promozione Sociale Genitori in fuga (https://www.genitoriinfuga.org/), and Associazione sportiva dilettantistica NCO Crew to AM and SB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors acknowledge Maria Elena Maccari (Freiburg, Germany) and Sven Kracker (Paris, France) for their assistance in the pAkt testing. We thank Istituto di Ricerca Pediatrica for supporting our scientific work.
1. Sipos F, Muzes G. Isolated lymphoid follicles in colon: switch points between inflammation and colorectal cancer? World J Gastroenterol. (2011) 17:1666–73. doi: 10.3748/wjg.v17.i13.1666
2. Albuquerque A. Nodular lymphoid hyperplasia in the gastrointestinal tract in adult patients: A review. World J Gastrointest Endosc. (2014) 6:534–40. doi: 10.4253/wjge.v6.i11.534
3. Mansueto P, Iacono G, Seidita A, D'Alcamo A, Sprini D, Carroccio A. Review article: intestinal lymphoid nodular hyperplasia in children–the relationship to food hypersensitivity. Aliment Pharmacol Ther. (2012) 35:1000–9. doi: 10.1111/j.1365-2036.2012.05062.x
4. Elkholy S, Mogawer S, Farag A. Nodular lymphoid hyperplasia of the gastrointestinal tract : a comprehensive review. Acta Gastroenterol Belg. (2017) 80:405–410.
5. Colón AR, DiPalma JS, Leftridge CA. Intestinal lymphonodular hyperplasia of childhood: patterns of presentation. J Clin Gastroenterol. (1991) 13:163–6. doi: 10.1097/00004836-199104000-00009
6. Piscaglia AC, Laterza L, Cesario V, Gerardi V, Landi R, Lopetuso LR, et al. Nodular lymphoid hyperplasia: A marker of low-grade inflammation in irritable bowel syndrome? World J Gastroenterol. (2016) 22:10198–209. doi: 10.3748/wjg.v22.i46.10198
7. Gurkan OE, Yilmaz G, Aksu AU, Demirtas Z, Akyol G, Dalgic B. Colonic lymphoid nodular hyperplasia in childhood: causes of familial Mediterranean fever need extra attention. J Pediatr Gastroenterol Nutr. (2013) 57:817–21. doi: 10.1097/MPG.0b013e3182a9083b
8. Marzollo A, Colavito D, Sartori S, Fanelli GN, Putti MC. Cerebral Lymphoproliferation in a Patient with Kabuki Syndrome. J Clin Immunol. (2018) 38:475–7. doi: 10.1007/s10875-018-0516-9
9. Shankar S, Rosenbaum J. Chronic diarrhoea in children: A practical algorithm-based approach. J Paediatr Child Health. (2020) 56:1029–38. doi: 10.1111/jpc.14986
10. Iacono G, Ravelli A, Di Prima L, Scalici C, Bolognini S, Chiappa S, et al. Colonic lymphoid nodular hyperplasia in children: relationship to food hypersensitivity. Clin Gastroenterol Hepatol. (2007) 5:361–6. doi: 10.1016/j.cgh.2006.12.010
11. Smedley D, Robinson PN. Phenotype-driven strategies for exome prioritization of human Mendelian disease genes. Genome Med. (2015) 7:81. doi: 10.1186/s13073-015-0199-2
12. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2020) 18:20. doi: 10.1007/s10875-019-00737-x
13. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol. (2014) 15:88–97. doi: 10.1038/ni.2771
14. Tsujita Y, Mitsui-Sekinaka K, Imai K, Yeh TW, Mitsuiki N, Asano T, Ohnishi H, Kato Z, Sekinaka Y, Zaha K, et al. Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase δ syndrome–like immunodeficiency. J Allergy Clin Immunol. (2016) 138:1672–80.e10. doi: 10.1016/j.jaci.2016.03.055
15. Jamee M, Moniri S, Zaki-Dizaji M, Olbrich P, Yazdani R, Jadidi-Niaragh F, et al. Clinical, immunological, and genetic features in patients with activated PI3Kδ syndrome (APDS): a systematic review. Clin Rev Allergy Immunol. (2020) 59:323–33. doi: 10.1007/s12016-019-08738-9
16. Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase gene mutation predisposes to respiratory infection and airway damage. Science (80-). (2013) 342:866–71. doi: 10.1126/science.1243292
17. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, Goodlad JR, Farmer G, Steele CL, Leahy TR, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: A large patient cohort study. J Allergy Clin Immunol. (2017) 139:597–606.e4. doi: 10.1016/j.jaci.2016.06.021
18. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med. (2014) 211:2537–47. doi: 10.1084/jem.20141759
19. Marzollo A, Maestrini G, La Starza R, Elia L, Malfona F, Pierini T, Tretti C, Coppe A, Bortoluzzi S, Biffi A, et al. A novel germline variant in PIK3R1 results in SHORT syndrome associated with TAL/LMO T-cell Acute Lymphoblastic Leukaemia. Am J Hematol. (2020) 95:E335–8. doi: 10.1002/ajh.25998
20. Edwards ESJ, Bier J, Cole TS, Wong M, Hsu P, Berglund LJ, Boztug K, Lau A, Gostick E, Price DA, et al. Activating PIK3CD mutations impair human cytotoxic lymphocyte differentiation and function and EBV immunity. J Allergy Clin Immunol. (2019) 143:276–291.e6. doi: 10.1016/j.jaci.2018.04.030
21. Asano T, Okada S, Tsumura M, Yeh T-W, Mitsui-Sekinaka K, Tsujita Y, et al. Enhanced AKT phosphorylation of circulating B cells in patients with activated PI3Kδ syndrome. Front Immunol. (2018) 9:568. doi: 10.3389/fimmu.2018.00568
22. Thauland TJ, Pellerin L, Ohgami RS, Bacchetta R, Butte MJ. Case study: mechanism for increased follicular helper T cell development in activated PI3K delta syndrome. Front Immunol. (2019) 10:753. doi: 10.3389/fimmu.2019.00753
23. Tesch VK, Abolhassani H, Shadur B, Zobel J, Mareika Y, Sharapova S, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol. (2020) 145:1452–63. doi: 10.1016/j.jaci.2019.12.896
24. Bosa L, Batura V, Colavito D, Fiedler K, Gaio P, Guo C, et al. Novel CARMIL2 loss-of-function variants are associated with pediatric inflammatory bowel disease. Sci Rep. (2021) 11:1–13. doi: 10.1038/s41598-021-85399-9
25. Maccari ME, Abolhassani H, Aghamohammadi A, Aiuti A, Aleinikova O, Bangs C, et al. Disease evolution and response to rapamycin in activated phosphoinositide 3-kinase δ syndrome: the european society for immunodeficiencies-activated phosphoinositide 3-kinase δ syndrome registry. Front Immunol. (2018) 9:543. doi: 10.3389/fimmu.2018.00543
26. Boisson B, Quartier P, Casanova J-L. Immunological loss-of-function due to genetic gain-of-function in humans: autosomal dominance of the third kind. Curr Opin Immunol. (2015) 32:90–105. doi: 10.1016/j.coi.2015.01.005
27. Rao VK, Webster S, Dalm VASH, Šedivá A, Van Hagen PM, Holland S, et al. Effective “activated PI3Kδ syndrome” –targeted therapy with the PI3Kδ inhibitor leniolisib. Blood. (2017) 130:2307–16. doi: 10.1182/blood-2017-08-801191
Keywords: inborn error of immunity, nodular lymphoid hyperplasia-GIT, activated PI3K-delta syndrome, novel variant, sirolimus, PIK3CD
Citation: Marzollo A, Bresolin S, Colavito D, Cani A, Gaio P, Bosa L, Mescoli C, Rossini L, Barzaghi F, Perilongo G, Leon A, Biffi A and Cananzi M (2021) Case Report: Intestinal Nodular Lymphoid Hyperplasia as First Manifestation of Activated PI3Kδ Syndrome Due to a Novel PIK3CD Variant. Front. Pediatr. 9:703056. doi: 10.3389/fped.2021.703056
Received: 30 April 2021; Accepted: 01 September 2021;
Published: 06 October 2021.
Edited by:
Caterina Cancrini, University of Rome Tor Vergata, ItalyReviewed by:
Vassilios Lougaris, University of Brescia, ItalyCopyright © 2021 Marzollo, Bresolin, Colavito, Cani, Gaio, Bosa, Mescoli, Rossini, Barzaghi, Perilongo, Leon, Biffi and Cananzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Marzollo, YW50b25pby5tYXJ6b2xsb0B1bmlwZC5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.